Alcohol Sensitivity as an Endophenotype of Alcohol Use Disorder: Exploring Its Translational Utility between Rodents and Humans


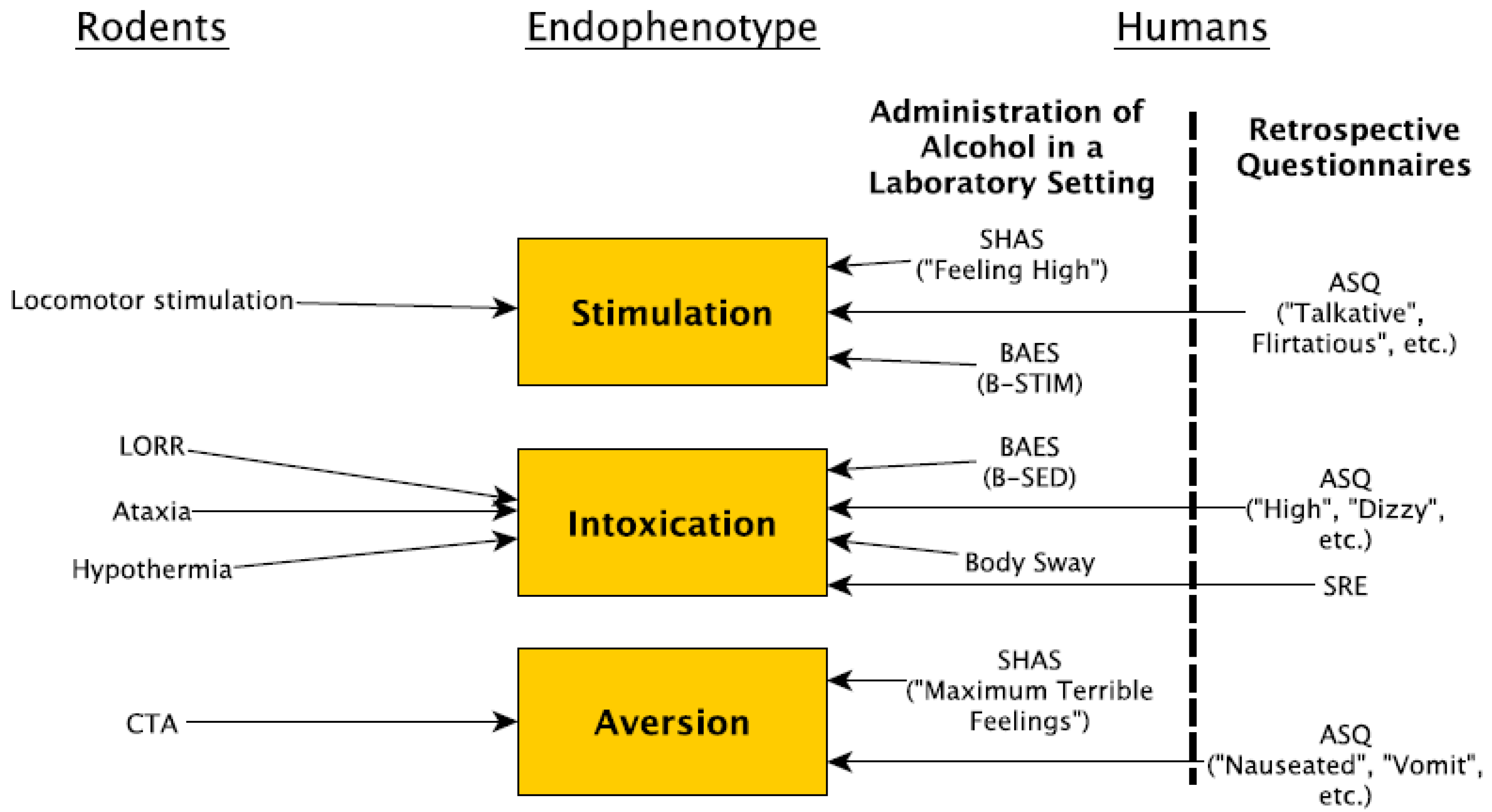{kind=link}
Abstract
:1. Introduction
2. Alcohol Sensitivity in Humans
3. Advantages of Rodent Models
4. Genetic Approaches in Rodent Models of AUDs
4.1. Forward Genetics
4.2. Reverse Genetics
5. Alcohol Sensitivity in Rodents
5.1. Ethanol-Induced Locomotor Stimulation
5.2. Ethanol-Induced Intoxication (Ataxia, LORR, Hypothermia)
5.2.1. Ataxia
5.2.2. LORR
5.2.3. Hypothermia
5.3. Ethanol-Induced Conditioned Taste Aversion (CTA)
6. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Degenhardt, L.; Charlson, F.; Ferrari, A.; Santomauro, D.; Erskine, H.; Mantilla-Herrara, A. GBD 2016 Alcohol and Drug Use Collaborators The global burden of disease attributable to alcohol and drug use in 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Psychiatry 2018, 5, 987–1012. [Google Scholar] [CrossRef] [Green Version]
- Ducci, F.; Goldman, D. Genetic approaches to addiction: Genes and alcohol. Addiction 2008, 103, 1414–1428. [Google Scholar] [CrossRef] [Green Version]
- Kendler, K.S.; Aggen, S.H.; Prescott, C.A.; Crabbe, J.; Neale, M.C. Evidence for multiple genetic factors underlying the DSM-IV criteria for alcohol dependence. Mol. Psychiatry 2012, 17, 1306–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhulst, B.; Neale, M.C.; Kendler, K.S. The heritability of alcohol use disorders: A meta-analysis of twin and adoption studies. Psychol. Med. 2015, 45, 1061–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelernter, J.; Kranzler, H.R.; Sherva, R.; Koesterer, R.; Almasy, L.; Zhao, H.; Farrer, L.A. Genome-wide association study of opioid dependence: Multiple associations mapped to calcium and potassium pathways. Biol. Psychiatry 2014, 76, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelernter, J.; Sun, N.; Polimanti, R.; Pietrzak, R.H.; Levey, D.F.; Lu, Q.; Hu, Y.; Li, B.; Radhakrishnan, K.; Aslan, M.; et al. Genome-wide Association Study of Maximum Habitual Alcohol Intake in >140,000 U.S. European and African American Veterans Yields Novel Risk Loci. Biol. Psychiatry 2019, 86, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Mbarek, H.; Milaneschi, Y.; Fedko, I.O.; Hottenga, J.-J.; de Moor, M.H.M.; Jansen, R.; Gelernter, J.; Sherva, R.; Willemsen, G.; Boomsma, D.I.; et al. The genetics of alcohol dependence: Twin and SNP-based heritability, and genome-wide association study based on AUDIT scores. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2015, 168, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Quillen, E.E.; Chen, X.-D.; Almasy, L.; Yang, F.; He, H.; Li, X.; Wang, X.-Y.; Liu, T.-Q.; Hao, W.; Deng, H.-W.; et al. ALDH2 is associated to alcohol dependence and is the major genetic determinant of “daily maximum drinks” in a GWAS study of an isolated rural Chinese sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165B, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Roige, S.; Palmer, A.A.; Fontanillas, P.; Elson, S.L.; 23andMe Research Team, the Substance Use Disorder Working Group of the Psychiatric Genomics Consortium; Adams, M.J.; Howard, D.M.; Edenberg, H.J.; Davies, G.; Crist, R.C.; et al. Genome-Wide Association Study Meta-Analysis of the Alcohol Use Disorders Identification Test (AUDIT) in Two Population-Based Cohorts. Am. J. Psychiatry 2019, 176, 107–118. [Google Scholar] [CrossRef]
- Sanchez-Roige, S.; Fontanillas, P.; Elson, S.L.; 23andMe Research Team; Gray, J.C.; de Wit, H.; Davis, L.K.; MacKillop, J.; Palmer, A.A. Genome-wide association study of alcohol use disorder identification test (AUDIT) scores in 20,328 research participants of European ancestry. Addict. Biol. 2019, 24, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Sealock, J.M.; Sanchez-Roige, S.; Clarke, T.-K.; Levey, D.F.; Cheng, Z.; Li, B.; Polimanti, R.; Kember, R.L.; Smith, R.V.; et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat. Neurosci. 2020, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.L.; Zhang, J.; Wang, J.C.; Su, J.; Kuo, S.I.; Kapoor, M.; Wetherill, L.; Bertelsen, S.; Lai, D.; Salvatore, J.E.; et al. An endophenotype approach to the genetics of alcohol dependence: A genome wide association study of fast beta EEG in families of African ancestry. Mol. Psychiatry 2017, 22, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.L.; Zhang, J.; Chorlian, D.B.; Pandey, A.K.; Kamarajan, C.; Wang, J.-C.; Wetherill, L.; Lai, D.; Chao, M.; Chan, G.; et al. A genome-wide association study of interhemispheric theta EEG coherence: Implications for neural connectivity and alcohol use behavior. Mol. Psychiatry 2020, 1–13. [Google Scholar] [CrossRef]
- Hart, A.B.; Kranzler, H.R. Alcohol dependence genetics: Lessons learned from Genome-Wide Association Studies (GWAS) and Post-GWAS Analyses. Alcohol. Clin. Exp. Res. 2015, 39, 1312–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [Green Version]
- Stranger, B.E.; Stahl, E.A.; Raj, T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics 2011, 187, 367–383. [Google Scholar] [CrossRef] [Green Version]
- Ward, L.D.; Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012, 30, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Roige, S.; Palmer, A.A.; Clarke, T.-K. Recent efforts to dissect the genetic basis of alcohol use and abuse. Biol. Psychiatry 2020, 87, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Farris, S.P.; Riley, B.P.; Williams, R.W.; Mulligan, M.K.; Miles, M.F.; Lopez, M.F.; Hitzemann, R.; Iancu, O.D.; Colville, A.; Walter, N.A.R.; et al. Cross-species molecular dissection across alcohol behavioral domains. Alcohol 2018, 72, 19–31. [Google Scholar] [CrossRef]
- Barr, P.B.; Dick, D.M. The genetics of externalizing problems; Springer: Berlin, Germany, 2019. [Google Scholar]
- Grotzinger, A.D.; Mallard, T.T.; Akingbuwa, W.A.; Ip, H.F.; Adams, M.J.; Lewis, C.M.; McIntosh, A.M.; Grove, J.; Dalsgaard, S.; Peter-Lesch, K.; et al. Genetic architecture of 11 major psychiatric disorders at biobehavioral, functional genomic, and molecular genetic levels of analysis. medRxiv 2020, arXiv:2020.09.22.20196089. [Google Scholar]
- Mallard, T.T.; Linnér, R.K.; Grotzinger, A.D.; Sanchez-Roige, S.; Seidlitz, J.; Okbay, A.; de Vlaming, R.; Meddens, S.F.W.; Consortium, B.D.W.G. of the P.G.; Palmer, A.A.; et al. Multivariate GWAS of psychiatric disorders and their cardinal symptoms reveal two dimensions of cross-cutting genetic liabilities. bioRxiv 2020, arXiv:603134. [Google Scholar]
- Waldman, I.D.; Poore, H.E.; Luningham, J.M.; Yang, J. Testing structural models of psychopathology at the genomic level. World Psychiatry Off. J. World Psychiatr. Assoc. 2020, 19, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Dick, D.M. Mapping risk from genes to behavior: The enduring and evolving influence of irving gottesman’s endophenotype concept. Twin Res. Human Genet. 2018, 21, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, I.I.; Gould, T.D. The endophenotype concept in psychiatry: Etymology and strategic intentions. Am. J. Psychiatry 2003, 160, 636–645. [Google Scholar] [CrossRef]
- Gould, T.D.; Gottesman, I.I. Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav. 2006, 5, 113–119. [Google Scholar] [CrossRef]
- Ramchandani, V.A.; Umhau, J.; Pavon, F.J.; Ruiz-Velasco, V.; Margas, W.; Sun, H.; Damadzic, R.; Eskay, R.; Schoor, M.; Thorsell, A.; et al. A genetic determinant of the striatal dopamine response to alcohol in men. Mol. Psychiatry 2011, 16, 809–817. [Google Scholar] [CrossRef]
- King, A.C.; de Wit, H.; McNamara, P.J.; Cao, D. Rewarding, stimulant, and sedative alcohol responses and relationship to future binge drinking. Arch. Gen. Psychiatry 2011, 68, 389–399. [Google Scholar] [CrossRef]
- Newlin, D.B.; Thomson, J.B. Alcohol challenge with sons of alcoholics: A critical review and analysis. Psychol. Bull. 1990, 108, 383–402. [Google Scholar] [CrossRef]
- Schuckit, M.A. Low level of response to alcohol as a predictor of future alcoholism. Am. J. Psychiatry 1994, 151, 184–189. [Google Scholar]
- Schuckit, M.A.; Smith, T.L. Onset and course of alcoholism over 25 years in middle class men. Drug Alcohol Depend 2011, 113, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Schuckit, M.A.; Smith, T.L.; Kalmijn, J.A. The patterns of drug and alcohol use and associated problems over 30 years in 397 men. Alcohol. Clin. Exp. Res. 2014, 38, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, B.F. The impact of a family history of alcoholism on the relationship between age at onset of alcohol use and DSM-IV alcohol dependence: Results from the National Longitudinal Alcohol Epidemiologic Survey. Alcohol Health Res. World 1998, 22, 144–147. [Google Scholar] [PubMed]
- Schuckit, M.A.; Smith, T.L. The relationships of a family history of alcohol dependence, a low level of response to alcohol and six domains of life functioning to the development of alcohol use disorders. J. Stud. Alcohol 2000, 61, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Schuckit, M.A.; Smith, T.L. A comparison of correlates of DSM-IV alcohol abuse or dependence among more than 400 sons of alcoholics and controls. Alcohol. Clin. Exp. Res. 2001, 25, 1–8. [Google Scholar] [CrossRef]
- Hendler, R.A.; Ramchandani, V.A.; Gilman, J.; Hommer, D.W. Stimulant and sedative effects of alcohol. Curr. Top Behav. Neurosci. 2013, 13, 489–509. [Google Scholar]
- Krystal, J.H.; Petrakis, I.L.; Mason, G.; Trevisan, L.; D’Souza, D.C. N-methyl-D-aspartate glutamate receptors and alcoholism: Reward, dependence, treatment, and vulnerability. Pharmacol. Ther. 2003, 99, 79–94. [Google Scholar] [CrossRef]
- Schuckit, M.A.; Kalmijn, J.A.; Smith, T.L.; Saunders, G.; Fromme, K. Structuring a college alcohol prevention program on the low level of response to alcohol model: A pilot study. Alcohol. Clin. Exp. Res. 2012, 36, 1244–1252. [Google Scholar] [CrossRef]
- Schuckit, M.A.; Smith, T.L.; Tipp, J.E. The Self-Rating of the Effects of alcohol (SRE) form as a retrospective measure of the risk for alcoholism. Addiction 1997, 92, 979–988. [Google Scholar] [CrossRef]
- Moresco, E.M.Y.; Li, X.; Beutler, B. Going forward with genetics: Recent technological advances and forward genetics in mice. Am. J. Pathol. 2013, 182, 1462–1473. [Google Scholar] [CrossRef] [Green Version]
- King, A.C.; Hasin, D.; O’Connor, S.J.; McNamara, P.J.; Cao, D. A prospective 5-year re-examination of alcohol response in heavy drinkers progressing in alcohol use disorder. Biol. Psychiatry 2016, 79, 489–498. [Google Scholar] [CrossRef] [Green Version]
- King, A.C.; Cao, D.; deWit, H.; O’Connor, S.J.; Hasin, D.S. The role of alcohol response phenotypes in the risk for alcohol use disorder. BJPsych Open 2019, 5, e38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, D.J.O.; Palmeri, M.D.; King, A.C. Acute alcohol response phenotype in heavy social drinkers is robust and reproducible. Alcohol. Clin. Exp. Res. 2014, 38, 844–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuckit, M.A. A critical review of methods and results in the search for genetic contributors to alcohol sensitivity. Alcohol. Clin. Exp. Res. 2018, 42, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.E.; Neale, Z.; Cho, S.B.; Hancock, L.; Kalmijn, J.A.; Smith, T.L.; Schuckit, M.A.; Donovan, K.K.; Dick, D.M. Level of response to alcohol as a factor for targeted prevention in college students. Alcohol. Clin. Exp. Res. 2015, 39, 2215–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuckit, M.A.; Smith, T.L.; Clausen, P.; Fromme, K.; Skidmore, J.; Shafir, A.; Kalmijn, J. The low level of response to alcohol-based heavy drinking prevention program: One-year follow-up. J. Stud. Alcohol Drugs 2016, 77, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuckit, M.A. Ethanol-induced changes in body sway in men at high alcoholism risk. Arch. Gen. Psychiatry 1985, 42, 375–379. [Google Scholar] [CrossRef]
- Quinn, P.D.; Fromme, K. Subjective response to alcohol challenge: A quantitative review. Alcohol. Clin. Exp. Res. 2011, 35, 1759–1770. [Google Scholar] [CrossRef] [Green Version]
- de Wit, H.; Phillips, T.J. Do initial responses to drugs predict future use or abuse? Neurosci. Biobehav. Rev. 2012, 36, 1565–1576. [Google Scholar] [CrossRef] [Green Version]
- Schuckit, M.A.; Edenberg, H.J.; Kalmijn, J.; Flury, L.; Smith, T.L.; Reich, T.; Bierut, L.; Goate, A.; Foroud, T. A genome-wide search for genes that relate to a low level of response to alcohol. Alcohol. Clin. Exp. Res. 2001, 25, 323–329. [Google Scholar] [CrossRef]
- Ehlers, C.L.; Gizer, I.R.; Schuckit, M.A.; Wilhelmsen, K.C. Genome-wide scan for self-rating of the effects of alcohol in American Indians. Psychiatr. Genet. 2010, 20, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Joslyn, G.; Ravindranathan, A.; Brush, G.; Schuckit, M.; White, R.L. Human variation in alcohol response is influenced by variation in neuronal signaling genes. Alcohol. Clin. Exp. Res. 2010, 34, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.C.; Deak, J.D.; Gizer, I.R.; Lai, D.; Chatzinakos, C.; Wilhelmsen, K.P.; Lindsay, J.; Heron, J.; Hickman, M.; Webb, B.T.; et al. Meta-analysis of genetic influences on initial alcohol sensitivity. Alcohol. Clin. Exp. Res. 2018, 42, 2349–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daeppen, J.B.; Landry, U.; Pécoud, A.; Decrey, H.; Yersin, B. A measure of the intensity of response to alcohol to screen for alcohol use disorders in primary care. Alcohol Alcohol. 2000, 35, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Kalu, N.; Ramchandani, V.A.; Marshall, V.; Scott, D.; Ferguson, C.; Cain, G.; Taylor, R. Heritability of level of response and association with recent drinking history in nonalcohol-dependent drinkers. Alcohol. Clin. Exp. Res. 2012, 36, 1034–1041. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, S.L.; McCarthy, D.M. Differences in acute response to alcohol between African Americans and European Americans. Alcohol. Clin. Exp. Res. 2013, 37, 1056–1063. [Google Scholar] [CrossRef] [Green Version]
- Schuckit, M.A.; Smith, T.L.; Danko, G.P.; Pierson, J.; Hesselbrock, V.; Bucholz, K.K.; Kramer, J.; Kuperman, S.; Dietiker, C.; Brandon, R.; et al. The ability of the Self-Rating of the Effects of Alcohol (SRE) Scale to predict alcohol-related outcomes five years later. J. Stud. Alcohol Drugs 2007, 68, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.C.; Pierre, C.L.S.; Meyers, J.L.; Aliev, F.; McCutcheon, V.V.; Lai, D.; Dick, D.M.; Goate, A.M.; Kramer, J.; Kuperman, S.; et al. The genetic relationship between alcohol consumption and aspects of problem drinking in an ascertained sample. Alcohol. Clin. Exp. Res. 2019, 43, 1113–1125. [Google Scholar] [CrossRef]
- Schuckit, M.A.; Wilhelmsen, K.; Smith, T.L.; Feiler, H.S.; Lind, P.; Lange, L.A.; Kalmijn, J. Autosomal linkage analysis for the level of response to alcohol. Alcohol. Clin. Exp. Res. 2005, 29, 1976–1982. [Google Scholar] [CrossRef]
- Schuckit, M.A.; Smith, T.L. An 8-year follow-up of 450 sons of alcoholic and control subjects. Arch. Gen. Psychiatry 1996, 53, 202–210. [Google Scholar] [CrossRef]
- Lai, D.; Wetherill, L.; Kapoor, M.; Johnson, E.C.; Schwandt, M.; Ramchandani, V.A.; Goldman, D.; Joslyn, G.; Rao, X.; Liu, Y.; et al. Genome-wide association studies of the self-rating of effects of ethanol (SRE). Addict. Biol. 2020, 25, e12800. [Google Scholar] [CrossRef]
- Parker, C.C.; Gopalakrishnan, S.; Carbonetto, P.; Gonzales, N.M.; Leung, E.; Park, Y.J.; Aryee, E.; Davis, J.; Blizard, D.A.; Ackert-Bicknell, C.L.; et al. Genome-wide association study of behavioral, physiological and gene expression traits in outbred CFW mice. Nat. Genet. 2016, 48, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicod, J.; Davies, R.W.; Cai, N.; Hassett, C.; Goodstadt, L.; Cosgrove, C.; Yee, B.K.; Lionikaite, V.; McIntyre, R.E.; Remme, C.A.; et al. Genome-wide association of multiple complex traits in outbred mice by ultra-low-coverage sequencing. Nat. Genet. 2016, 48, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Keele, G.R.; Prokop, J.W.; He, H.; Holl, K.; Littrell, J.; Deal, A.; Francic, S.; Cui, L.; Gatti, D.M.; Broman, K.W.; et al. Genetic fine-mapping and identification of candidate genes and variants for adiposity traits in outbred rats. Obesity 2018, 26, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Chitre, A.S.; Polesskaya, O.; Holl, K.; Gao, J.; Cheng, R.; Bimschleger, H.; Martinez, A.G.; George, T.; Gileta, A.F.; Han, W.; et al. Genome-wide association study in 3,173 outbred rats identifies multiple loci for body weight, adiposity, and fasting glucose. Obesity 2020, 28, 1964–1973. [Google Scholar] [CrossRef]
- Crabbe, J.C.; Wahlsten, D.; Dudek, B.C. Genetics of mouse behavior: Interactions with laboratory environment. Science 1999, 284, 1670–1672. [Google Scholar] [CrossRef] [Green Version]
- Lawson, H.A.; Cheverud, J.M. Metabolic syndrome components in murine models. Endocr. Metab. Immune. Disord. Drug Targets 2010, 10, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, R.A.; Weinstock, G.M.; Metzker, M.L.; Muzny, D.M.; Sodergren, E.J.; Scherer, S.; Scott, G.; Steffen, D.; Worley, K.C.; Burch, P.E.; et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 2004, 428, 493–521. [Google Scholar]
- Mouse Genome Sequencing, C.; Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef]
- Ramdas, S.; Ozel, A.B.; Treutelaar, M.K.; Holl, K.; Mandel, M.; Woods, L.C.S.; Li, J.Z. Extended regions of suspected mis-assembly in the rat reference genome. Sci. Data 2019, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Keane, T.M.; Goodstadt, L.; Danecek, P.; White, M.A.; Wong, K.; Yalcin, B.; Heger, A.; Agam, A.; Slater, G.; Goodson, M.; et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 2011, 477, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, B.; Wong, K.; Agam, A.; Goodson, M.; Keane, T.M.; Gan, X.; Nellåker, C.; Goodstadt, L.; Nicod, J.; Bhomra, A.; et al. Sequence-based characterization of structural variation in the mouse genome. Nature 2011, 477, 326–329. [Google Scholar] [CrossRef] [Green Version]
- Agapite, J.; Albou, L.-P.; Aleksander, S.; Argasinska, J.; Arnaboldi, V.; Attrill, H.; Bello, S.M.; Blake, J.A.; Blodgett, O.; Bradford, Y.M.; et al. Alliance of Genome Resources Portal: Unified model organism research platform. Nucleic Acids Res. 2020, 48, D650–D658. [Google Scholar]
- Hu, Y.; Flockhart, I.; Vinayagam, A.; Bergwitz, C.; Berger, B.; Perrimon, N.; Mohr, S.E. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinform. 2011, 12, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.L.; Hauser, S.R.; Liang, T.; Sari, Y.; Maldonado-Devincci, A.; Rodd, Z.A. Rat animal models for screening medications to treat alcohol use disorders. Neuropharmacology 2017, 122, 201–243. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Jiang, Z.; Georgel, P.; Crozat, K.; Croker, B.; Rutschmann, S.; Du, X.; Hoebe, K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu. Rev. Immunol. 2006, 24, 353–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, F.W.; Kruglyak, L. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 2015, 16, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Crabbe, J.C. Chapter 5—Use of Animal Models of Alcohol-Related Behavior. In Handbook of Clinical Neurology; Sullivan, E.V., Pfefferbaum, A., Eds.; Alcohol and the Nervous System; Elsevier: Amsterdam, The Netherlands, 2014; Volume 125, pp. 71–86. [Google Scholar]
- Milner, L.C.; Buck, K.J. Identifying Quantitative Trait Loci (QTLs) and Genes (QTGs) for Alcohol-Related Phenotypes in Mice. In International Review of Neurobiology; Reilly, M.T., Lovinger, D.M., Eds.; Functional plasticity and genetic variation: Insights into the neurobiology of alcoholism; Academic Press: Cambridge, MA, USA, 2010; Volume 91, pp. 173–204. [Google Scholar]
- Crabbe, J.C.; Young, E.R.; Deutsch, C.M.; Tam, B.R.; Kosobud, A. Mice genetically selected for differences in open-field activity after ethanol. Pharmacol. Biochem. Behav. 1987, 27, 577–581. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Hallett, C.L.; Niehus, D.R.; Hunter, J.S.; Nouth, L.; Risinger, F.O. Assessment of ethanol’s hedonic effects in mice selectively bred for sensitivity to ethanol-induced hypothermia. Psychopharmacology 1991, 105, 84–92. [Google Scholar] [CrossRef]
- Friedman, H.J.; Bass, M.B.; Lester, D. Ethanol-induced analgesia in rats selectively bred for ethanol sensitivity. Pharmacol. Biochem. Behav. 1980, 13, 773–776. [Google Scholar] [CrossRef]
- Kulkosky, P.J.; Carr, B.A.; Flores, R.K.; LaHeist, A.F.; Hopkins, L.M. Conditioned taste aversions induced by alcohol and lithium in rats selectively bred for ethanol neurosensitivity. Alcohol. Clin. Exp. Res. 1995, 19, 945–950. [Google Scholar] [CrossRef]
- Mayer, J.M.; Khanna, J.M.; Kim, C.; Kalant, H. Differential pharmacological responses to ethanol, pentobarbital and morphine in rats selectively bred for ethanol sensitivity. Psychopharmacology 1983, 81, 6–9. [Google Scholar] [CrossRef] [PubMed]
- McClearn, G.; Kakihana, R. Selective breeding for ethanol sensitivity in mice. Behav. Genet. 1973, 3, 409–410. [Google Scholar]
- Palmer, A.A.; McKinnon, C.S.; Bergstrom, H.C.; Phillips, T.J. Locomotor activity responses to ethanol, other alcohols, and GABA-A acting compounds in forward- and reverse-selected FAST and SLOW mouse lines. Behav. Neurosci. 2002, 116, 958–967. [Google Scholar] [CrossRef]
- Crabbe, J.C.; Kosobud, A.; Tam, B.R.; Young, E.R.; Deutsch, C.M. Genetic selection of mouse lines sensitive (cold) and resistant (hot) to acute ethanol hypothermia. Alcohol Drug Res. 1987, 7, 163–174. [Google Scholar] [PubMed]
- Riley, E.P.; Worsham, E.D.; Lester, D.; Freed, E.X. Selective breeding of rats for differences in reactivity to alcohol. An approach to an animal model of alcoholism. II. Behavioral measures. J. Stud. Alcohol 1977, 38, 1705–1717. [Google Scholar] [CrossRef]
- Riley, E.P.; Freed, E.X.; Lester, D. Selective breeding of rats for differences in reactivity to alcohol. An approach to an animal model of alcoholism. I. General procedures. J. Stud. Alcohol 1976, 37, 1535–1547. [Google Scholar] [CrossRef]
- Baud, A.; Flint, J. Identifying genes for neurobehavioural traits in rodents: Progress and pitfalls. Dis. Models Mech. 2017, 10, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Baud, A.; Guryev, V.; Hummel, O.; Johannesson, M.; Rat Genome Sequencing and Mapping Consortium; Flint, J. Genomes and phenomes of a population of outbred rats and its progenitors. Sci. Data 2014, 1, 140011. [Google Scholar] [CrossRef]
- Bennett, B.J.; Farber, C.R.; Orozco, L.; Kang, H.M.; Ghazalpour, A.; Siemers, N.; Neubauer, M.; Neuhaus, I.; Yordanova, R.; Guan, B.; et al. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010, 20, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Churchill, G.A.; Airey, D.C.; Allayee, H.; Angel, J.M.; Attie, A.D.; Beatty, J.; Beavis, W.D.; Belknap, J.K.; Bennett, B.; Berrettini, W.; et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 2004, 36, 1133–1137. [Google Scholar]
- Crabbe, J.C.; Belknap, J.K.; Mitchell, S.R.; Crawshaw, L.I. Quantitative trait loci mapping of genes that influence the sensitivity and tolerance to ethanol-induced hypothermia in BXD recombinant inbred mice. J. Pharmacol. Exp. Ther. 1994, 269, 184–192. [Google Scholar] [PubMed]
- Hansen, C.; Spuhler, K. Development of the National Institutes of Health Genetically Heterogeneous Rat Stock. Alcohol. Clin. Exp. Res. 1984, 8, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Mott, R.; Flint, J. Simultaneous detection and fine mapping of quantitative trait loci in mice using heterogeneous stocks. Genetics 2002, 160, 1609–1618. [Google Scholar] [PubMed]
- Philip, V.M.; Sokoloff, G.; Ackert-Bicknell, C.L.; Striz, M.; Branstetter, L.; Beckmann, M.A.; Spence, J.S.; Jackson, B.L.; Galloway, L.D.; Barker, P.; et al. Genetic analysis in the Collaborative Cross breeding population. Genome Res. 2011, 21, 1223–1238. [Google Scholar] [CrossRef] [Green Version]
- Soller, M.; Abu-Toamih Atamni, H.J.; Binenbaum, I.; Chatziioannou, A.; Iraqi, F.A. Designing a QTL Mapping Study for implementation in the realized collaborative cross genetic reference population. Curr. Protoc. Mouse Biol. 2019, 9, e66. [Google Scholar] [CrossRef]
- Tabakoff, B.; Smith, H.; Vanderlinden, L.A.; Hoffman, P.L.; Saba, L.M. Networking in Biology: The Hybrid Rat Diversity Panel. In Rat Genomics; Hayman, G.T., Smith, J.R., Dwinell, M.R., Shimoyama, M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 213–231. ISBN 978-1-4939-9581-3. [Google Scholar]
- Valdar, W.; Solberg, L.C.; Gauguier, D.; Burnett, S.; Klenerman, P.; Cookson, W.O.; Taylor, M.S.; Rawlins, J.N.P.; Mott, R.; Flint, J. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat. Genet. 2006, 38, 879–887. [Google Scholar] [CrossRef]
- Yalcin, B.; Flint, J.; Mott, R. Using progenitor strain information to identify quantitative trait nucleotides in outbred mice. Genetics 2005, 171, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Churchill, G.A.; Gatti, D.M.; Munger, S.C.; Svenson, K.L. The Diversity Outbred mouse population. Mamm. Genome 2012, 23, 713–718. [Google Scholar] [CrossRef] [Green Version]
- Collaborative Cross Consortium. The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics 2012, 190, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.W.; Williams, E.G. Resources for Systems Genetics. In Systems Genetics: Methods and Protocols; Schughart, K., Williams, R.W., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 3–29. ISBN 978-1-4939-6427-7. [Google Scholar]
- Goldowitz, D.; Frankel, W.N.; Takahashi, J.S.; Holtz-Vitaterna, M.; Bult, C.; Kibbe, W.A.; Snoddy, J.; Li, Y.; Pretel, S.; Yates, J.; et al. Large-scale mutagenesis of the mouse to understand the genetic bases of nervous system structure and function. Mol. Brain Res. 2004, 132, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Bryant, C.D.; Smith, D.J.; Kantak, K.M.; Nowak, T.S.; Williams, R.W.; Damaj, M.I.; Redei, E.E.; Chen, H.; Mulligan, M.K. Facilitating complex trait analysis via reduced complexity crosses. Trends Genet. 2020, 36, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, N.M.; Palmer, A.A. Fine-mapping QTLs in advanced intercross lines and other outbred populations. Mamm. Genome 2014, 25, 271–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, B.; Larson, C.; Richmond, P.A.; Odell, A.T.; Saba, L.M.; Tabakoff, B.; Dowell, R.; Radcliffe, R.A. Quantitative trait locus mapping of acute functional tolerance in the LXS recombinant inbred strains. Alcohol. Clin. Exp. Res. 2015, 39, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Crabbe, J.C.; Kosobud, A.; Young, E.R.; Janowsky, J.S. Polygenic and single-gene determination of responses to ethanol in BXD/Ty recombinant inbred mouse strains. Neurobehav. Toxicol Teratol. 1983, 5, 181–187. [Google Scholar] [PubMed]
- Hitzemann, R.; Phillips, T.J.; Lockwood, D.R.; Darakjian, P.; Searles, R.P. Phenotypic and gene expression features associated with variation in chronic ethanol consumption in heterogeneous stock collaborative cross mice. Genomics 2020, 112, 4516–4524. [Google Scholar] [CrossRef]
- Hoffman, P.L.; Saba, L.M.; Vanderlinden, L.A.; Tabakoff, B. Voluntary exposure to a toxin: The genetic influence on ethanol consumption. Mamm. Genome. 2018, 29, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Lusk, R.; Saba, L.M.; Vanderlinden, L.A.; Zidek, V.; Silhavy, J.; Pravenec, M.; Hoffman, P.L.; Tabakoff, B. Unsupervised, statistically based systems biology approach for unraveling the genetics of complex traits: A demonstration with ethanol metabolism. Alcohol. Clin. Exp. Res. 2018, 42, 1177–1191. [Google Scholar] [CrossRef]
- Mulligan, M.K.; Lu, L.; Cavigelli, S.A.; Mormède, P.; Terenina, E.; Zhao, W.; Williams, R.W.; Jones, B.C. Impact of genetic variation on stress-related ethanol consumption. Alcohol. Clin. Exp. Res. 2019, 43, 1391–1402. [Google Scholar] [CrossRef]
- Pravenec, M.; Klír, P.; Kren, V.; Zicha, J.; Kunes, J. An analysis of spontaneous hypertension in spontaneously hypertensive rats by means of new recombinant inbred strains. J. Hypertens. 1989, 7, 217–221. [Google Scholar] [CrossRef]
- Putman, A.H.; Wolen, A.R.; Harenza, J.L.; Yordanova, R.K.; Webb, B.T.; Chesler, E.J.; Miles, M.F. Identification of quantitative trait loci and candidate genes for an anxiolytic-like response to ethanol in BXD recombinant inbred strains. Genes Brain Behav. 2016, 15, 367–381. [Google Scholar] [CrossRef] [Green Version]
- Radcliffe, R.A.; Larson, C.; Bennett, B. Genetic studies of acute tolerance, rapid tolerance, and drinking in the dark in the LXS recombinant inbred strains. Alcohol. Clin. Exp. Res. 2013, 37, 2019–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, L.M.; Flink, S.C.; Vanderlinden, L.A.; Israel, Y.; Tampier, L.; Colombo, G.; Kiianmaa, K.; Bell, R.L.; Printz, M.P.; Flodman, P.; et al. The sequenced rat brain transcriptome—Its use in identifying networks predisposing alcohol consumption. FEBS J. 2015, 282, 3556–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théberge, E.T.; Baker, J.A.; Dubose, C.; Boyle, J.K.; Balce, K.; Goldowitz, D.; Hamre, K.M. Genetic influences on the amount of cell death in the neural tube of bxd mice exposed to acute ethanol at midgestation. Alcohol. Clin. Exp. Res. 2019, 43, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.W.; Bennett, B.; Lu, L.; Gu, J.; DeFries, J.C.; Carosone–Link, P.J.; Rikke, B.A.; Belknap, J.K.; Johnson, T.E. Genetic structure of the LXS panel of recombinant inbred mouse strains: A powerful resource for complex trait analysis. Mamm. Genome. 2004, 15, 637–647. [Google Scholar] [CrossRef]
- Goodson, M.; Rust, M.B.; Witke, W.; Bannerman, D.; Mott, R.; Ponting, C.P.; Flint, J. Cofilin-1: A modulator of anxiety in mice. PLoS Genet. 2012, 8, e1002970. [Google Scholar] [CrossRef] [Green Version]
- Hettema, J.M.; Webb, B.T.; Guo, A.-Y.; Zhao, Z.; Maher, B.S.; Chen, X.; An, S.-S.; Sun, C.; Aggen, S.H.; Kendler, K.S.; et al. Prioritization and association analysis of murine-derived candidate genes in anxiety-spectrum disorders. Biol. Psychiatry 2011, 70, 888–896. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.H.; Schultz, L.E.; Gulati, A.; Su, A.I.; Pletcher, M.T. Phenotypic characterization of a genetically diverse panel of mice for behavioral despair and anxiety. PLoS ONE 2010, 5, e14458. [Google Scholar] [CrossRef]
- Ghazalpour, A.; Doss, S.; Kang, H.; Farber, C.; Wen, P.-Z.; Brozell, A.; Castellanos, R.; Eskin, E.; Smith, D.J.; Drake, T.A.; et al. High-resolution mapping of gene expression using association in an outbred mouse stock. PLoS Genet. 2008, 4, e1000149. [Google Scholar] [CrossRef]
- Díaz-Morán, S.; Palència, M.; Mont-Cardona, C.; Cañete, T.; Blázquez, G.; Martínez-Membrives, E.; López-Aumatell, R.; Sabariego, M.; Donaire, R.; Morón, I.; et al. Gene expression in amygdala as a function of differential trait anxiety levels in genetically heterogeneous NIH-HS rats. Behav. Brain Res. 2013, 252, 422–431. [Google Scholar] [CrossRef]
- Hughson, A.R.; Horvath, A.P.; Holl, K.; Palmer, A.A.; Solberg Woods, L.C.; Robinson, T.E.; Flagel, S.B. Incentive salience attribution, “sensation-seeking” and “novelty-seeking” are independent traits in a large sample of male and female heterogeneous stock rats. Sci. Rep. 2019, 9, 2351. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.G.; Auwerx, J. The convergence of systems and reductionist approaches in complex trait analysis. Cell 2015, 162, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deisseroth, K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat. Neurosci. 2015, 18, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rost, B.R.; Schneider-Warme, F.; Schmitz, D.; Hegemann, P. Optogenetic tools for subcellular applications in neuroscience. Neuron 2017, 96, 572–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieger, K.; Belbellaa, B.; le Guiner, C.; Moullier, P.; Rolling, F. In vivo gene regulation using tetracycline-regulatable systems. Adv. Drug Deliv. Rev. 2009, 61, 527–541. [Google Scholar] [CrossRef]
- Phillips, T.J.; Hen, R.; Crabbe, J.C. Complications associated with genetic background effects in research using knockout mice. Psychopharmacology 1999, 147, 5–7. [Google Scholar] [CrossRef]
- Cui, X.; Ji, D.; Fisher, D.A.; Wu, Y.; Briner, D.M.; Weinstein, E.J. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat. Biotechnol. 2011, 29, 64–67. [Google Scholar] [CrossRef]
- Geurts, A.M.; Cost, G.J.; Freyvert, Y.; Zeitler, B.; Miller, J.C.; Choi, V.M.; Jenkins, S.S.; Wood, A.; Cui, X.; Meng, X.; et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 2009, 325, 433. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef]
- Wood, A.J.; Lo, T.-W.; Zeitler, B.; Pickle, C.S.; Ralston, E.J.; Lee, A.H.; Amora, R.; Miller, J.C.; Leung, E.; Meng, X.; et al. Targeted genome editing across species using ZFNs and TALENs. Science 2011, 333, 307. [Google Scholar] [CrossRef] [Green Version]
- Aitman, T.; Dhillon, P.; Geurts, A.M. A RATional choice for translational research? Dis. Model Mech. 2016, 9, 1069–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, R.A.; Bozarth, M.A. A psychomotor stimulant theory of addiction. Psychol. Rev. 1987, 94, 469–492. [Google Scholar] [CrossRef] [PubMed]
- Di Chiara, G.; Imperato, A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 1988, 85, 5274–5278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boileau, I.; Assaad, J.-M.; Pihl, R.O.; Benkelfat, C.; Leyton, M.; Diksic, M.; Tremblay, R.E.; Dagher, A. Alcohol promotes dopamine release in the human nucleus accumbens. Synapse 2003, 49, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Gilman, J.M.; Ramchandani, V.A.; Davis, M.B.; Bjork, J.M.; Hommer, D.W. Why we like to drink: A functional magnetic resonance imaging study of the rewarding and anxiolytic effects of alcohol. J. Neurosci. 2008, 28, 4583–4591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.J.; Volkow, N.D.; Franceschi, D.; Fowler, J.S.; Thanos, P.K.; Scherbaum, N.; Pappas, N.; Wong, C.T.; Hitzemann, R.J.; Felder, C.A. Regional brain metabolism during alcohol intoxication. Alcohol. Clin. Exp. Res. 2000, 24, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Yoder, K.K.; Kareken, D.A.; Seyoum, R.A.; O’connor, S.J.; Wang, C.; Zheng, Q.-H.; Mock, B.; Morris, E.D. Dopamine D(2) receptor availability is associated with subjective responses to alcohol. Alcohol. Clin. Exp. Res. 2005, 29, 965–970. [Google Scholar] [CrossRef] [Green Version]
- Martinez, D.; Slifstein, M.; Broft, A.; Mawlawi, O.; Hwang, D.-R.; Huang, Y.; Cooper, T.; Kegeles, L.; Zarahn, E.; Abi-Dargham, A.; et al. Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: Amphetamine-induced dopamine release in the functional subdivisions of the striatum. J. Cereb. Blood Flow Metab. 2003, 23, 285–300. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Gatley, S.J.; Ding, Y.S.; Logan, J.; Dewey, S.L.; Hitzemann, R.; Lieberman, J. Relationship between psychostimulant-induced “high” and dopamine transporter occupancy. Proc. Natl. Acad. Sci. USA 1996, 93, 10388–10392. [Google Scholar] [CrossRef] [Green Version]
- Yoder, K.K.; Constantinescu, C.C.; Kareken, D.A.; Normandin, M.D.; Cheng, T.-E.; O’Connor, S.J.; Morris, E.D. Heterogeneous effects of alcohol on dopamine release in the striatum: A PET study. Alcohol. Clin. Exp. Res. 2007, 31, 965–973. [Google Scholar] [CrossRef]
- Demarest, K.; Koyner, J.; McCaughran, J.; Cipp, L.; Hitzemann, R. Further characterization and high-resolution mapping of quantitative trait loci for ethanol-induced locomotor activity. Behav. Genet. 2001, 31, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Downing, C.; Rodd-Henricks, K.K.; Flaherty, L.; Dudek, B.C. Genetic analysis of the psychomotor stimulant effect of ethanol. Genes. Brain Behav. 2003, 2, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Downing, C.; Carosone-Link, P.; Bennett, B.; Johnson, T. QTL mapping for low-dose ethanol activation in the LXS recombinant inbred strains. Alcohol. Clin. Exp. Res. 2006, 30, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- DuBose, C.S.; Chesler, E.J.; Goldowitz, D.; Hamre, K.M. Use of the expanded panel of BXD mice narrow QTL regions in ethanol-induced locomotor activation and motor incoordination. Alcohol. Clin. Exp. Res. 2013, 37, 170–183. [Google Scholar] [CrossRef]
- Hitzemann, R.; Cipp, L.; Demarest, K.; Mahjubi, E.; McCaughran, J. Genetics of ethanol-induced locomotor activation: Detection of QTLs in a C57BL/6J x DBA/2J F2 intercross. Mamm. Genome 1998, 9, 956–962. [Google Scholar] [CrossRef]
- Hitzemann, R.; Malmanger, B.; Cooper, S.; Coulombe, S.; Reed, C.; Demarest, K.; Koyner, J.; Cipp, L.; Flint, J.; Talbot, C.; et al. Multiple cross mapping (MCM) markedly improves the localization of a QTL for ethanol-induced activation. Genes Brain Behav. 2002, 1, 214–222. [Google Scholar] [CrossRef]
- Palmer, A.A.; Lessov-Schlaggar, C.N.; Ponder, C.A.; McKinnon, C.S.; Phillips, T.J. Sensitivity to the locomotor-stimulant effects of ethanol and allopregnanolone: A quantitative trait locus study of common genetic influence. Genes Brain Behav. 2006, 5, 506–517. [Google Scholar] [CrossRef]
- Phillips, T.J.; Huson, M.; Gwiazdon, C.; Burkhart-Kasch, S.; Shen, E.H. Effects of acute and repeated ethanol exposures on the locomotor activity of BXD recombinant inbred mice. Alcohol. Clin. Exp. Res. 1995, 19, 269–278. [Google Scholar] [CrossRef]
- Risinger, F.O.; Malott, D.H.; Prather, L.K.; Niehus, D.R.; Cunningham, C.L. Motivational properties of ethanol in mice selectively bred for ethanol-induced locomotor differences. Psychopharmacology 1994, 116, 207–216. [Google Scholar] [CrossRef]
- Sanchez, F.P.; Dickenson, L.; George, F.R. Ethanol self-administration is genetically independent of locomotor stimulation in fast and slow mice. Alcohol 1996, 13, 79–84. [Google Scholar] [CrossRef]
- Agabio, R.; Carai, M.A.; Lobina, C.; Pani, M.; Reali, R.; Vacca, G.; Gessa, G.L.; Colombo, G. Alcohol stimulates motor activity in selectively bred Sardinian alcohol-preferring (sP), but not in Sardinian alcohol-nonpreferring (sNP), rats. Alcohol 2001, 23, 123–126. [Google Scholar] [CrossRef]
- Quintanilla, M.E. Effect of low doses of ethanol on spontaneous locomotor activity in UChB and UChA rats. Addict. Biol. 1999, 4, 443–448. [Google Scholar] [CrossRef]
- Rodd, Z.A.; Bell, R.L.; McKinzie, D.L.; Webster, A.A.; Murphy, J.M.; Lumeng, L.; Li, T.-K.; McBride, W.J. Low-dose stimulatory effects of ethanol during adolescence in rat lines selectively bred for high alcohol intake. Alcohol. Clin. Exp. Res. 2004, 28, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Krimmer, E.C.; Schechter, M.D. HAD and LAD rats respond differently to stimulating effect but not discriminative effects of ethanol. Alcohol 1992, 9, 71–74. [Google Scholar] [CrossRef]
- Waller, M.B.; Murphy, J.M.; McBride, W.J.; Lumeng, L.; Li, T.K. Effect of low dose ethanol on spontaneous motor activity in alcohol-preferring and -nonpreferring lines of rats. Pharmacol. Biochem. Behav. 1986, 24, 617–623. [Google Scholar] [CrossRef]
- Phillips, T.J.; Broadbent, J.; Burkhart-Kasch, S.; Henderson, C.; Wenger, C.D.; McMullin, C.; McKinnon, C.S.; Cunningham, C.L. Genetic correlational analyses of ethanol reward and aversion phenotypes in short-term selected mouse lines bred for ethanol drinking or ethanol-induced conditioned taste aversion. Behav. Neurosci. 2005, 119, 892–910. [Google Scholar] [CrossRef]
- Bettinger, J.C.; Davies, A.G. The role of the BK channel in ethanol response behaviors: Evidence from model organism and human studies. Front. Physiol. 2014, 5, 346. [Google Scholar] [CrossRef] [Green Version]
- Torres, Y.P.; Morera, F.J.; Carvacho, I.; Latorre, R. A Marriage of convenience: β-subunits and voltage-dependent K+ channels. J. Biol. Chem. 2007, 282, 24485–24489. [Google Scholar] [CrossRef] [Green Version]
- Pongs, O.; Schwarz, J.R. Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 2010, 90, 755–796. [Google Scholar] [CrossRef] [Green Version]
- Kerns, R.T.; Ravindranathan, A.; Hassan, S.; Cage, M.P.; York, T.; Sikela, J.M.; Williams, R.W.; Miles, M.F. Ethanol-responsive brain region expression networks: Implications for behavioral responses to acute ethanol in DBA/2J versus C57BL/6J mice. J. Neurosci. 2005, 25, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Crawley, J.N.; Belknap, J.K.; Collins, A.; Crabbe, J.C.; Frankel, W.; Henderson, N.; Hitzemann, R.J.; Maxson, S.C.; Miner, L.L.; Silva, A.J.; et al. Behavioral phenotypes of inbred mouse strains: Implications and recommendations for molecular studies. Psychopharmacology 1997, 132, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Wolen, A.R.; Phillips, C.A.; Langston, M.A.; Putman, A.H.; Vorster, P.J.; Bruce, N.A.; York, T.P.; Williams, R.W.; Miles, M.F. Genetic dissection of acute ethanol responsive gene networks in prefrontal cortex: Functional and mechanistic implications. PLoS ONE 2012, 7, e33575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendler, K.S.; Kalsi, G.; Holmans, P.A.; Sanders, A.R.; Aggen, S.H.; Dick, D.M.; Aliev, F.; Shi, J.; Levinson, D.F.; Gejman, P.V. Genomewide association analysis of symptoms of alcohol dependence in the molecular genetics of schizophrenia (MGS2) control sample. Alcohol. Clin. Exp. Res. 2011, 35, 963–975. [Google Scholar] [CrossRef]
- Edenberg, H.J.; Koller, D.L.; Xuei, X.; Wetherill, L.; McClintick, J.N.; Almasy, L.; Bierut, L.J.; Bucholz, K.K.; Goate, A.; Aliev, F.; et al. Genome-wide association study of alcohol dependence implicates a region on chromosome 11. Alcohol. Clin. Exp. Res. 2010, 34, 840–852. [Google Scholar] [CrossRef]
- Han, S.; Yang, B.-Z.; Kranzler, H.R.; Liu, X.; Zhao, H.; Farrer, L.A.; Boerwinkle, E.; Potash, J.B.; Gelernter, J. Integrating GWASs and human protein interaction networks identifies a gene subnetwork underlying alcohol dependence. Am. J. Hum. Genet. 2013, 93, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.E.; Hendrickson, L.M.; Penta, K.L.; Friesen, R.M.; Pietrzykowski, A.Z.; Tapper, A.R.; Treistman, S.N. Identification of a BK channel auxiliary protein controlling molecular and behavioral tolerance to alcohol. Proc. Natl. Acad. Sci. USA 2008, 105, 17543–17548. [Google Scholar] [CrossRef] [Green Version]
- Kreifeldt, M.; Le, D.; Treistman, S.N.; Koob, G.F.; Contet, C. BK channel β1 and β4 auxiliary subunits exert opposite influences on escalated ethanol drinking in dependent mice. Front. Integr. Neurosci. 2013, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Crabbe, J.C.; Cameron, A.J.; Munn, E.; Bunning, M.; Wahlsten, D. Overview of mouse assays of ethanol intoxication. Curr. Protoc. Neurosci. 2008, 42, 9–26. [Google Scholar] [CrossRef]
- Crabbe, J.C.; Metten, P.; Cameron, A.J.; Wahlsten, D. An analysis of the genetics of alcohol intoxication in inbred mice. Neurosci. Biobehav. Rev. 2005, 28, 785–802. [Google Scholar] [CrossRef] [Green Version]
- Morean, M.E.; Corbin, W.R. Subjective response to alcohol: A critical review of the literature. Alcohol. Clin. Exp. Res. 2010, 34, 385–395. [Google Scholar] [CrossRef]
- LeBlanc, A.E.; Kalant, H.; Gibbins, R.J. Acute tolerance to ethanol in the rat. Psychopharmacologia 1975, 41, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Vogel-Sprott, M.; Fillmore, M.T. Impairment and recovery under repeated doses of alcohol: Effects of response-outcomes. Pharmacol. Biochem. Behav. 1993, 45, 59–63. [Google Scholar] [CrossRef]
- Vogel-Sprott, M.; Chipperfield, B. Family history of problem drinking among young male social drinkers: Behavioral effects of alcohol. J. Stud. Alcohol 1987, 48, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Boecker, H.; Wills, A.J.; Ceballos-Baumann, A.; Samuel, M.; Thompson, P.D.; Findley, L.J.; Brooks, D.J. The effect of ethanol on alcohol-responsive essential tremor: A positron emission tomography study. Ann. Neurol. 1996, 39, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Hanchar, H.J.; Dodson, P.D.; Olsen, R.W.; Otis, T.S.; Wallner, M. Alcohol-induced motor impairment caused by increased extrasynaptic GABA(A) receptor activity. Nat. Neurosci. 2005, 8, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Mullani, N.; Gould, L.; Adler, S.S.; Guynn, R.W.; Overall, J.E.; Dewey, S. Effects of acute alcohol intoxication on cerebral blood flow measured with PET. Psychiatry Res. 1988, 24, 201–209. [Google Scholar] [CrossRef]
- Browman, K.E.; Crabbe, J.C. Quantitative trait loci affecting ethanol sensitivity in BXD recombinant inbred mice. Alcohol. Clin. Exp. Res. 2000, 24, 17–23. [Google Scholar] [CrossRef]
- Chesler, E.J.; Plitt, A.; Fisher, D.; Hurd, B.; Lederle, L.; Bubier, J.A.; Kiselycznyk, C.; Holmes, A. Quantitative trait loci for sensitivity to ethanol intoxication in a C57BL/6J×129S1/SvImJ inbred mouse cross. Mamm. Genome 2012, 23, 305–321. [Google Scholar] [CrossRef] [Green Version]
- Gehle, V.M.; Erwin, V.G. The genetics of acute functional tolerance and initial sensitivity to ethanol for an ataxia test in the LSxSS RI strains. Alcohol. Clin. Exp. Res. 2000, 24, 579–587. [Google Scholar] [CrossRef]
- Radcliffe, R.A.; Erwin, V.G.; Bludeau, P.; Deng, X.; Fay, T.; Floyd, K.L.; Deitrich, R.A. A major QTL for acute ethanol sensitivity in the alcohol tolerant and non-tolerant selected rat lines. Genes Brain Behav. 2009, 8, 611–625. [Google Scholar] [CrossRef] [Green Version]
- Crabbe, J.C.; Bell, R.L.; Ehlers, C.L. Human and laboratory rodent low response to alcohol: Is better consilience possible? Addict. Biol. 2010, 15, 125–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, B.M.; Grahame, N.J.; Boehm, S.L. Selection for high alcohol preference drinking in mice results in heightened sensitivity and rapid development of acute functional tolerance to alcohol’s ataxic effects. Genes Brain Behav. 2013, 12, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, B.M.; Cordero, K.A.; Barkley-Levenson, A.M.; Metten, P.; Crabbe, J.C.; Boehm, S.L. Genetic relationship between predisposition for binge alcohol consumption and blunted sensitivity to adverse effects of alcohol in mice. Alcohol. Clin. Exp. Res. 2014, 38, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Barker, J.S.; Hines, R.M. Regulation of GABAA receptor subunit expression in substance use disorders. Int. J. Mol. Sci. 2020, 21, 4445. [Google Scholar] [CrossRef] [PubMed]
- Blednov, Y.A.; Borghese, C.M.; McCracken, M.L.; Benavidez, J.M.; Geil, C.R.; Osterndorff-Kahanek, E.; Werner, D.F.; Iyer, S.; Swihart, A.; Harrison, N.L.; et al. Loss of ethanol conditioned taste aversion and motor stimulation in knockin mice with ethanol-insensitive α2-containing GABA(A) receptors. J. Pharmacol. Exp. Ther. 2011, 336, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Blednov, Y.A.; Benavidez, J.M.; Black, M.; Chandra, D.; Homanics, G.E.; Rudolph, U.; Harris, R.A. Linking GABA(A) receptor subunits to alcohol-induced conditioned taste aversion and recovery from acute alcohol intoxication. Neuropharmacology 2013, 67, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, S.L.; Ponomarev, I.; Jennings, A.W.; Whiting, P.J.; Rosahl, T.W.; Garrett, E.M.; Blednov, Y.A.; Harris, R.A. gamma-Aminobutyric acid A receptor subunit mutant mice: New perspectives on alcohol actions. Biochem. Pharmacol. 2004, 68, 1581–1602. [Google Scholar] [CrossRef] [PubMed]
- Mihic, S.J.; Harris, R.A. Inhibition of rho1 receptor GABAergic currents by alcohols and volatile anesthetics. J. Pharmacol. Exp. Ther. 1996, 277, 411–416. [Google Scholar]
- Xuei, X.; Flury-Wetherill, L.; Dick, D.; Goate, A.; Tischfield, J.; Nurnberger, J.; Schuckit, M.; Kramer, J.; Kuperman, S.; Hesselbrock, V.; et al. GABRR1 and GABRR2, encoding the GABA-A receptor subunits rho1 and rho2, are associated with alcohol dependence. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153, 418–427. [Google Scholar] [CrossRef] [Green Version]
- Blednov, Y.A.; Borghese, C.M.; Ruiz, C.I.; Cullins, M.A.; Da Costa, A.; Osterndorff-Kahanek, E.A.; Homanics, G.E.; Harris, R.A. Mutation of the inhibitory ethanol site in GABAA ρ1 receptors promotes tolerance to ethanol-induced motor incoordination. Neuropharmacology 2017, 123, 201–209. [Google Scholar] [CrossRef]
- Gill, K.; Liu, Y.; Deitrich, R.A. Voluntary alcohol consumption in BXD recombinant inbred mice: Relationship to alcohol metabolism. Alcohol. Clin. Exp. Res. 1996, 20, 185–190. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Benavidez, J.M.; Black, M.; Leiter, C.R.; Osterndorff-Kahanek, E.; Johnson, D.; Borghese, C.M.; Hanrahan, J.R.; Johnston, G.A.R.; Chebib, M.; et al. GABAA receptors containing ρ1 subunits contribute to in vivo effects of ethanol in mice. PLoS ONE 2014, 9, e85525. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.; Beeson, M.; Gordon, L.; Johnson, T.E. Quick method for confirmation of quantitative trait loci. Alcohol. Clin. Exp. Res. 1997, 21, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.; Carosone-Link, P.; Zahniser, N.R.; Johnson, T.E. Confirmation and fine mapping of ethanol sensitivity quantitative trait loci, and candidate gene testing in the LXS recombinant inbred mice. J. Pharmacol. Exp. Ther. 2006, 319, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.; Carosone-Link, P.; Beeson, M.; Gordon, L.; Phares-Zook, N.; Johnson, T.E. Genetic dissection of quantitative trait locus for ethanol sensitivity in long- and short-sleep mice. Genes Brain Behav. 2008, 7, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.C.; Johnson, T.E.; Markel, P.D.; Clark, V.J.; Fulker, D.W.; Corley, R.P.; Collins, A.C.; Wehner, J.M. Quantitative trait locus analyses of sleep-times induced by sedative-hypnotics in LSXSS recombinant inbred strains of mice. Alcohol. Clin. Exp. Res. 1996, 20, 543–550. [Google Scholar] [CrossRef] [PubMed]
- MacLaren, E.J.; Bennett, B.; Johnson, T.E.; Sikela, J.M. Expression profiling identifies novel candidate genes for ethanol sensitivity QTLs. Mamm. Genome 2006, 17, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Mandt, B.H.; Larson, C.; Fay, T.; Bludeau, P.; Allen, R.M.; Deitrich, R.A.; Radcliffe, R.A. Quantitative trait loci for sensitivity to acute ethanol and ethanol consummatory behaviors in rats. Alcohol 2018, 66, 55–67. [Google Scholar] [CrossRef]
- Markel, P.D.; Bennett, B.; Beeson, M.; Gordon, L.; Johnson, T.E. Confirmation of quantitative trait loci for ethanol sensitivity in long-sleep and short-sleep mice. Genome Res. 1997, 7, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Radcliffe, R.A.; Bohl, M.L.; Lowe, M.V.; Cycowski, C.S.; Wehner, J.M. Mapping of quantitative trait loci for hypnotic sensitivity to ethanol in crosses derived from the C57BL/6 and DBA/2 mouse strains. Alcohol. Clin. Exp. Res. 2000, 24, 1335–1342. [Google Scholar] [CrossRef]
- Radcliffe, R.A.; Bludeau, P.; Deng, X.-S.; Erwin, V.G.; Deitrich, R.A. Short-term selection for acute ethanol tolerance and sensitization from an F2 population derived from the high and low alcohol-sensitive selectively bred rat lines. Alcohol 2007, 41, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wit, H.; Metz, J.; Wagner, N.; Cooper, M. Behavioral and subjective effects of ethanol: Relationship to cerebral metabolism using PET. Alcohol. Clin. Exp. Res. 1990, 14, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Beleslin, D.B.; DjokanoviΔ, N.; JovanoviΔ-MiΔiΔ, D.; SamardæiΔ, R. Opposite effects of GABAA and NMDA receptor antagonists on ethanol-induced behavioral sleep in rats. Alcohol 1997, 14, 167–173. [Google Scholar] [CrossRef]
- Schuckit, M.A.; Tipp, J.E.; Smith, T.L.; Wiesbeck, G.A.; Kalmijn, J. The relationship between self-rating of the effects of alcohol and alcohol challenge results in ninety-eight young men. J. Stud. Alcohol 1997, 58, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Radcliffe, R.A.; Floyd, K.L.; Lee, M.J. Rapid ethanol tolerance mediated by adaptations in acute tolerance in inbred mouse strains. Pharmacol. Biochem. Behav. 2006, 84, 524–534. [Google Scholar] [CrossRef]
- Blednov, Y.A.; Bajo, M.; Roberts, A.J.; Da Costa, A.J.; Black, M.; Edmunds, S.; Mayfield, J.; Roberto, M.; Homanics, G.E.; Lasek, A.W.; et al. Scn4b regulates the hypnotic effects of ethanol and other sedative drugs. Genes Brain Behav. 2019, 18, e12562. [Google Scholar] [CrossRef]
- Farris, S.P.; Arasappan, D.; Hunicke-Smith, S.; Harris, R.A.; Mayfield, R.D. Transcriptome organization for chronic alcohol abuse in human brain. Mol. Psychiatry 2015, 20, 1438–1447. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, M.K.; Ponomarev, I.; Hitzemann, R.J.; Belknap, J.K.; Tabakoff, B.; Harris, R.A.; Crabbe, J.C.; Blednov, Y.A.; Grahame, N.J.; Phillips, T.J.; et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc. Natl. Acad. Sci. USA 2006, 103, 6368–6373. [Google Scholar] [CrossRef] [Green Version]
- Tabakoff, B.; Saba, L.; Kechris, K.; Hu, W.; Bhave, S.V.; Finn, D.A.; Grahame, N.J.; Hoffman, P.L. The genomic determinants of alcohol preference in mice. Mamm. Genome 2008, 19, 352–365. [Google Scholar] [CrossRef] [Green Version]
- Kalant, H.; Lê, A.D. Effects of ethanol on thermoregulation. Pharmacol. Ther. 1983, 23, 313–364. [Google Scholar] [CrossRef]
- Freund, B.J.; O’brien, C.; Young, A.J. Alcohol ingestion and temperature regulation during cold exposure. J. Wilderness Med. 1994, 5, 88–98. [Google Scholar] [CrossRef]
- Cunningham, C.L. Modulation of ethanol reinforcement by conditioned hyperthermia. Psychopharmacology 1994, 115, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.L.; Hawks, D.M.; Niehus, D.R. Role of hypothermia in ethanol-induced conditioned taste aversion. Psychopharmacology 1988, 95, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.L.; Niehus, D.R. Effect of ingestion-contingent hypothermia on ethanol self-administration. Alcohol 1989, 6, 377–380. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Niehus, J.S. Drug-induced hypothermia and conditioned place aversion. Behav. Neurosci. 1993, 107, 468–479. [Google Scholar] [CrossRef]
- Chang, S.L.; Patel, N.A.; Romero, A.A. Activation and desensitization of Fos immunoreactivity in the rat brain following ethanol administration. Brain Res. 1995, 679, 89–98. [Google Scholar] [CrossRef]
- Bubier, J.A.; Wilcox, T.D.; Jay, J.J.; Langston, M.A.; Baker, E.J.; Chesler, E.J. Cross-species integrative functional genomics in geneweaver reveals a role for Pafah1b1 in altered response to alcohol. Front. Behav. Neurosci. 2016, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Baker, E.J.; Jay, J.J.; Bubier, J.A.; Langston, M.A.; Chesler, E.J. GeneWeaver: A web-based system for integrative functional genomics. Nucleic Acids Res. 2012, 40, D1067–D1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.L.; Eppig, J.T. The mammalian phenotype ontology: Enabling robust annotation and comparative analysis. Wiley Interdiscip Rev. Syst. Biol. Med. 2009, 1, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Byrne, K.A.; Otto, A.R.; Pang, B.; Patrick, C.J.; Worthy, D.A. Substance use is associated with reduced devaluation sensitivity. Cogn. Affect Behav. Neurosci. 2019, 19, 40–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudebeck, P.H.; Murray, E.A. Amygdala and orbitofrontal cortex lesions differentially influence choices during object reversal learning. J. Neurosci. 2008, 28, 8338–8343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broadbent, J.; Linder, H.V.; Cunningham, C.L. Genetic differences in naloxone enhancement of ethanol-induced conditioned taste aversion. Psychopharmacology 1996, 126, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, J.; Muccino, K.J.; Cunningham, C.L. Ethanol-induced conditioned taste aversion in 15 inbred mouse strains. Behav. Neurosci. 2002, 116, 138–148. [Google Scholar] [CrossRef]
- Cannon, D.S.; Carrell, L.E. Rat strain differences in ethanol self-administration and taste aversion learning. Pharmacol. Biochem. Behav. 1987, 28, 57–63. [Google Scholar] [CrossRef]
- Risinger, F.O.; Cunningham, C.L. Ethanol-induced conditioned taste aversion in BXD recombinant inbred mice. Alcohol. Clin. Exp. Res. 1998, 22, 1234–1244. [Google Scholar] [CrossRef]
- Faure, A.; Reynolds, S.M.; Richard, J.M.; Berridge, K.C. Mesolimbic dopamine in desire and dread: Enabling motivation to be generated by localized glutamate disruptions in nucleus accumbens. J. Neurosci. 2008, 28, 7184–7192. [Google Scholar] [CrossRef]
- Wenzel, J.M.; Rauscher, N.A.; Cheer, J.F.; Oleson, E.B. A role for phasic dopamine release within the nucleus accumbens in encoding aversion: A review of the neurochemical literature. ACS Chem. Neurosci. 2015, 6, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Zweifel, L.S.; Fadok, J.P.; Argilli, E.; Garelick, M.G.; Jones, G.L.; Dickerson, T.M.K.; Allen, J.M.; Mizumori, S.J.Y.; Bonci, A.; Palmiter, R.D. Activation of dopamine neurons is critical for aversive conditioning and prevention of generalized anxiety. Nat. Neurosci. 2011, 14, 620–626. [Google Scholar] [CrossRef] [Green Version]
- Haack, A.K.; Sheth, C.; Schwager, A.L.; Sinclair, M.S.; Tandon, S.; Taha, S.A. Institute, for the B. Lesions of the lateral habenula increase voluntary ethanol consumption and operant self-administration, block yohimbine-induced reinstatement of ethanol seeking, and attenuate ethanol-induced conditioned taste aversion. PLoS ONE 2014, 9, e92701. [Google Scholar] [CrossRef]
- Barkley-Levenson, A.M.; Cunningham, C.L.; Smitasin, P.J.; Crabbe, J.C. Rewarding and aversive effects of ethanol in high drinking in the dark selectively bred mice. Addict. Biol. 2015, 20, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, G.; Carai, M.A.M.; Lobina, C.; Melis, S.; Serra, S.; Vacca, G.; Gessa, G.L.; Colombo, G. Differences in ethanol-induced conditioned taste aversion in Sardinian alcohol-preferring and Sardinian alcohol-nonpreferring rats. Alcohol 2002, 26, 167–172. [Google Scholar] [CrossRef]
- Chester, J.A.; Lumeng, L.; Li, T.-K.; Grahame, N.J. High- and low-alcohol-preferring mice show differences in conditioned taste aversion to alcohol. Alcohol. Clin. Exp. Res. 2003, 27, 12–18. [Google Scholar] [PubMed]
- Crabbe, J.C.; Metten, P.; Savarese, A.M.; Ozburn, A.R.; Schlumbohm, J.P.; Spence, S.E.; Hack, W.R. Ethanol conditioned taste aversion in high drinking in the dark mice. Brain Sci. 2019, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyr, W.; Wyszogrodzka, E.; Paterak, J.; Siwińska-Ziółkowska, A.; Małkowska, A.; Polak, P. Ethanol-induced conditioned taste aversion in Warsaw Alcohol High-Preferring (WHP) and Warsaw Alcohol Low-Preferring (WLP) rats. Alcohol 2016, 51, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, J.C.; Harts, J.; Lumeng, L.; Li, T.K. Differences in response to the aversive properties of ethanol in rats selectively bred for oral ethanol preference. Pharmacol. Biochem. Behav. 1988, 31, 215–222. [Google Scholar] [CrossRef]
- Green, A.S.; Grahame, N.J. Ethanol drinking in rodents: Is free-choice drinking related to the reinforcing effects of ethanol? Alcohol 2008, 42, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla, M.E.; Callejas, O.; Tampier, L. Differences in sensitivity to the aversive effects of ethanol in low-alcohol drinking (UChA) and high-alcohol drinking (UChB) rats. Alcohol 2001, 23, 177–182. [Google Scholar] [CrossRef]
- Fernández, M.S.; Báez, B.; Bordón, A.; Espinosa, L.; Martínez, E.; Pautassi, R.M. Short-term selection for high and low ethanol intake yields differential sensitivity to ethanol’s motivational effects and anxiety-like responses in adolescent Wistar rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 220–233. [Google Scholar] [CrossRef]
- Edenberg, H.J.; McClintick, J.N. Alcohol Dehydrogenases, Aldehyde Dehydrogenases, and Alcohol Use Disorders: A Critical Review. Alcohol. Clin. Exp. Res. 2018, 42, 2281–2297. [Google Scholar] [CrossRef]
- Whitfield, J.B. Alcohol dehydrogenase and alcohol dependence: Variation in genotype-associated risk between populations. Am. J. Hum. Genet. 2002, 71, 1247–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, C.A.; Romaniuk, H.; Salinger, J.; Staiger, P.K.; Bonomo, Y.; Hulbert, C.; Patton, G.C. Drinking patterns of adolescents who develop alcohol use disorders: Results from the Victorian Adolescent Health Cohort Study. BMJ Open 2016, 6, e010455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spear, L.P. Effects of adolescent alcohol consumption on the brain and behaviour. Nat. Rev. Neurosci. 2018, 19, 197–214. [Google Scholar] [CrossRef] [PubMed]
- DeWit, D.J.; Adlaf, E.M.; Offord, D.R.; Ogborne, A.C. Age at first alcohol use: A risk factor for the development of alcohol disorders. Am. J. Psychiatry 2000, 157, 745–750. [Google Scholar] [CrossRef]
- Ehlers, C.L.; Slutske, W.S.; Gilder, D.A.; Lau, P.; Wilhelmsen, K.C. Age at first intoxication and alcohol use disorders in Southwest California Indians. Alcohol. Clin. Exp. Res. 2006, 30, 1856–1865. [Google Scholar] [CrossRef]
- Dawson, D.A.; Goldstein, R.B.; Chou, S.P.; Ruan, W.J.; Grant, B.F. Age at first drink and the first incidence of adult-onset DSM-IV alcohol use disorders. Alcohol. Clin. Exp. Res. 2008, 32, 2149–2160. [Google Scholar] [CrossRef]
- Morean, M.E.; Kong, G.; Camenga, D.R.; Cavallo, D.A.; Connell, C.; Krishnan-Sarin, S. First drink to first drunk: Age of onset and delay to intoxication are associated with adolescent alcohol use and binge drinking. Alcohol. Clin. Exp. Res. 2014, 38, 2615–2621. [Google Scholar] [CrossRef]
- Morean, M.E.; L’Insalata, A.; Butler, E.R.; McKee, A.; Krishnan-Sarin, S. Age at drinking onset, age at first intoxication, and delay to first intoxication: Assessing the concurrent validity of measures of drinking initiation with alcohol use and related problems. Addict. Behav. 2018, 79, 195–200. [Google Scholar] [CrossRef]
- Kuntsche, E.; Rossow, I.; Engels, R.; Kuntsche, S. Is “age at first drink” a useful concept in alcohol research and prevention? We doubt that. Addiction 2016, 111, 957–965. [Google Scholar] [CrossRef]
- Patrick, M.E.; Evans-Polce, R.; Terry-McElrath, Y.M. Faster escalation from first drink to first intoxication as a risk factor for binge and high-intensity drinking among adolescents. Addict. Behav. 2019, 92, 199–202. [Google Scholar] [CrossRef]
- Prescott, C.A.; Kendler, K.S. Age at first drink and risk for alcoholism: A noncausal association. Alcohol. Clin. Exp. Res. 1999, 23, 101–107. [Google Scholar] [CrossRef]
- Holstein, S.E.; Spanos, M.; Hodge, C.W. Adolescent C57BL/6J mice show elevated alcohol intake, but reduced taste aversion, as compared to adult mice: A potential behavioral mechanism for binge drinking. Alcohol. Clin. Exp. Res. 2011, 35, 1842–1851. [Google Scholar] [CrossRef]
- Pautassi, R.M.; Godoy, J.C.; Molina, J.C. Adolescent rats are resistant to the development of ethanol-induced chronic tolerance and ethanol-induced conditioned aversion. Pharmacol. Biochem. Behav. 2015, 138, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Schramm-Sapyta, N.L.; DiFeliceantonio, A.G.; Foscue, E.; Glowacz, S.; Haseeb, N.; Wang, N.; Zhou, C.; Kuhn, C.M. Aversive effects of ethanol in adolescent versus adult rats: Potential causes and implication for future drinking. Alcohol. Clin. Exp. Res. 2010, 34, 2061–2069. [Google Scholar] [CrossRef] [PubMed]
- Schramm-Sapyta, N.L.; Francis, R.; MacDonald, A.; Keistler, C.; O’Neill, L.; Kuhn, C.M. Effect of sex on ethanol consumption and conditioned taste aversion in adolescent and adult rats. Psychopharmacology 2014, 231, 1831–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.I.; Varlinskaya, E.I.; Spear, L.P. Ethanol-induced conditioned taste aversion in male sprague-dawley rats: Impact of age and stress. Alcohol. Clin. Exp. Res. 2010, 34, 2106–2115. [Google Scholar] [CrossRef]
- Saalfield, J.; Spear, L. The ontogeny of ethanol aversion. Physiol. Behav. 2016, 156, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Vetter-O’Hagen, C.; Varlinskaya, E.; Spear, L. Sex differences in ethanol intake and sensitivity to aversive effects during adolescence and adulthood. Alcohol Alcohol. 2009, 44, 547–554. [Google Scholar] [CrossRef]
- Diaz-Granados, J.L.; Graham, D.L. The effects of continuous and intermittent ethanol exposure in adolesence on the aversive properties of ethanol during adulthood. Alcohol. Clin. Exp. Res. 2007, 31, 2020–2027. [Google Scholar] [CrossRef]
- Saalfield, J.; Spear, L. Consequences of repeated ethanol exposure during early or late adolescence on conditioned taste aversions in rats. Dev. Cogn. Neurosci. 2015, 16, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Sherrill, L.K.; Berthold, C.; Koss, W.A.; Juraska, J.M.; Gulley, J.M. Sex differences in the effects of ethanol pre-exposure during adolescence on ethanol-induced conditioned taste aversion in adult rats. Behav. Brain Res. 2011, 225, 104–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaux-Cantin, S.; Warnault, V.; Legastelois, R.; Botia, B.; Pierrefiche, O.; Vilpoux, C.; Naassila, M. Alcohol intoxications during adolescence increase motivation for alcohol in adult rats and induce neuroadaptations in the nucleus accumbens. Neuropharmacology 2013, 67, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Towner, T.T.; Varlinskaya, E.I. Adolescent ethanol exposure: Anxiety-like behavioral alterations, ethanol intake, and sensitivity. Front. Behav. Neurosci. 2020, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.M.; Forrest, R.D.; Boehm, S.L. Genotype modulates age-related alterations in sensitivity to the aversive effects of ethanol: An eight inbred strain analysis of conditioned taste aversion. Genes Brain Behav. 2013, 12, 70–77. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, C.C.; Lusk, R.; Saba, L.M. Alcohol Sensitivity as an Endophenotype of Alcohol Use Disorder: Exploring Its Translational Utility between Rodents and Humans. Brain Sci. 2020, 10, 725. https://doi.org/10.3390/brainsci10100725
Parker CC, Lusk R, Saba LM. Alcohol Sensitivity as an Endophenotype of Alcohol Use Disorder: Exploring Its Translational Utility between Rodents and Humans. Brain Sciences. 2020; 10(10):725. https://doi.org/10.3390/brainsci10100725
Chicago/Turabian StyleParker, Clarissa C., Ryan Lusk, and Laura M. Saba. 2020. "Alcohol Sensitivity as an Endophenotype of Alcohol Use Disorder: Exploring Its Translational Utility between Rodents and Humans" Brain Sciences 10, no. 10: 725. https://doi.org/10.3390/brainsci10100725
APA StyleParker, C. C., Lusk, R., & Saba, L. M. (2020). Alcohol Sensitivity as an Endophenotype of Alcohol Use Disorder: Exploring Its Translational Utility between Rodents and Humans. Brain Sciences, 10(10), 725. https://doi.org/10.3390/brainsci10100725