A Simple and Efficient Microfluidic System for Reverse Chemical Synthesis (5′-3′) of a Short-Chain Oligonucleotide Without Inert Atmosphere


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
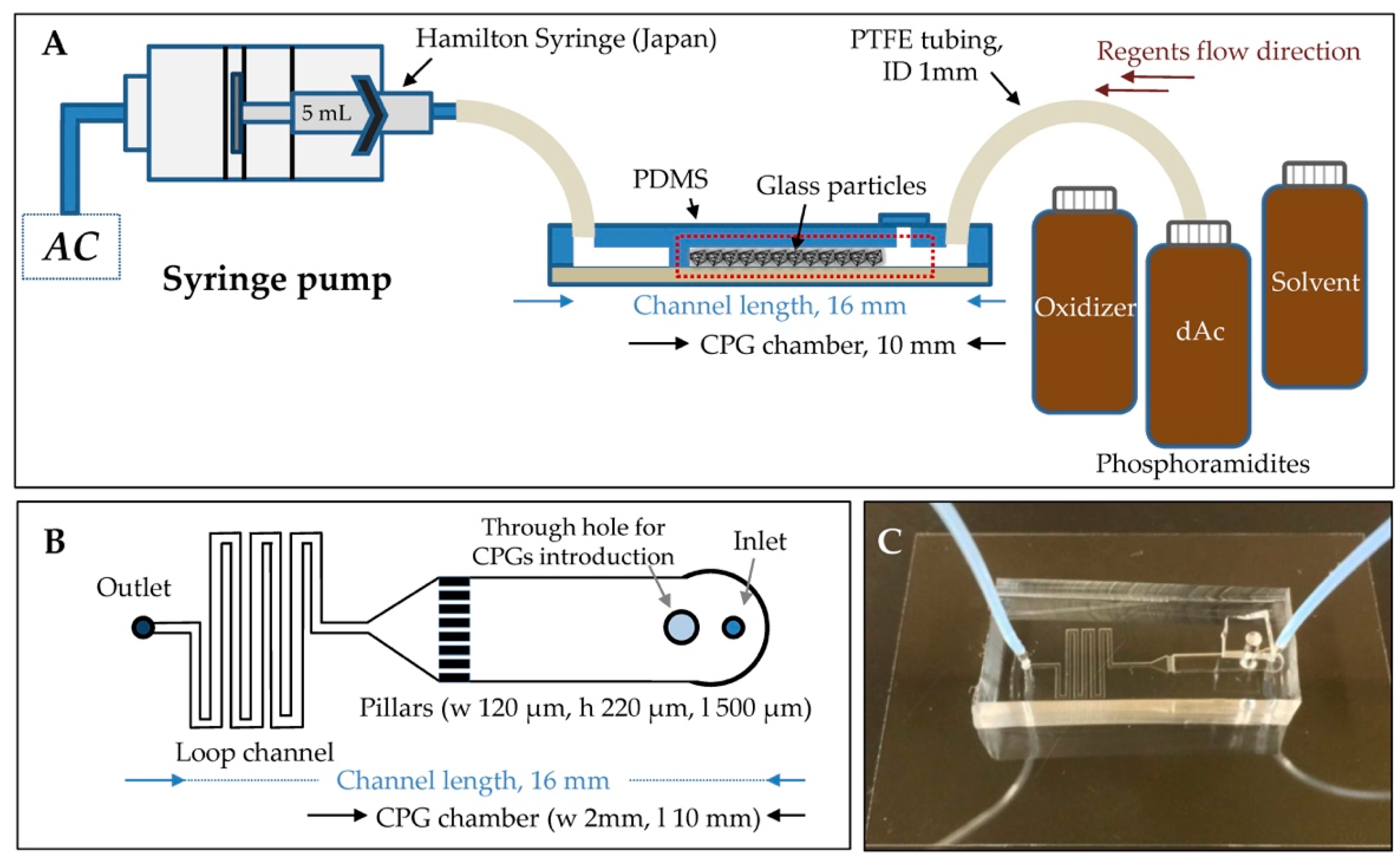
2.2. Fabrication of the PDMS Microfluidic Chip
2.3. Operation of the Microfluidic Chip
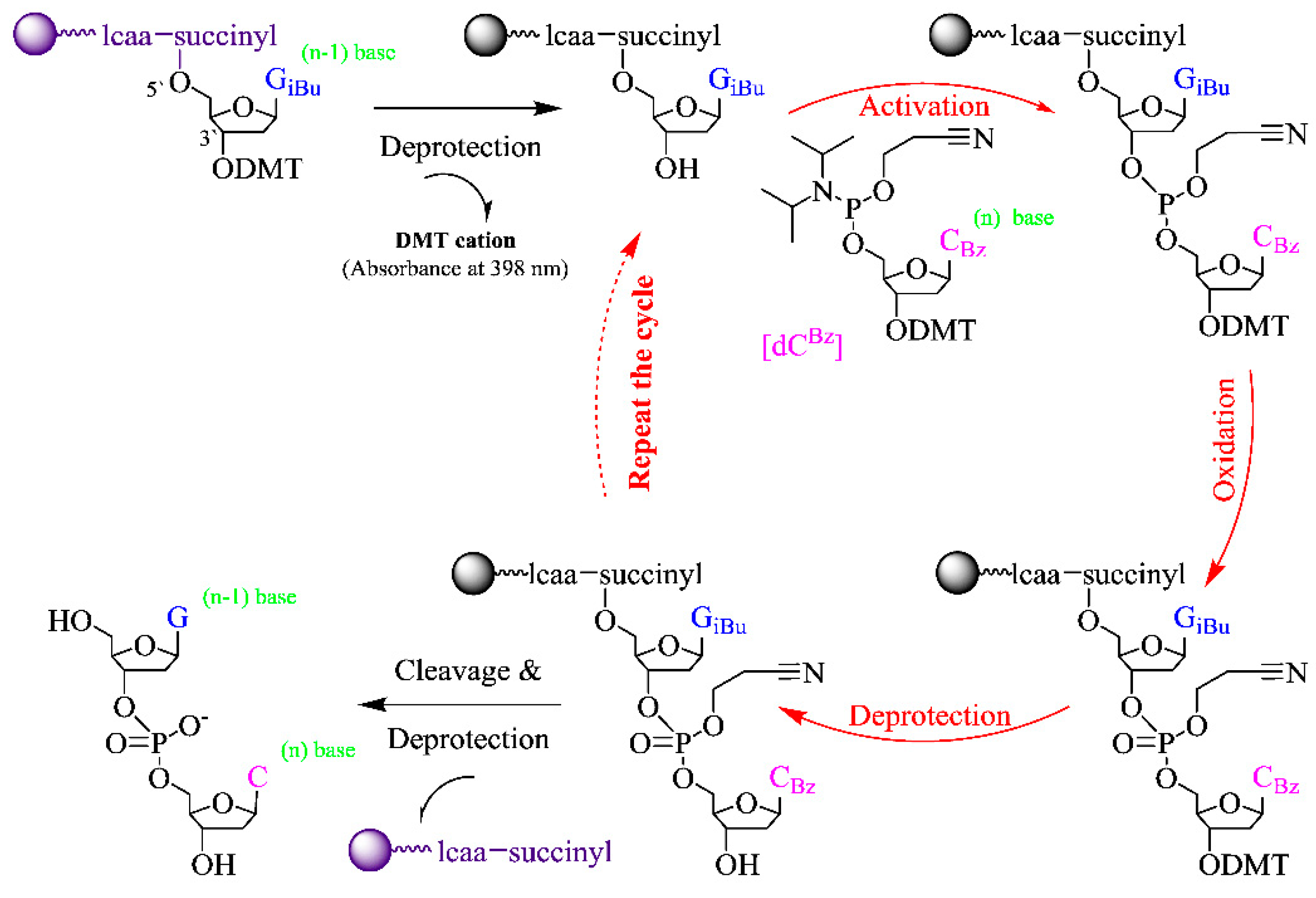
2.4. On-Chip Chemical Syntheses of Reverse Oligonucleotides (5′-3′)
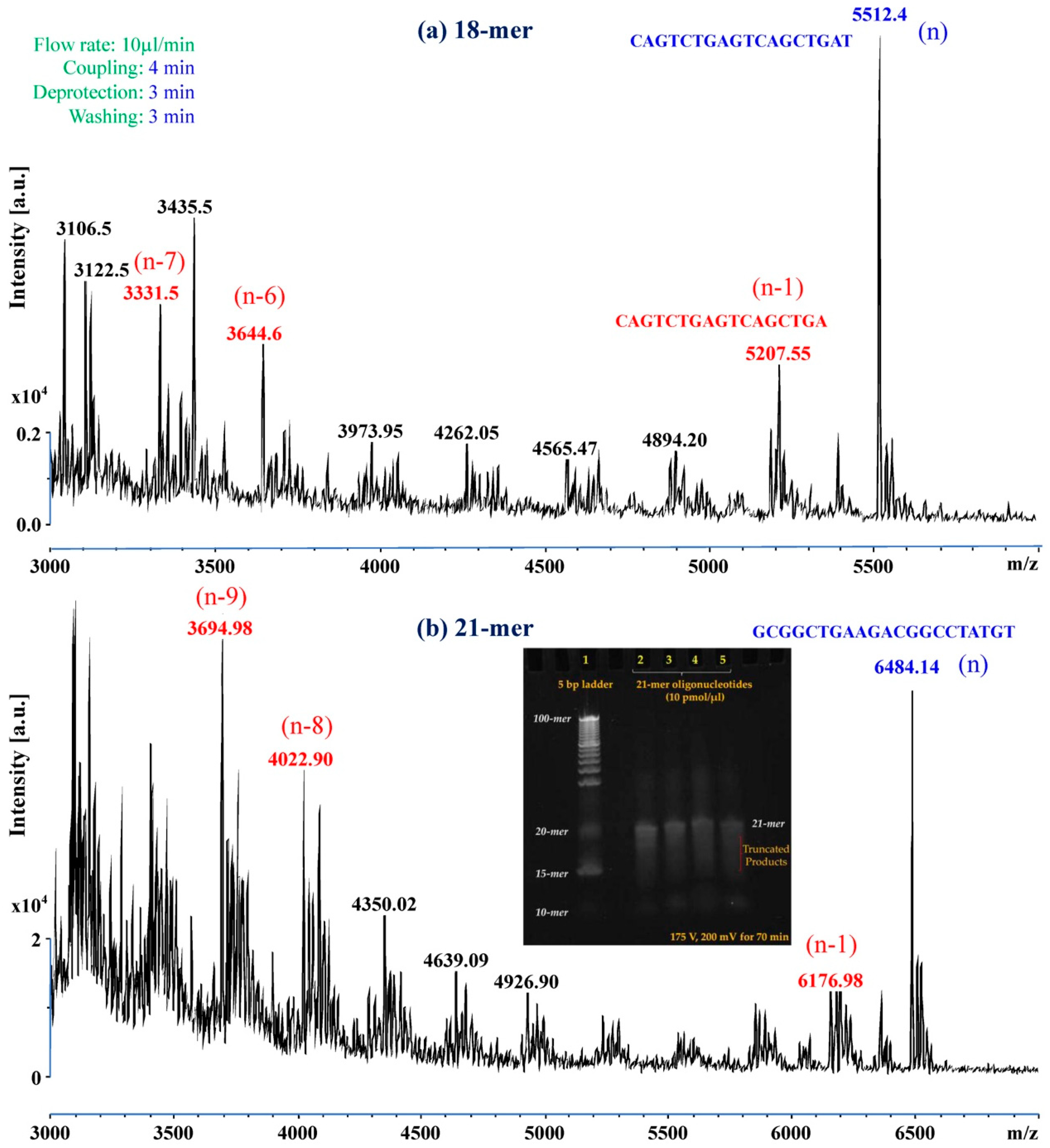
2.5. Characterization Using Mass Spectroscopy (MS)
2.6. Electrophoresis
3. Results and Discussions
On-Chip Reverse Synthesis of Oligonucleotides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Church, G.M.; Gao, Y.; Kosuri, S. Next-generation digital information storage in DNA. Science 2012, 337, 1628. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Högberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 2009, 459, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Linko, V.; Ora, A.; Kostiainen, M.A. DNA nanostructures as smart drug-delivery vehicles and molecular devices. Trends Biotechnol. 2015, 33, 586–594. [Google Scholar] [CrossRef]
- Pinheiro, A.V.; Han, D.; Shih, W.M.; Yan, H. Challenges and opportunities for structural DNA nanotechnology. Nat. Nanotechnol. 2011, 6, 763–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H. DNA-templated self-assembly of protein arrays and highly conductive nanowires. Science 2003, 301, 1882–1884. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, M.D.; Caruthers, M.H. Synthesis of deoxyoligonucleotides on a polymer support. J. Am. Chem. Soc. 1981, 103, 3185–3191. [Google Scholar] [CrossRef]
- Caruthers, M.H. The chemical synthesis of DNA/rna: Our gift to science. J. Biol. Chem. 2013, 288, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Claeboe, C.D.; Gao, R.; Hecht, S.M. 3′-modified oligonucleotides by reverse DNA synthesis. Nucleic Acids Res. 2003, 31, 5685–5691. [Google Scholar] [CrossRef]
- Koga, M.; Moore, M.F.; Beaucage, S.L. Alternating.Alpha.,.Beta.-oligothymidylates with alternating (3′.Fwdarw.3′)- and (5′.Fwdarw.5′)-internucleotidic phosphodiester linkages as models for antisense oligodeoxyribonucleotides. J. Org. Chem. 1991, 56, 3757–3759. [Google Scholar] [CrossRef]
- Koga, M.; Wilk, A.; Moore, M.F.; Scremin, C.L.; Zhou, L.; Beaucage, S.L. Synthesis and physicochemical properties of alternating. α,β-oligodeoxyribonucleotides with alternating (3′.Fwdarw.3′)- and (5′.Fwdarw.5′)-internucleotidic phosphodiester linkages. J. Org. Chem. 1995, 60, 1520–1530. [Google Scholar] [CrossRef]
- Wagner, T.; Pfleiderer, W. Nucleotides, part lxv, synthesis of 2′-deoxyribonucleoside 5′-phosphoramidites: New building blocks for the inverse (5′-3′)-oligonucleotide approach. Helv. Chim. Acta 2000, 83, 2023–2035. [Google Scholar] [CrossRef]
- Gierahn, T.M.; Wadsworth, M.H.; Hughes, T.K.; Bryson, B.D.; Butler, A.; Satija, R.; Fortune, S.; Love, J.C.; Shalek, A.K. Seq-well: Portable, low-cost rna sequencing of single cells at high throughput. Nat. Methods 2017, 14, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Zilionis, R.; Nainys, J.; Veres, A.; Savova, V.; Zemmour, D.; Klein, A.M.; Mazutis, L. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 2016, 12, 44–73. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef]
- Elvira, K.S.; i Solvas, X.C.; Wootton, R.C.R.; deMello, A.J. The past, present and potential for microfluidic reactor technology in chemical synthesis. Nat. Chem. 2013, 5, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C. Multistep synthesis of a radiolabeled imaging probe using integrated microfluidics. Science 2005, 310, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sui, G.; Mocharla, V.P.; Lin, R.J.; Phelps, M.E.; Kolb, H.C.; Tseng, H.-R. Integrated microfluidics for parallel screening of an in situ click chemistry library. Angew. Chem. Int. Ed. 2006, 45, 5276–5281. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Xia, Y.; Srivannavit, O.; Rouillard, J.-M.; Zhou, X.; Gao, X.; Gulari, E. A versatile microreactor platform featuring a chemical-resistant microvalve array for addressable multiplex syntheses and assays. J. Micromech. Microeng. 2006, 16, 1433–1443. [Google Scholar] [CrossRef]
- Huang, Y.; Castrataro, P.; Lee, C.-C.; Quake, S.R. Solvent resistant microfluidic DNA synthesizer. Lab Chip 2007, 7, 24–26. [Google Scholar] [CrossRef]
- Rolland, J.P.; Van Dam, R.M.; Schorzman, D.A.; Quake, S.R.; DeSimone, J.M. Solvent-resistant photocurable “liquid teflon” for microfluidic device fabrication. J. Am. Chem. Soc. 2004, 126, 2322–2323. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Snyder, T.M.; Quake, S.R. A microfluidic oligonucleotide synthesizer. Nucleic Acids Res. 2010, 38, 2514–2521. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, R.; Lightson, N.; Ukita, Y.; Takamura, Y. Development of oligopeptide-based novel biosensor by solid-phase peptide synthesis on microchip. Sensors Actuators B Chem. 2014, 192, 818–825. [Google Scholar] [CrossRef]
- Beaucage, S.L.; Caruthers, M.H. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981, 22, 1859–1862. [Google Scholar] [CrossRef]
- Matteucci, M.D.; Caruthers, M.H. The synthesis of oligodeoxyprimidines on a polymer support. Tetrahedron Lett. 1980, 21, 719–722. [Google Scholar] [CrossRef]
- Guzaev, A.P.; Pon, R.T. Attachment of nucleosides and other linkers to solid-phase supports for oligonucleotide synthesis. Curr. Protoc. Nucleic Acid Chem. 2013, 52, 3.2.1–3.2.23. [Google Scholar]
- LeProust, E.M.; Peck, B.J.; Spirin, K.; McCuen, H.B.; Moore, B.; Namsaraev, E.; Caruthers, M.H. Synthesis of high-quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process. Nucleic Acids Res. 2010, 38, 2522–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pon, R.T.; Buck, G.A.; Hager, K.M.; Naeve, C.W.; Niece, R.L.; Robertson, M.; Smith, A.J. Multi-facility survey of oligonucleotide synthesis and an examination of the performance of unpurified primers in automated DNA sequencing. Biotechniques 1996, 21, 680–685. [Google Scholar]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhardwaj, R.; Tue, P.T.; Biyani, M.; Takamura, Y. A Simple and Efficient Microfluidic System for Reverse Chemical Synthesis (5′-3′) of a Short-Chain Oligonucleotide Without Inert Atmosphere. Appl. Sci. 2019, 9, 1357. https://doi.org/10.3390/app9071357
Bhardwaj R, Tue PT, Biyani M, Takamura Y. A Simple and Efficient Microfluidic System for Reverse Chemical Synthesis (5′-3′) of a Short-Chain Oligonucleotide Without Inert Atmosphere. Applied Sciences. 2019; 9(7):1357. https://doi.org/10.3390/app9071357
Chicago/Turabian StyleBhardwaj, Rahul, Phan T. Tue, Manish Biyani, and Yuzuru Takamura. 2019. "A Simple and Efficient Microfluidic System for Reverse Chemical Synthesis (5′-3′) of a Short-Chain Oligonucleotide Without Inert Atmosphere" Applied Sciences 9, no. 7: 1357. https://doi.org/10.3390/app9071357
APA StyleBhardwaj, R., Tue, P. T., Biyani, M., & Takamura, Y. (2019). A Simple and Efficient Microfluidic System for Reverse Chemical Synthesis (5′-3′) of a Short-Chain Oligonucleotide Without Inert Atmosphere. Applied Sciences, 9(7), 1357. https://doi.org/10.3390/app9071357