Survey of Pharmacological Activity and Pharmacokinetics of Selected ?-Adrenergic Blockers in Regard to Their Stereochemistry

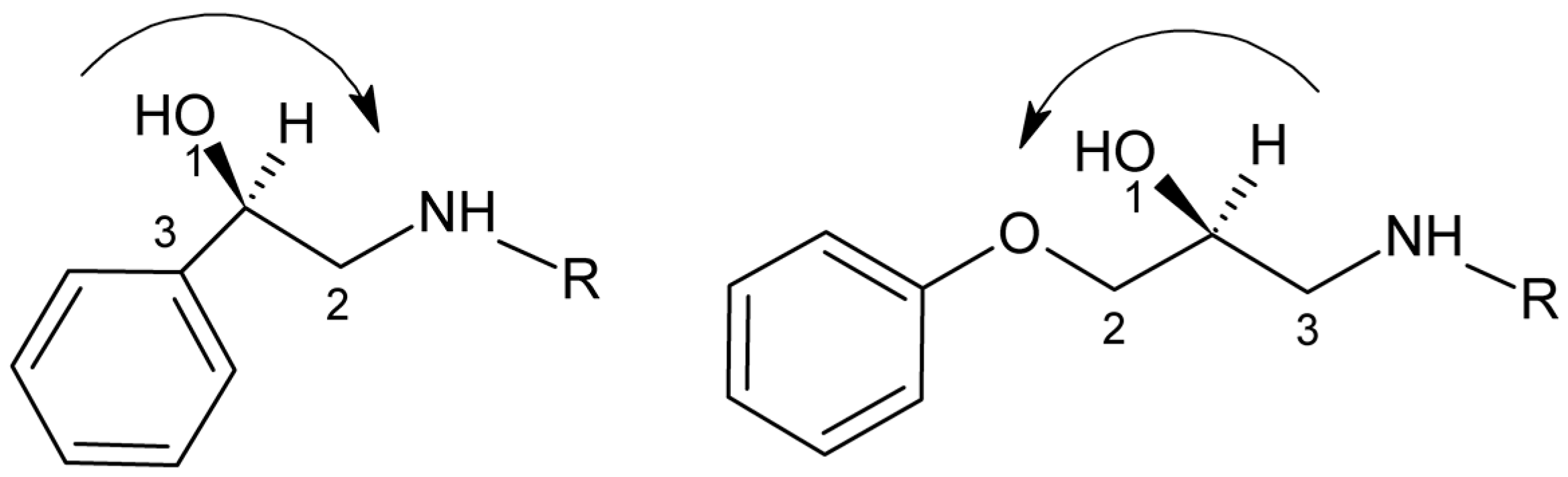{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
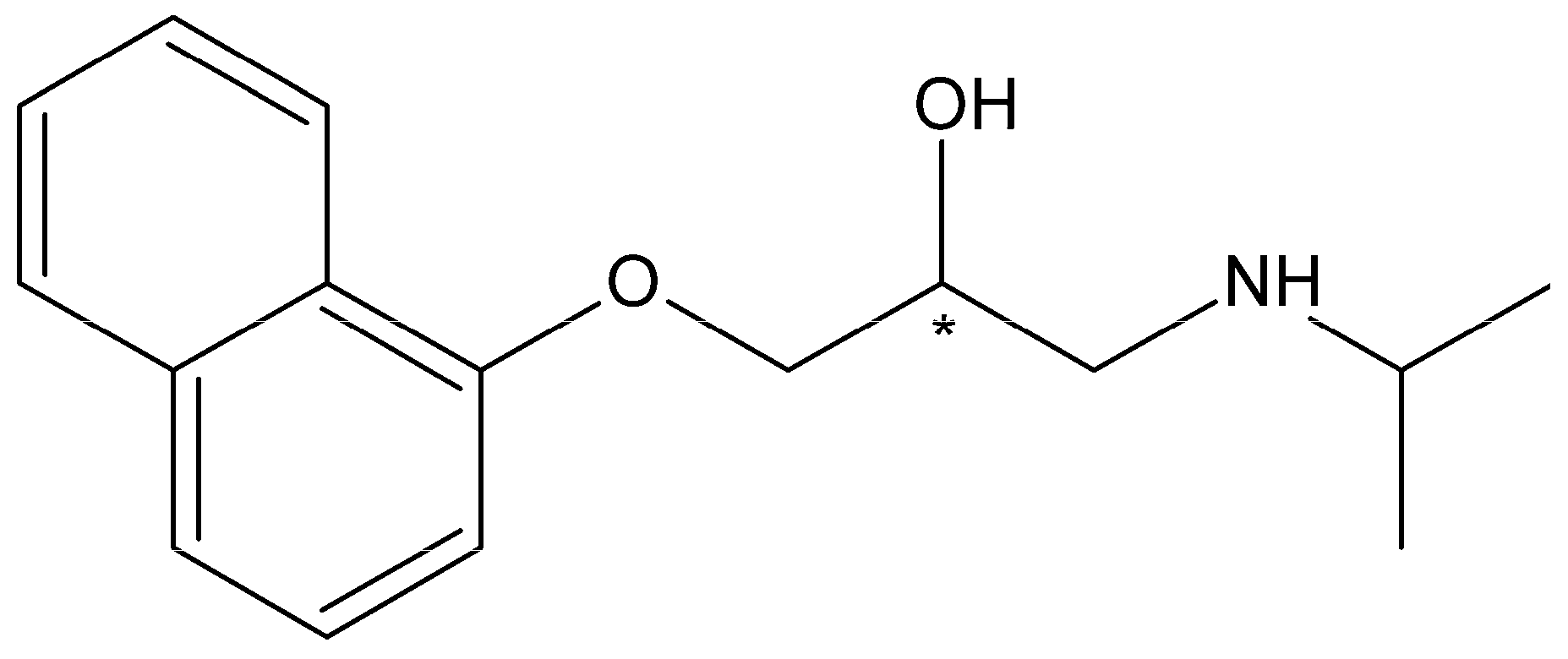{kind=link}
{kind=link}
{kind=link}
{kind=link}
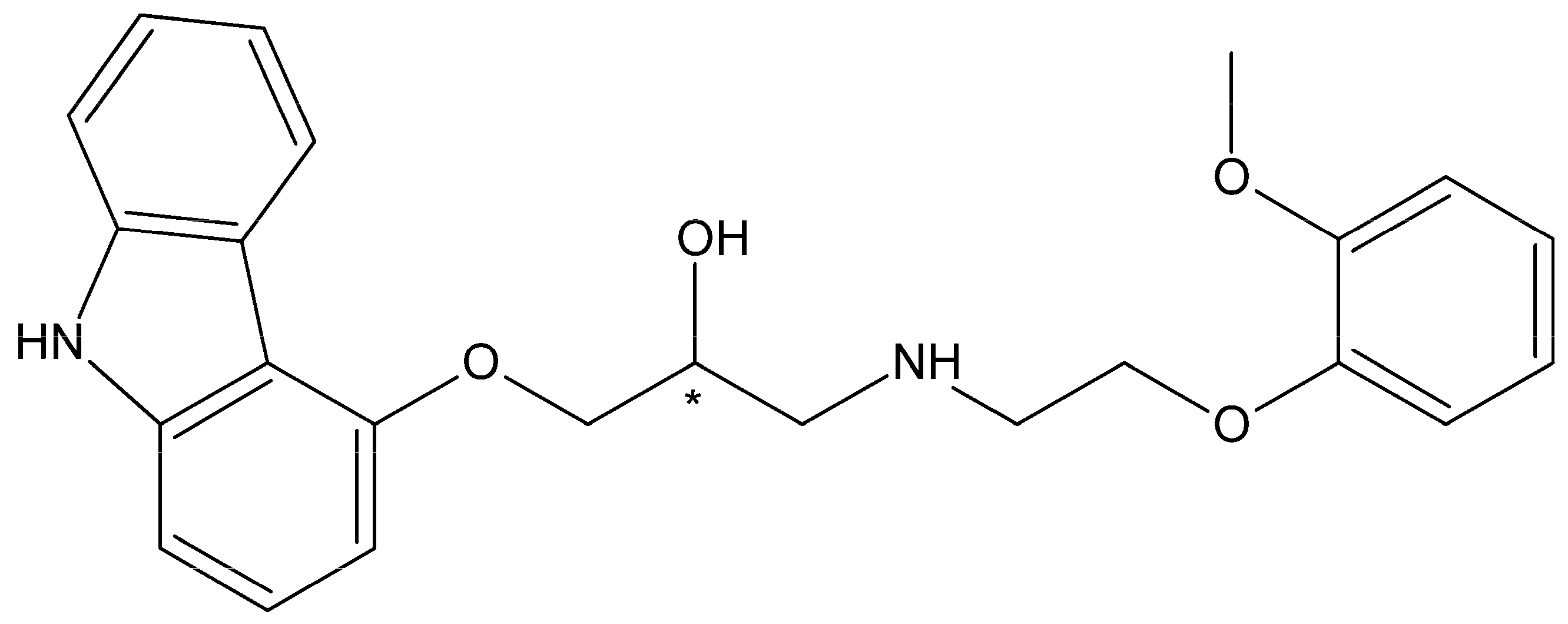{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- A.
- Cardioselective β-adrenolytics without ISA.
- B.
- Cardioselective β-adrenolytics with ISA.
- C.
- Non-selective β-adrenolytics without ISA.
- D.
- Non-selective β-adrenolytics with ISA.
- E.
- β-adrenolytics blocking simultaneously α- and β-receptors.
- F.
- Lipophilic β-adrenolytics.
- G.
- Hydrophilic β-adrenolytics.
2. Chirality of β-Blockers
2.1. Interaction of β-Blockers With the Receptor Protein
2.2. The Arylaminoethanol Group of β-Blockers
2.2.1. Pronethalol
2.2.2. Sotalol
2.2.3. Labetalol
2.2.4. Bufuranol
2.3. The Aryloxyaminopropanol Group of β-Blockers
2.3.1. Monocyclic Compounds
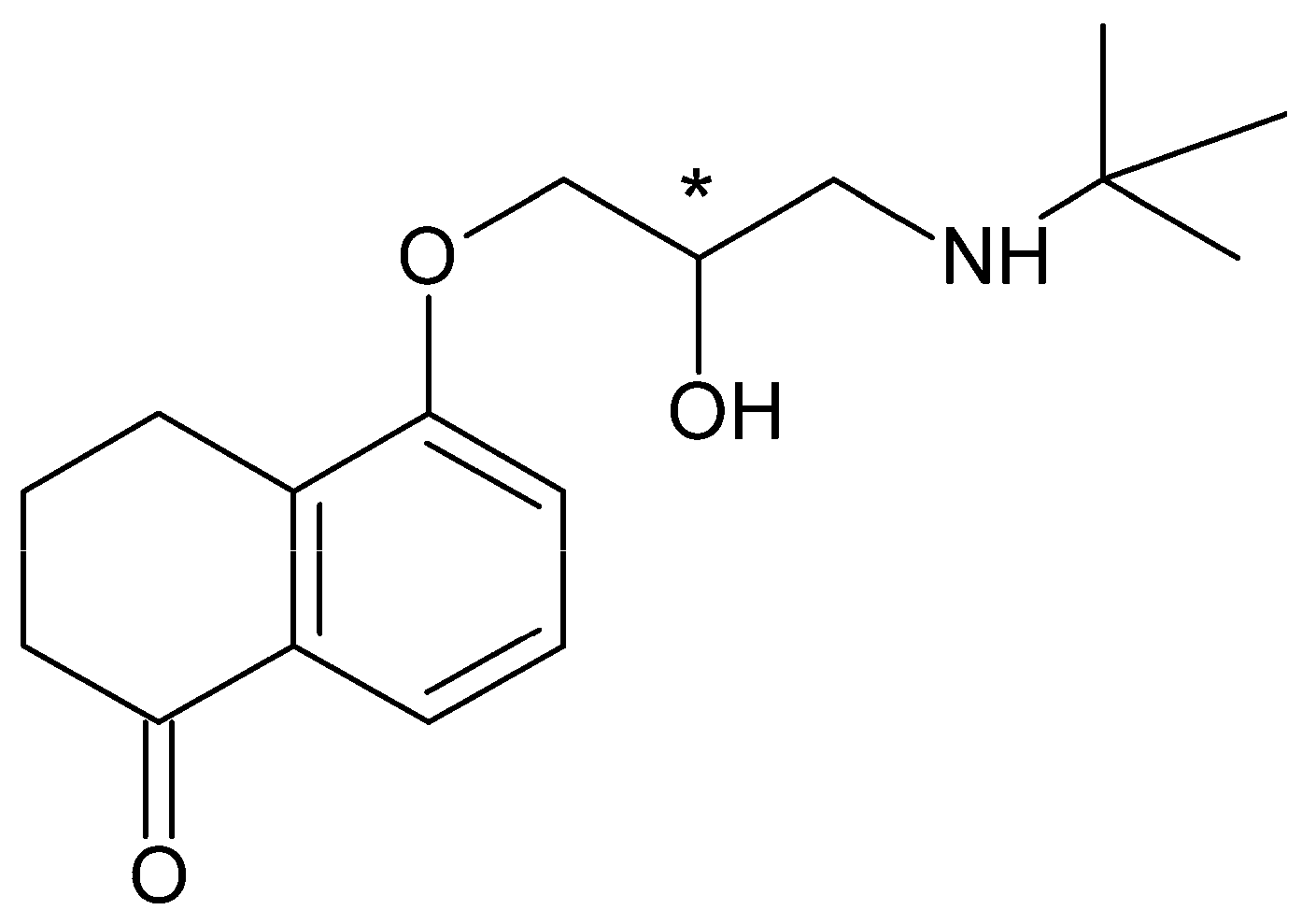
Betaxolol
Esmolol
2.3.2. Monocyclic Derivatives with Several Substituents
2.3.3. Bicyclic Derivatives
Propranolol
Nadolol
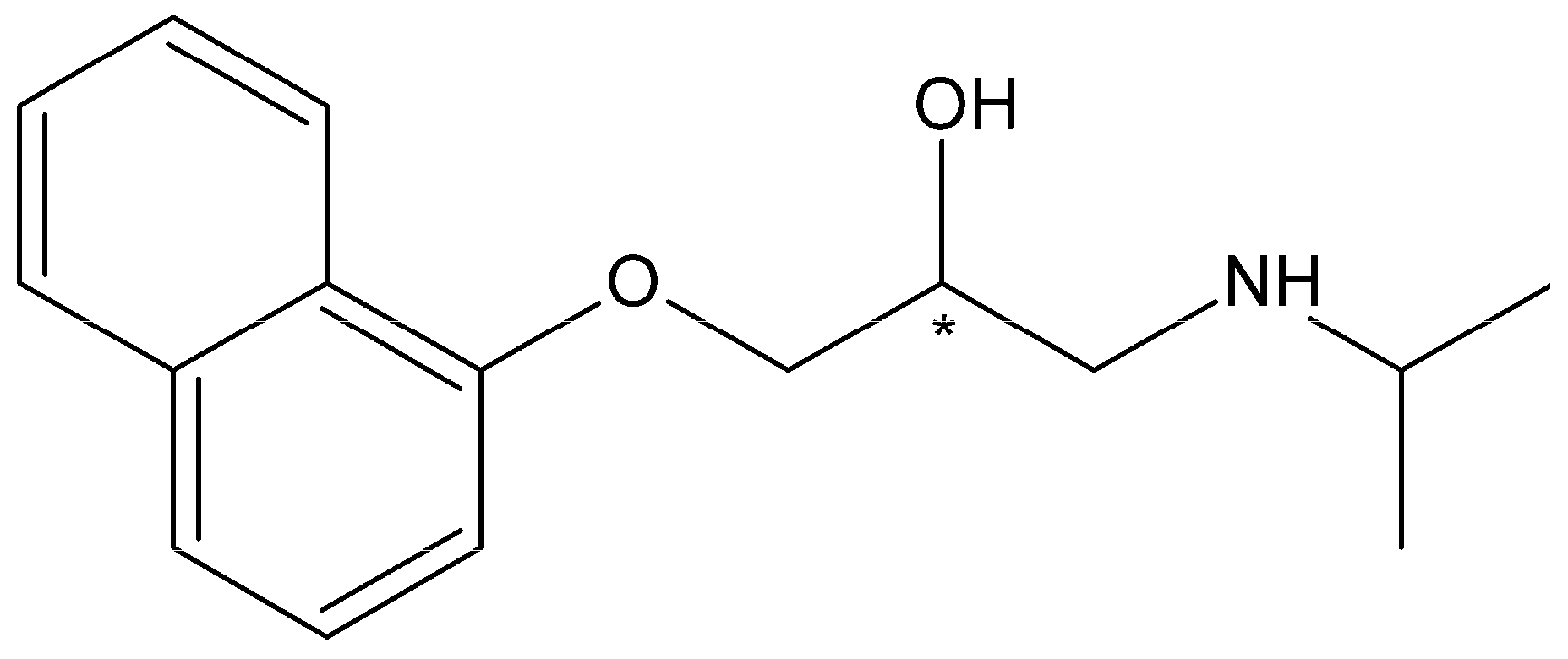
Bunolol
2.3.4. Heterocyclic Derivatives
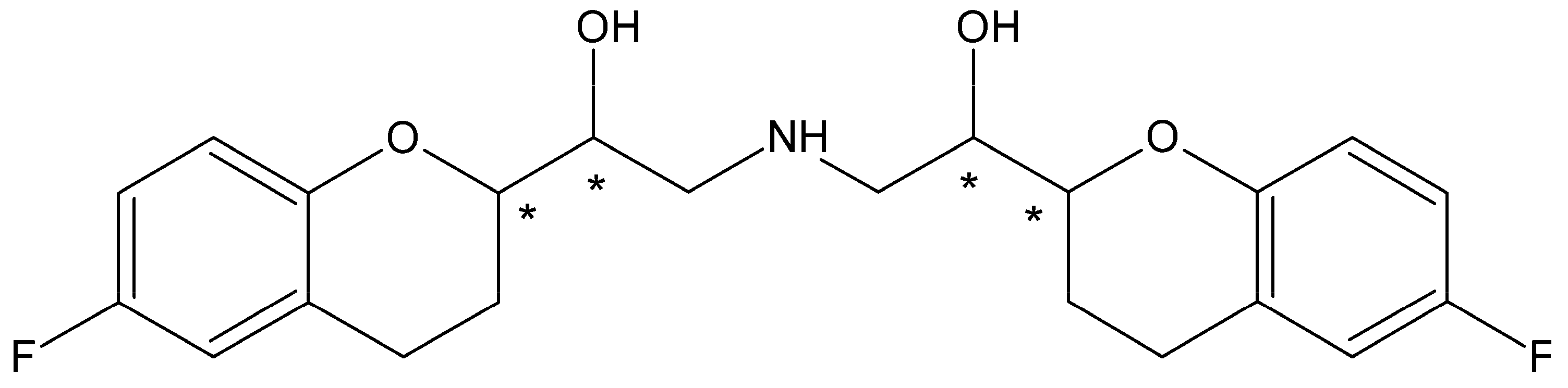
Timolol
Carvedilol
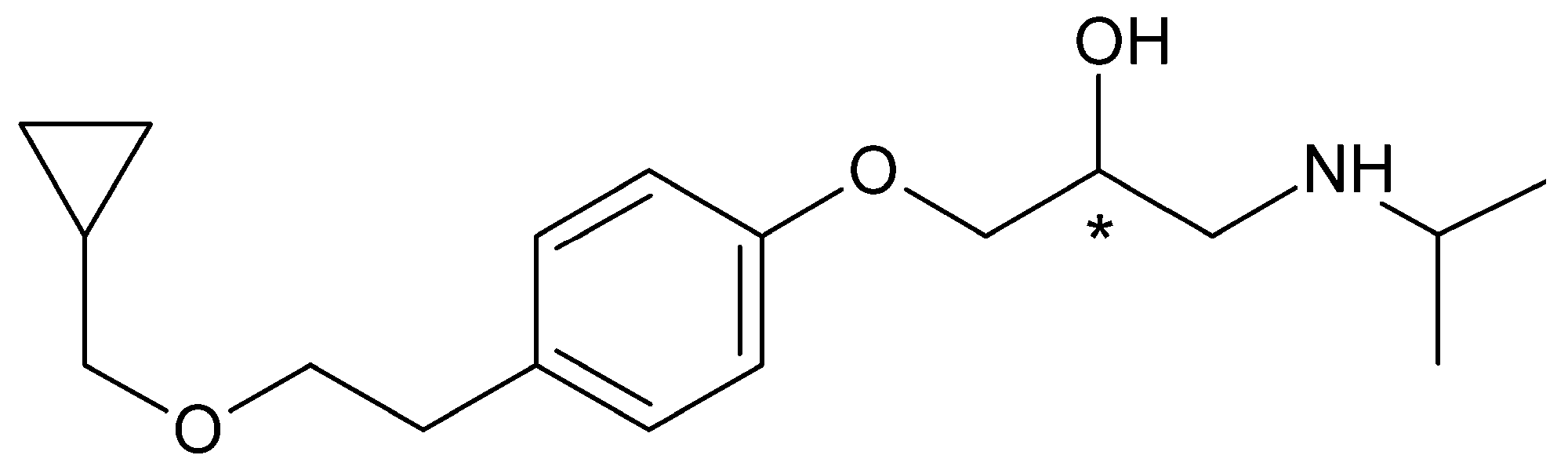
Nebivolol
3. Pharmacokinetics of Chiral β-Blockers
3.1. Resorption
3.2. Distribution
3.3. Metabolism
3.4. Renal Excretion
4. Stereoselective Analytical Methods in the Study of Pharmacokinetics of β-Blockers
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lechat, P.; Volta, S.D.; Cannon, C.P. Clinical pharmacology of beta-blockers in cardiology trial results and clinical application. Hot Top. Cardiol. 2008, 10, 7–44. [Google Scholar] [CrossRef]
- Saxena, R.; Prakash, J.; Mathur, P.; Gupta, S.K. Pharmacotherapy of glaucoma. Ind. J. Pharmacol. 2002, 34, 71–85. [Google Scholar]
- Sharma, R.; Shastri, N.; Sadhotra, P. β-Blockers as glaucoma therapy. JK Sci. 2007, 9, 42–45. [Google Scholar]
- Dooley, T.P. Treating anxiety with either beta blockers or antiemetic antimuscarinic drugs: A review. Ment. Health Fam. Med. 2015, 11, 89–99. [Google Scholar] [CrossRef]
- Feely, J.; Peden, N. Use of beta-adrenoceptor blocking drugs in hyperthyroidism. Drugs 1984, 27, 425–446. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Castrillón, J.L.; De Luis, D.A.; Duenas-Laita, A. Are beta-blockers useful in the prevencion of osteoporotic fractures? Eur. Rev. Med. Pharmacol. Sci. 2009, 13, 157–162. [Google Scholar] [PubMed]
- Migliazzo, C.V.; Hagan, J.C. Beta blocker eye drops for treatment of acute migraine. Mo. Med. 2014, 111, 283–288. [Google Scholar]
- Rehsia, N.S.; Dhalla, N.S. Mechanisms of the beneficial effects of beta-adrenoceptor antagonists in congestive heart failure. Exp. Clin. Cardiol. 2010, 15, e86–e95. [Google Scholar] [PubMed]
- Weber, M.A. The role of the new beta-blockers in treating cardiovascular disease. Am. J. Hypertens. 2005, 18, 169S–176S. [Google Scholar] [CrossRef]
- Emmirati, E.; Contri, R.; Coppini, R.; Ceachi, F.; Frigerio, M.; Olivotto, I. Pharmacological treatment of hypertrophic cardiomyopathy: Current practice and novel perspectives. Eur. J. Heart Fail. 2016, 18, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Tobe, S.W. β-adrenergic receptor blockers in hypertension. Can. J. Cardiol. 2014, 30, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, P.; Tobe, S.W.; Lacourcière, Y. β-blockers in hypertension: Studies and meta-analyses over the years. Can. J. Cardiol. 2014, 30, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Weir, M.R. Beta-blockers in the treatment of hypertension: Are there clinically relevant differences? Postgrad. Med. 2009, 121, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, J.M. Beta-blockers and heart failure. Indian Heart J. 2010, 62, 101–110. [Google Scholar] [PubMed]
- Bruchatá, K.; Čižmáriková, R. New derivatives of aryloxyaminopropanol—Structure biological activity relationship (slovak). Farm. Obzor 2010, 79, 237–293. [Google Scholar]
- Poirier, L.; Tobe, S.W. Contemporary use of β-blockers: Clinical relevance of subclassification. Can. J. Cardiol. 2014, 30 (Suppl. 5), S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Cahn, R.S.; Ingold, C.K.; Prelog, V. Specification of molecular chirality. Angew. Chem. Int. Ed. 1966, 5, 385–415. [Google Scholar] [CrossRef]
- Prelog, V.; Helmchen, G. Basic principles of the CIP-system and proposals for a revision. Angew. Chem. Int. Ed. 1982, 21, 567–583. [Google Scholar] [CrossRef]
- Ariëns, E.J. Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology. Eur. J. Clin. Pharmacol. 1984, 26, 663–668. [Google Scholar] [CrossRef]
- Lehmann, F.P.A.; Rodriques de Miranda, J.F.; Ariëns, E.J. Stereoselectivity and affinity in molecular pharmacology. III. Structural aspects in the mode of action of natural and synthetic auxins. Chem. Biol. Interact. 1978, 20, 101–142. [Google Scholar] [CrossRef]
- Ariëns, E.J. Racemic therapeutics - ethical and regulatory aspects. Eur. J. Clin. Pharmacol. 1991, 41, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Čižmáriková, R. β-Adrenergic receptor blockers—The group of chiral drugs: Different effects of individual enantiomers. Čes. Slov. Farm. 2002, 51, 121–128. [Google Scholar]
- Agustian, J.; Kamaruddin, A.H.; Bhatia, S. Enantiomeric β-blockers—The existing technologies. Process Biochem. 2010, 45, 1587–1604. [Google Scholar] [CrossRef]
- Nagatomo, T.; Koike, K. Recent advance in structure, binding sites with ligands and pharmacological function of β-adrenoceptors obtained by molecular biology and molecular modeling. Life Sci. 2000, 66, 2419–2426. [Google Scholar] [CrossRef]
- Glover, W.E.; Greenfield, A.D.M.; Shanks, R.G. Effect of dichloroisoprenaline on the peripheral vascular responses to adrenaline in man. Br. J. Pharmacol. Chemother. 1962, 19, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Almirante, L.; Murmann, W. Relationship between configuration and adrenergic β-receptor blocking activity of optical isomers of 1-(4-nitrophenyl)-2-isopropylaminoethanol (INPEA). J. Med. Chem. 1966, 9, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Petrongolo, C.; Tomasi, J.; Macchia, B.; Macchia, M. Molecular orbital studies on the mechanism of drug-receptor interaction. 1. Adrenergic drugs. Conformation and reactivity of isoproterenol and 1-(p-nitrophenyl)-2-isopropylaminoethanol. J. Med. Chem. 1974, 17, 501–507. [Google Scholar] [CrossRef]
- Black, J.W.; Stephenson, J.S. Pharmacology of a new adrenergic beta-receptor-blocking compound (Nethalide). Lancet 1962, 280, 311–314. [Google Scholar] [CrossRef]
- Howe, R.; Rao, B.S. Beta-adrenergic blocking agents. III. The optical isomers of pronethalol, propranolol, and several related compounds. J. Med. Chem. 1968, 11, 1118–1121. [Google Scholar] [CrossRef]
- Lucchesi, B.R. The effects of pronethalol and its dextro isomer upon experimental cardiac arrhythmias. J. Pharmac. Exp. Ther. 1965, 148, 94–99. [Google Scholar]
- Paget, G.E. Carcinogenic action of pronethalol. Br. J. Pharmacol. 1963, 2, 1266–1267. [Google Scholar] [CrossRef]
- Cho, B.T.; Kang, S.K.; Yang, W.K. Convenient synthesis of enantiopure β-adrenergic blockers: (R)-nifenalol, (R)-denopamine, (R)-dichloroisoproterenol and (R)-pronethalol. Bull. Korean Chem. Soc. 2002, 23, 1328–1330. [Google Scholar] [CrossRef]
- Saddique, F.A.; Zahoor, A.F.; Yousaf, M.; Irfan, M.; Ahmad, M.; Mansha, A.; Khan, Z.A.; Naqvi, S.A.R. Synthetic approaches towards the synthesis of beta-blockers (betaxolol, metoprolol, sotalol, and timolol). Turk. J. Chem. 2016, 40, 193–224. [Google Scholar] [CrossRef]
- Funck-Brentano, C. Pharmacokinetic and pharmacodynamic profiles of d-sotalol and d,l-sotalol. Eur. Heart J. 1993, 14 (Suppl. H), 30–35. [Google Scholar] [CrossRef] [PubMed]
- Salazar, D.E.; Much, D.R.; Nichola, P.S.; Seibold, J.R.; Shindler, D.; Slugg, P.H. A pharmacokinetic-pharmacodynamic model of d-sotalol Q-Tc prolongation during intravenous administration to healthy subjects. J. Clin. Pharmacol. 1997, 37, 799–809. [Google Scholar] [CrossRef]
- Waldo, A.; Camm, A.; de Ruyter, H.; Friedman, P.; MacNeil, D.; Pauls, J.; Pitt, B.; Pratt, C.; Schwartz, P.; Veltri, E. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. Lancet 1996, 348, 7–12. [Google Scholar] [CrossRef]
- Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Labetalol: A review of its pharmacology and therapeutic use in hypertension. Drugs 1978, 15, 251–270. [Google Scholar] [CrossRef]
- Riva, E.; Mennini, T.; Latini, R. The alpha- and beta-adrenoceptor blocking activities of labetalol and its RR-SR (50:50) stereoisomers. Br. J. Pharmacol. 1991, 104, 823–828. [Google Scholar] [CrossRef]
- Nagy, B.; Dima, N.; Paizs, C.; Brem, J.; Irimie, F.D.; Toşa, M.I. New chemo-enzymatic approaches for the synthesis of (R)- and (S)-bufuralol. Tetrahedron Asymmetry 2014, 25, 1316–1322. [Google Scholar] [CrossRef]
- Pringle, T.H.; Francis, R.J.; East, P.B.; Shanks, R.G. Pharmacodynamic and pharmacokinetic studies on bufuralol in man. Br. J. Clin. Pharmacol. 1986, 22, 527–534. [Google Scholar] [CrossRef]
- Hamilton, T.C.; Parkes, M.W. Bufuralol a new β-adrenoceptor blocking agent in a series of benzofuran-2-ethanolamines. Arzneim. Forsch. (Drug Res.) 1977, 27, 1410–1417. [Google Scholar]
- Forthegill, G.A.; Osbond, J.M.; Wickens, J.C. Bufuranol, a new β-adrenoceptor blocking agent. Part 1: Synthesis and structure activity studies in a series of benzofuran-2-ethanol amines. Arzneim. Forsch. (Drug Res.) 1977, 27, 978–981. [Google Scholar]
- Meier, M.; Hedwall, P.R.; Imhof, P.; Wilhelm, M.; Brunner, H. Untersuchungen mit den optischen Antipoden der adrenergischen β-Receptoren Blockers l-(o-Allyloxy-phenoxy)-3-isopropylamino-2-propranolol. Arzneim. Forsch. (Drug Res.) 1970, 20, 1890–1896. [Google Scholar]
- Buckner, C.K.; Patil, P.N. The rate of onset of β-adrenergic blockade by the optical isomers of alprenolol. Eur. J. Pharmacol. 1971, 14, 308–311. [Google Scholar] [CrossRef]
- Belliveau, R.E.; Covino, B.G. Effects of a new β-adrenergic receptor blocking agent, alprenolol, and its optical isomers. Arch. Int. Pharmacodyn. 1969, 180, 341–349. [Google Scholar] [PubMed]
- Duce, B.R.; Garberg, L.; Smith, E.R. Effects of (±)-propranolol, (±)-, (+)-, and (-)-alprenolol on unanaesthetized dogs with ventricular arrhythmias resulting from coronary artery ligation. Br. J. Pharmacol. 1970, 39, 809–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunamäki, K. Studies on the anti-arrhythmic effect of dextro-alprenolol. Investigation of the anti-arrhythmic effect of dextro-alprenolol. Eur. J. Clin. Pharmacol. 1970, 3, 23–31. [Google Scholar] [CrossRef]
- Vohra, J.K.; Thompson, P.L.; Sloman, J.G. Clinical experience with dextro-alprenolol. Br. Med. J. 1970, 1, 791–792. [Google Scholar] [CrossRef]
- Porciatti, F.; Cerbai, E.; Masini, I.; Mugelli, A. Electrophysiological evaluation of the beta-blocking properties and direct membrane effects of l-moprolol and its enantiomer d-moprolol. Arch. Int. Pharmacodyn. Ther. 1989, 299, 200–209. [Google Scholar]
- Hartfelder, G.; Lessenich, H.; Schmitt, K. Penbutolol (Hoe 893 d), ein neues, stark wirksames Sympatholytikum mit langer Wirkungsdauer. Arzneim. Forsch. (Drug Res.) 1972, 22, 930–932. [Google Scholar]
- Dogrell, S.A. Effects of (+/-)- (+)- and (-)-metoprolol, (+/-)- (+)- and (-)-pindolol, (+/-)-mepindolol and (+/-)-bopindolol on the rat left atria and portal vein. Gen. Pharmacol. 1991, 22, 1169–1177. [Google Scholar] [CrossRef]
- Mohan, J.C.; Shah, S.N.; Chinchansurkar, S.; Dey, A.; Jain, R. Rediscovering chirality - role of S-metoprolol in cardiovascular disease management. J. Assoc. Physicians India 2017, 65, 74–79. [Google Scholar] [PubMed]
- Sharif, N.A.; Xu, S.X.; Crider, J.Y.; McLaughlin, M.; Davis, T.L. Levobetaxolol (Betaxon) and other beta-adrenergic antagonists: Preclinical pharmacology, IOP-lowering activity and sites of action in human eyes. J. Ocul. Pharmacol. Ther. 2001, 17, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Wood, J.P.; Chidlow, G.; Casson, R.; DeSantis, L.; Schmidt, K.G. Effectiveness of levobetaxolol and timolol at blunting retinal ischaemia is related to their calcium and sodium blocking activities: Relevance to glaucoma. Brain Res. Bull. 2004, 62, 525–528. [Google Scholar] [CrossRef]
- Quaranta, L.; Turano, R.; Pizzolante, T. Levobetaxolol hydrochloride: A review of its pharmacology and use in the treatment of chronic open-angle glaucoma and ocular hypertension. Clin. Ophthalmol. 2007, 1, 93–97. [Google Scholar] [PubMed]
- Sum, C.Y.; Yacobi, A.; Kartzinel, R.; Stampfli, H.; Davis, C.S.; Lai, C.M. Kinetics of esmolol, an ultra-short-acting beta blocker, and of its major metabolite. Clin. Pharmacol. Ther. 1983, 34, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Volz-Zang, C.; Eckrich, B.; Jahn, P.; Schneidrowski, B.; Schulte, B.; Palm, D. Esmolol, an ultrashort-acting, selective beta 1-adrenoceptor antagonist: Pharmacodynamic and pharmacokinetic properties. Eur. J. Clin. Pharmacol. 1994, 46, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, P.; Drici, M. Recent antiarrhythmic drugs. Am. J. Cardiol. 1989, 64, 65J–69J. [Google Scholar] [CrossRef]
- Deng, C.Y.; Lin, S.G.; Zhang, W.C.; Kuang, S.J.; Qian, W.M.; Wu, S.L.; Shan, Z.X.; Yang, M.; Yu, X.Y. Esmolol inhibits Na+ current in rat ventricular myocytes. Methods Find. Exp. Clin. Pharmacol. 2006, 28, 697–702. [Google Scholar] [CrossRef]
- Quon, C.Y.; Mai, K.; Patil, G.; Stampfli, H.F. Species differences in the stereoselective hydrolysis of esmolol by blood esterases. Drug Metab. Dispos. 1988, 16, 425–428. [Google Scholar]
- Okamura, M.; Kumagai, M.; Murasaki, Y.; Ohkura, T.; Miyamoto, Y.; Kawai, Y.; Tamura, T.; Takariki, Y.; Tomisawa, H. Stereoselective pharmacokinetics of esmolol enantiomers. Xenobio. Metabol. Dispos. 2001, 16, 427–435. [Google Scholar] [CrossRef]
- Dogrel, S.A. Effects of (+/-)-, (+)- and (-)-celiprolol on the rat left atria and portal vein. J. Pharm. Pharmacol. 1992, 44, 239–243. [Google Scholar] [CrossRef]
- Howe, R.; Shanks, R.G. Optical isomers of propranolol. Nature 1966, 210, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Stoschitzky, K.; Lindner, W.; Kiowski, W. Stereoselective vascular effects of the (R)- and (S)-enantiomers of propranolol and atenolol. J. Cardiovasc. Pharmacol. 1995, 25, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.M.; Cullum, V.A. The biological properties of the optical isomers of propranolol and their effect on cardiac arrhythmias. Br. J. Pharmacol. 1968, 34, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Bode, W.; Toet, A.E.; Stolker, A.A.; van Ginkel, L.A.; Groen, K.; Wermer, J.; de Wildt, D.J. Toxicokinetics of a single intravenous dose of rac-propranolol versus optically pure propranolol in the rat. Chirality 1995, 7, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Toet, A.E.; van de Kuil, A.; Vleeming, W.; Wemer, J.; Bode, W.; Meulenbelt, J.; de Wildt, D.J. Toxic doses of rac-, (-)-(S)- and (+)-(R )-propranolol in rats and rabbits. Chirality 1996, 8, 411–417. [Google Scholar] [CrossRef]
- Stanley, J.K.; Ramirez, A.J.; Mottaleb, M.; Chambliss, C.K.; Brooks, B.W. Enantiospecific toxicity of the beta-blocker propranolol to Daphnia magna and Pimephales promelas. Environ. Toxicol. Chem. 2006, 25, 1780–1786. [Google Scholar] [CrossRef]
- Midha, K.; McKay, G.; Rawson, M.J.; Hubbard, J.W. The impact of stereoisomerism in bioequivalence studies. J. Pharm. Sci. 1998, 87, 797–802. [Google Scholar] [CrossRef]
- Evans, D.B.; Peschka, M.T.; Lee, R.J.; Laffan, R.J. Anti-arrhythmic action of nadolol, a beta-adrenergic receptor blocking agent. Eur. J. Pharmacol. 1976, 35, 17–27. [Google Scholar] [CrossRef]
- Wheeldon, N.M.; McDevitt, D.G.; Lipworth, B.J. The effects of lower than conventional doses of oral nadolol on relative beta 1/beta 2-adrenoceptor blockade. Br. J. Clin. Pharmacol. 1994, 38, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Quast, U.; Vollmer, K.O. Binding of beta-adrenoceptor antagonists to rat and rabbit lung: Special reference to levobunolol. Arzneim. Forsch. (Drug. Res.) 1984, 34, 579–584. [Google Scholar]
- Dong, Y.; Ishikawa, H.; Wu, Y.; Yoshitomi, T. Vasodilatory mechanism of levobunolol on vascular smooth muscle cells. Exp. Eye Res. 2007, 84, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.P.; Clissold, S.P. Ocular levobunolol. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 1987, 34, 648–661. [Google Scholar] [CrossRef]
- Karhuvaara, S.; Kaila, T.; Huupponen, R. Beta-adrenoceptor antagonist activities and binding affinities of timolol enantiomers in rat atria. J. Pharm. Pharmacol. 1989, 41, 649–650. [Google Scholar] [CrossRef] [PubMed]
- Brown, C. Chirality in Drug Design and Synthesis; Academic Press: New York, NY, USA, 1990; pp. 10–11. [Google Scholar]
- Tenero, D.; Boike, S.; Boyle, D.; Ilson, B.; Fesniak, H.F.; Brozena, S.; Jorkasky, D. Steady-state pharmacokinetics of carvedilol and its enantiomers in patients with congestive heart failure. J. Clin. Pharmacol. 2000, 40, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Siebert, C.D.; Hänsicke, A.; Nagel, T. Stereochemical comparison of nebivolol with other beta-blockers. Chirality 2008, 20, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J. Different pharmacological properties of two enantiomers in a unique beta-blocker, nebivolol. Cardiovasc. Ther. 2008, 26, 115–134. [Google Scholar] [CrossRef]
- Sacco, G.; Evangelista, S.; Criscuoli, M.; Goso, C.; Bigioni, M.; Binaschi, M.; Manzini, S.; Maggi, C.A. Involvement of nitric oxide in both central and peripheral haemodynamic effect of D/L-nebivolol and its enantiomers in rats. Eur. J. Pharmacol. 2005, 511, 167–174. [Google Scholar] [CrossRef]
- De Groot, A.A.; Mathy, M.J.; van Zwieten, P.A.; Peters, S.L. Antioxidant activity of nebivolol in the rat aorta. J. Cardiovasc. Pharmacol. 2004, 43, 148–153. [Google Scholar] [CrossRef]
- Mason, R.P.; Kubant, R.; Jacob, R.F.; Walter, M.F.; Boychuk, B.; Malinski, T. Effect of nebivolol on endothelial nitric oxide and peroxynitrite release in hypertensive animals: Role of antioxidant activity. J. Cardiovasc. Pharmacol. 2006, 48, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, M.S.; Whomsley, R.; Poggesi, I.; Cawello, W.; Mathy, F.-X.; Delporte, M.-L.; Papeleu, P.; Watelet, J.-B. Drug metabolism and pharmacokinetics. Drug. Metab. Rev. 2009, 41, 344–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Račanská, E.; Švec, P. Využitie β-blokátorov v súčasnej terapii a perspektívy ich ďalšieho vývoja. Farm. Obzor 1997, 66, 63–66. [Google Scholar]
- Taylor, D.C.; Pownall, R.; Burke, W. The absorption of beta-adrenoceptor antagonists in rat in-situ small intestine; the effect of lipophilicity. J. Pharm. Pharmacol. 1985, 37, 280–283. [Google Scholar] [CrossRef]
- Mehvar, R.; Brocks, D.R. Stereospecific pharmacokinetics and pharmacodynamics of beta-adrenergic blockers in humans. J. Pharm. Pharm. Sci. 2001, 4, 185–200. [Google Scholar] [PubMed]
- Mehvar, R.; Brocks, D.R. Stereospecific pharmacokinetics and pharmacodynamics: Cardiovascular drugs. In Chirality in Drug Design and Development; Reddy, I.K., Mehvar, R., Eds.; Marcel Dekker: New York, NY, USA, 2004; pp. 281–350. [Google Scholar]
- Takahashi, H.; Ogata, H.; Kanno, S.; Taskeuchi, H. Plasma protein binding of propranolol enantiomers as a major determinant of their stereoselective tissue distribution in rats. J. Pharmacol. Exp. Ther. 1990, 252, 272–278. [Google Scholar] [PubMed]
- Yan, H.; Levander, T. Differential tissue distribution of the enantiomers of racemic pindolol in the rat. Eur. Neuropsychopharmacol. 1999, 10, 59–62. [Google Scholar] [CrossRef]
- Rodgers, T.; Leahy, D.; Rowland, M. Tissue distribution of basic drugs: Accounting for enantiomeric compound and regional differences amongst beta-blocking drugs in rat. J. Pharm. Sci. 2005, 94, 1237–1248. [Google Scholar] [CrossRef]
- Ďuricová, J.; Grundmann, M. CYP2D6 a jeho klinický význam. Klin. Farmakol. Farm. 2007, 21, 133–136. [Google Scholar]
- Narimatsu, S.; Nakata, T.; Shimizudani, T.; Nagaoka, K.; Nakura, H.; Masuda, K.; Katsu, T.; Koeda, A.; Naito, S.; Yamano, S.; et al. Regio- and stereoselective oxidation of propranolol enantiomers by human CYP2D6, cynomolgus monkey CYP2D17 and marmoset CYP2D19. Chem. Biol. Interact. 2011, 189, 146–152. [Google Scholar] [CrossRef]
- Shimizudani, T.; Nagaoka, K.; Hanioka, N.; Yamano, S.; Narimatsu, S. Comparative study of the oxidation of propranolol enantiomers in hepatic and small intestinal microsomes from cynomolgus and marmoset monkeys. Chem. Biol. Interact. 2010, 183, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Oian, M.; Liu, Y.; Yao, T.; Zeng, S. Stereoselective metabolism of propranolol glucuronidation by human UDP-glucuronosyltransferases 2B7 and 1A9. Chirality 2010, 22, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, K.; Echizen, H.; Chiba, K.; Tani, M.; Ishizaki, T. Identification of human CYP isoform involved in the metabolism of propranolol enantiomers- N-desisopropylation is mediated mainly by CYP1A2. Br. J. Clin. Pharmacol. 1995, 39, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Donn, K.; Powell, J.; Weiner, I. Stereoselectivity of cimetidine inhibition of propranolol oral clearance. Clin. Pharmacol. Ther. 1985, 37, 191. [Google Scholar]
- Zhou, H.H.; Anthony, L.B.; Roden, D.M.; Wood, A.J. Quinidine reduces clearance of (+)-propranolol more than (-)-propranolol through marked reduction in 4-hydroxylation. Clin. Pharmacol. Ther. 1990, 47, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Tongwei, Y.; Yan, C. Selective role of cytochrome P450 isoenzymes in metabolism-depend binding of β-adrenoceptor antagonist to liver microsomal protein in rats. Zhejang Daxue Xuebao Yixueban 2001, 30, 197–200. [Google Scholar]
- Walle, T.; Walle, U.; Conradi, E. Pathway-selective sex differences in the metabolic clearance of propranolol in human subjects. Clin. Pharmacol. Ther. 1989, 46, 257–263. [Google Scholar] [CrossRef]
- Mistry, B.; Leslie, J.I.; Eddington, N.D. Influence of input rate on the stereospecific and nonstereospecific first pass metabolism and pharmacokinetics of metoprolol extended release formulations. Chirality 2002, 14, 297–304. [Google Scholar] [CrossRef]
- Cergueira, P.M.; Cesarino, E.J.; Bertucci, C.; Bonato, P.S.; Lanchote, V.L. Stereoselective metabolism of metoprolol: Enantioselectivity of α-hydroxymetoprolol in plasma and urine. Chirality 2003, 15, 542–549. [Google Scholar] [CrossRef]
- Boralli, V.B.; Coelho, E.B.; Cerqueira, P.M.; Lanchote, V.L. Stereoselective analysis of metoprolol and its metabolite in rat plasma with application to oxidative metabolism. J. Chromatogr. B 2005, 823, 195–202. [Google Scholar] [CrossRef]
- Seeringer, A.; Brockmöller, J.; Bauer, S.; Kirchheiner, J. Enantiospecific pharmacokinetics of metoprolol in CYP2D6 ultra-rapid metabolizers and correlation with exercise-induced heart rate. Eur. J. Clin. Pharmacol. 2008, 64, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Brodde, O.E.; Kroemer, H.K. Drug-drug interaction of β-adrenoceptor blockers. Arzneim. Forsch. (Drug. Res.) 2003, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Peřinová, I.; Ďuricová, J.; Brozmanová, H.; Kacířová, I.; Grundmann, M. Stanovení metoprololu a jeho metabolitu alfa-hydroxymetoprololu v sére metódou HPLC s fluorescenční detekcí. Česk. Slov. Farm. 2008, 57, 254–259. [Google Scholar]
- Zhou, H.H.; Wood, A.J. Stereoselective disposition of carvedilol is determined by CYP2D6. Clin. Pharmacol. Ther. 1995, 57, 518–524. [Google Scholar] [CrossRef]
- Hanioka, N.; Tanaka, S.; Moriguchi, Y.; Narimatsu, S. Stereoselective glucuronidation of carvedilol in human liver and intestinal microsomes. Pharmacology 2012, 90, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Graff, D.W.; Williamson, K.M.; Pieper, J.A.; Carson, S.W.; Adams, K.F.; Cascio, W.E.; Patterson, J.H. Effect of fluoxetine on carvedilol pharmacokinetics, CYP2D6 activity, and autonomic balance in heart failure patients. J. Clin. Pharmacol. 2001, 41, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Piquette-Miller, M.; Foster, R.T.; Kappagoda, C.T.; Jamali, F. Pharmacokinetics of acebutolol enantiomers in humans. J. Pharm. Sci. 1991, 80, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Horikiri, Y.; Suzuki, T.; Mizobe, M. Stereoselective pharmacokinetics of bisoprolol after intravenous and oral administration in beagle dogs. J. Pharm. Sci. 1997, 86, 560–564. [Google Scholar] [CrossRef]
- Horikiri, Y.; Suzuki, T.; Mizobe, M. Pharmacokinetics and metabolism of bisoprolol enantiomers in humans. J. Pharm. Sci. 1998, 87, 289–294. [Google Scholar] [CrossRef]
- Kirsch, W.; Rose, I.; Klingmann, I.; Pabst, J.; Ohnhaus, E.E. Interaction of bisoprolol with cimetidine and rifampicin. Eur. J. Clin. Pharmacol. 1986, 31, 59–62. [Google Scholar] [CrossRef]
- Nieminen, T.; Uusitalo, H.; Mäenpää, J.; Turjanmaa, V.; Rane, A.; Lundgren, S.; Ropo, A.; Rontu, R.; Lehtimäki, T.; Kähönen, M. Polymorphisms of genes CYP2D6, ADRB1 and GNAS1 in pharmacokinetics and systemic effects of ophthalmic timolol. A pilot study. Eur. J. Clin. Pharmacol. 2005, 61, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Volotinen, M.; Turpeinen, M.; Tolonen, A.; Uusitalo, J.; Mäenpää, J.; Pelkonen, O. Timolol metabolism in human liver microsomes is mediated principally by CYP2D6. Drug Metab. Dispos. 2007, 35, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Volotinen, M.; Hakkola, J.; Relkonen, O.; Vapaatalo, H.; Mäenpää, J. Metabolism of ophthalmic timolol: New aspects of an old drug. Basic Clin. Pharmacol. Toxicol. 2011, 108, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Gvozdják, J.; Gvozdjaková, A.; Kucharská, J.; Bada, V.; Baranyai, A.; Drímal, J. The effect of trimepranol on metabolism in the ischemic heart muscle. Vnitr. Lek. 1985, 31, 209–215. [Google Scholar] [PubMed]
- Abshagen, U.; Betzien, G.; Kaufmann, B.; Endele, G. Pharmacokinetics of metipranolol in normal man. Eur. J. Clin. Pharmacol. 1982, 21, 293–301. [Google Scholar] [CrossRef]
- Maffei Facino, R.; Bertuletti, R.; Carini, M.; Tofanetti, O. In vitro metabolism of methylpranolol by rat liver. Anal. Chem. Symp. Ser. 1980, 4, 217–223. [Google Scholar]
- Kirch, W.; Görg, K.G. Clinical pharmacokinetics of atenolol—A review. Eur. J. Drug Met. Pharmacokin. 1982, 7, 81–91. [Google Scholar] [CrossRef]
- Stoschitzky, K.; Egginger, G.; Zernig, G.; Klein, W.; Lindner, W. Stereoselective features of (R) and (S)-atenolol: Clinical pharmacological, pharmacokinetic, and radioligand binding studies. Chirality 1993, 5, 15–19. [Google Scholar] [CrossRef]
- Manoury, P.M.; Binet, J.L.; Rousseau, J.; Lefebre-Borg, F.M.; Cavero, I.G. Synthesis of a series compounds related to betaxolol, a new β1-adrenoceptor antagonist with a pharmacological and pharmacokinetic profile optimized for the treatment of chronic cardiovascular diseases. J. Med. Chem. 1987, 30, 1003–1011. [Google Scholar] [CrossRef]
- Beresford, R.; Heel, R.C. Betaxolol of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in hypertension. Drugs 1986, 31, 6–28. [Google Scholar] [CrossRef]
- Marko, V. Determination of Beta-Blockers in Biological Material; Elsevier Science Publishers: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Goncalves, P.V.; Mattes, A.C.; Da Cunha, S.P.; Lanchote, V.L. Enantioselectivity in the steady-state pharmacokinetics and transplacental distribution of pindolol at delivery in pregnancy-induced hypertension. Chirality 2002, 14, 683–687. [Google Scholar] [CrossRef] [PubMed]
- MacCarthy, E.P.; Bloomfield, S.S. Labetalol: A review of its pharmacology, pharmacokinetics, clinical uses and adverse effects. Pharmacotherapy 1983, 3, 193–219. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Akers, W.S.; Herring, V.L.; Wolfe, M.S.; Sullivan, J.M. Gender differences in labetalol kinetic: Importance of determing stereoisomers kinetics for racemic drugs. Pharmacotherapy 2000, 20, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T.M.; Cavalli, R.C.; Marques, M.P.; Da Cunha, S.P.; Baraldi, C.O.; Lanchote, V.L. Stereoselective analysis of labetalol in human plasma by LC-MS/MS: Application to pharmacokinetics. Chirality 2009, 21, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Weerawarna, S.A.; Geisshusler, S.M.; Murthy, S.S.; Nelson, W.L. Enantioselective and diastereoselective hydroxylation of bufuranol. Absolute configuration of the 7-(1-hydroxyethyl)-2-[1-hydroxy-2-(tert-butylamino)ethyl]benzofurans, the benzylic hydroxylation metabolites. J. Med. Chem. 1991, 34, 3091–3097. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, S.; Satoh, T.; Meyer, U.A.; Gonzales, F.J. Stereoselective metabolism of bufuralol racemate and enantiomers in human liver microsomes. J. Pharmacol. Exp. Ther. 2002, 303, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Tamagake, K.; Okuda, Y.; Torigoe, F.; Tsuzuki, D.; Isobe, T.; Hichiya, H.; Hanioka, N.; Yamamoto, S.; Narimatsu, S. Change in enantioselectivity in bufuralol 1‶-hydroxylation by the substitution of phenylalanine-120 by alanine in cytochrome P450 2D6. Chirality 2005, 17, 37–43. [Google Scholar] [CrossRef]
- Hillas, O.; Ezzo, D. Nebivolol (Bystolic), a novel beta blocker for hypertension. Pharm. Ther. 2009, 34, 188–192. [Google Scholar]
- Sanaee, F.; Neves, D.V.; Lanchote, V.L.; Jamali, F. Pharmacokinetics of nebivolol in the rat: Low oral absorption, loss in the gut and systemic stereoselectivity. Biopharm. Drug Dispos. 2013, 34, 312–320. [Google Scholar] [CrossRef]
- Jensen, B.P.; Sharp, C.F.; Gardiner, S.J.; Begg, E.J. Development and validation of a stereoselective liquid chromatography-tandem mass spectrometry assay for quantification of S- and R- metoprolol in human plasma. J. Chromatogr. B 2008, 865, 48–54. [Google Scholar] [CrossRef]
- Poggi, J.C.; Da Silva, F.G.; Coelho, E.B.; Marques, M.P.; Bertucci, C.; Lanchote, V.L. Analysis of carvedilol enantiomers in human plasma using chiral stationary phase column and liquid chromatography with tandem mass spectrometry. Chirality 2012, 24, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Al-Omar, M.A. Stereoselective HPLC assay of acebutolol enantiomers with fluorescence detection and its application to a pharmacokinetic study. World Appl. Sci. J. 2010, 8, 1309–1316. [Google Scholar]
- Hefnawy, M.M.; Al-Shehry, M.M.; Abounassif, A.; Mostafa, A.E. Enantioselective quantification of atenolol in mouse plasma by high-performance liquid chromatography using a chiral stationary phase: Application to a pharmacokinetic study. J. AOAC Int. 2013, 96, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Neves, D.V.; Vieira, C.P.; Coelho, E.B.; Marques, M.P.; Lanchote, V.L. Stereoselective analysis of nebivolol isomers in human plasma by high-performance liquid chromatography-tandem mass spectrometry: Application in pharmacokinetics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 1, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Randlett, C.; Junga, H.; Jiang, X.; Ji, Q. Using supported liquid extraction together with cellobiohydrolase chiral stationary phases-based liquid chromatography with tandem mass spectrometry for enantioselective determination of acebutolol and its active metabolite diacetol in spiked human plasma. J. Chromatogr. B 2009, 877, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Oleksiak, J.S.; Walczak, M.; Bojarski, J.; Aboul-Enein, H.Y. Enatioselective high performance liquid chromatographic assay of acebutolol and its active metabolite diacetol in human serum. Chirality 1999, 11, 267–271. [Google Scholar] [CrossRef]
- De Antunes, N.J.; Cavalli, R.C.; Marques, M.P.; Lanchote, V.L. Stereoselective determination of metoprolol and its metabolite α-hydroxymetoprolol in plasma by LC-MS/MS: Application to pharmacokinetics during pregnancy. Chirality 2013, 25, 1–7. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čižmáriková, R.; Habala, L.; Valentová, J.; Markuliak, M. Survey of Pharmacological Activity and Pharmacokinetics of Selected ?-Adrenergic Blockers in Regard to Their Stereochemistry. Appl. Sci. 2019, 9, 625. https://doi.org/10.3390/app9040625
Čižmáriková R, Habala L, Valentová J, Markuliak M. Survey of Pharmacological Activity and Pharmacokinetics of Selected ?-Adrenergic Blockers in Regard to Their Stereochemistry. Applied Sciences. 2019; 9(4):625. https://doi.org/10.3390/app9040625
Chicago/Turabian StyleČižmáriková, Ružena, Ladislav Habala, Jindra Valentová, and Mário Markuliak. 2019. "Survey of Pharmacological Activity and Pharmacokinetics of Selected ?-Adrenergic Blockers in Regard to Their Stereochemistry" Applied Sciences 9, no. 4: 625. https://doi.org/10.3390/app9040625
APA StyleČižmáriková, R., Habala, L., Valentová, J., & Markuliak, M. (2019). Survey of Pharmacological Activity and Pharmacokinetics of Selected ?-Adrenergic Blockers in Regard to Their Stereochemistry. Applied Sciences, 9(4), 625. https://doi.org/10.3390/app9040625