Blood-Based Biomarkers Predictive of Metformin Target Engagement in Fragile X Syndrome

 , and
, and
Abstract
:1. Introduction
2. Method
2.1. Participants
2.2. FMR1 Molecular Measures
2.3. MMP9 Expression Levels
2.4. Western Blotting
2.5. Clinical Measures
2.6. Statistical Analysis
3. Results
3.1. Participants
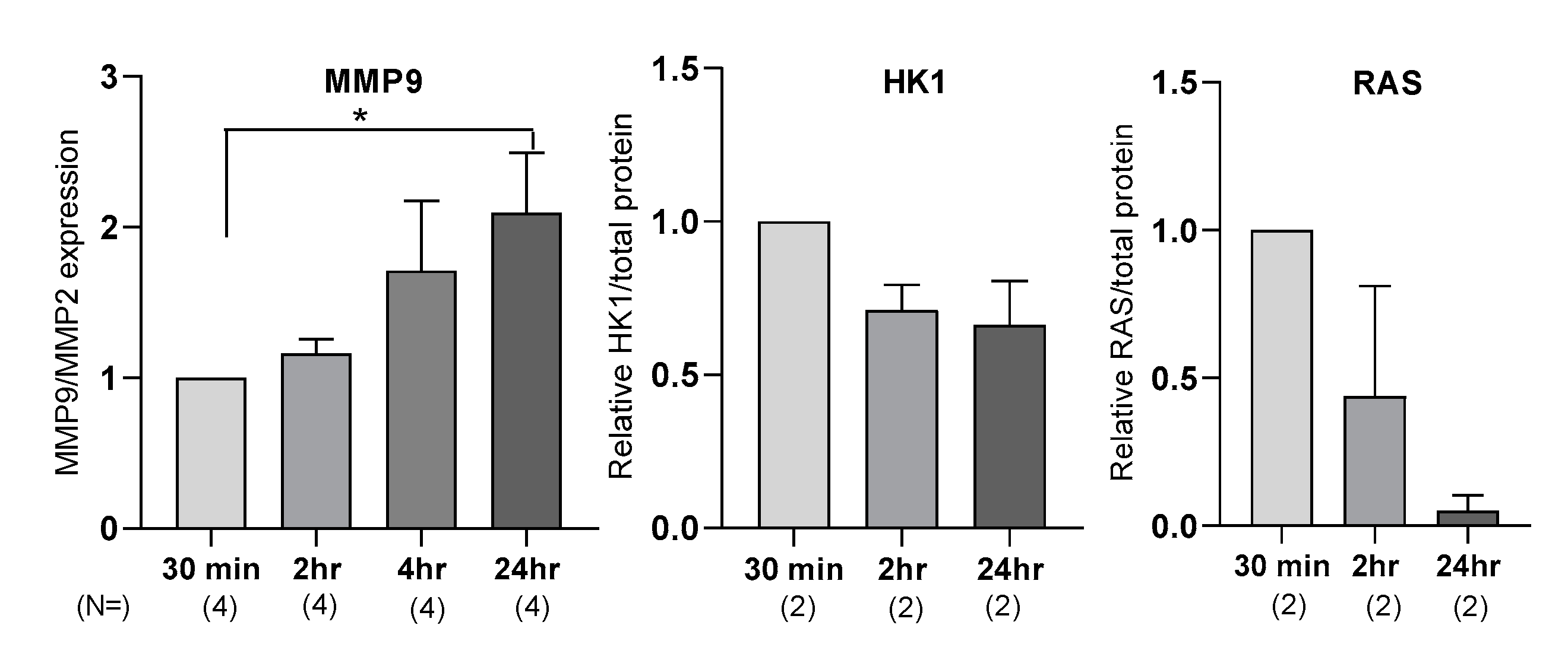
3.2. MMP9, HK1, and RAS Display Different Response to Metformin-Treatment
3.3. Correlation of Molecular Measures with Clinical Measures
3.4. Plasma Proteins Levels Are Affected by Processing Time
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bailey, D.B.; Mesibov, G.B.; Hatton, D.D.; Clark, R.D.; Roberts, J.E.; Mayhew, L. Autistic behavior in young boys with fragile X syndrome. J. Autism Dev. Disord. 1998, 28, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Baumgardner, T.L.; Reiss, A.L.; Freund, L.S.; Abrams, M.T. Specification of the neurobehavioral phenotype in males with fragile X syndrome. Pediatrics 1995, 95, 744–752. [Google Scholar] [PubMed]
- Clifford, S.; Dissanayake, C.; Bui, Q.M.; Huggins, R.; Taylor, A.K.; Loesch, D.Z. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord. 2007, 37, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Gabriels, R.L.; Hill, D.E.; Pierce, R.A.; Rogers, S.J.; Wehner, B. Predictors of treatment outcome in young children with autism: A retrospective study. Autism 2001, 5, 407–429. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.W.; Hessl, D.; Goodlin-Jones, B.; Ferranti, J.; Bacalman, S.; Barbato, I.; Tassone, F.; Hagerman, P.J.; Herman, K.; Hagerman, R.J. Autism profiles of males with fragile X syndrome. Am. J. Ment. Retard. 2008, 113, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitas, A.; McBogg, P.; Hagerman, R. Behavioral dysfunction in the fragile X syndrome. In The Fragile X Syndrome: Diagnosis, Biochemistry, and Intervention; Spectra Publishing Company: New York, NY, USA, 1983; pp. 153–173. [Google Scholar]
- McLennan, Y.; Polussa, J.; Tassone, F.; Hagerman, R. Fragile X syndrome. Curr. Genom. 2011, 12, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, S.T.; Tassone, F.; Ono, M.Y.; Ferranti, J.; Croquette, M.F.; Goodlin-Jones, B.; Hagerman, R.J. The Prader-Willi phenotype of fragile X syndrome. J. Dev. Behav. Pediatr. 2007, 28, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Dziembowska, M.; Pretto, D.I.; Janusz, A.; Kaczmarek, L.; Leigh, M.J.; Gabriel, N.; Durbin-Johnson, B.; Hagerman, R.J.; Tassone, F. High MMP-9 activity levels in fragile X syndrome are lowered by minocycline. Am. J. Med. Genet. Part A 2013, 161, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Gkogkas, C.G.; Khoutorsky, A.; Cao, R.; Jafarnejad, S.M.; Prager-Khoutorsky, M.; Giannakas, N.; Kaminari, A.; Fragkouli, A.; Nader, K.; Price, T.J. Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep. 2014, 9, 1742–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhu, H.; Dansie, L.E.; Hickmott, P.W.; Ethell, D.W.; Ethell, I.M. Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J. Neurosci. 2014, 34, 9867–9879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlOlaby, R.R.; Sweha, S.R.; Silva, M.; Durbin-Johnson, B.; Yrigollen, C.M.; Pretto, D.; Hagerman, R.J.; Tassone, F. Molecular biomarkers predictive of sertraline treatment response in young children with fragile X syndrome. Brain Dev. 2017, 39, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, S.M.; Razak, K.; Ethell, I.M. A delicate balance: role of MMP-9 in brain development and pathophysiology of neurodevelopmental disorders. Front. Cell. Neurosci. 2015, 9, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, A.; Spiller, K.J.; Towne, C.; Kanning, K.C.; Choe, G.T.; Geber, A.; Akay, T.; Aebischer, P.; Henderson, C.E. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron 2014, 81, 333–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vos, C.M.; Gartner, S.; Ransohoff, R.M.; McArthur, J.C.; Wahl, L.; Sjulson, L.; Hunter, E.; Conant, K. Matrix metalloprotease-9 release from monocytes increases as a function of differentiation: implications for neuroinflammation and neurodegeneration. J. Neuroimmunol. 2000, 109, 221–227. [Google Scholar] [CrossRef]
- Bilousova, T.; Dansie, L.; Ngo, M.; Aye, J.; Charles, J.R.; Ethell, D.W.; Ethell, I.M. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J. Med. Genet. 2009, 46, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Rotschafer, S.E.; Trujillo, M.S.; Dansie, L.E.; Ethell, I.M.; Razak, K.A. Minocycline treatment reverses ultrasonic vocalization production deficit in a mouse model of Fragile X Syndrome. Brain Res. 2012, 1439, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Dansie, L.E.; Phommahaxay, K.; Okusanya, A.G.; Uwadia, J.; Huang, M.; Rotschafer, S.E.; Razak, K.A.; Ethell, D.W.; Ethell, I.M. Long-lasting effects of minocycline on behavior in young but not adult Fragile X mice. Neuroscience 2013, 246, 186–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paribello, C.; Tao, L.; Folino, A.; Berry-Kravis, E.; Tranfaglia, M.; Ethell, I.M.; Ethell, D.W. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 2010, 10, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utari, A.; Chonchaiya, W.; Rivera, S.M.; Schneider, A.; Hagerman, R.J.; Faradz, S.M.; Ethell, I.M.; Nguyen, D.V. Side effects of minocycline treatment in patients with fragile X syndrome and exploration of outcome measures. Am. J. Intellect. Dev. Disabil. 2010, 115, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, M.J.S.; Nguyen, D.V.; Mu, Y.; Winarni, T.I.; Schneider, A.; Chechi, T.; Polussa, J.; Doucet, P.; Tassone, F.; Rivera, S.M. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J. Dev. Behav. Pediatr. 2013, 34, 147. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Bhattacharya, A. Modelling Protein Synthesis as A Biomarker in Fragile X Syndrome Patient-Derived Cells. Brain Sci. 2019, 9, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utami, K.H.; Yusof, N.A.B.M.; Kwa, J.E.; Peteri, U.-K.; Castrén, M.L.; Pouladi, M.A. Elevated de novo protein synthesis in FMRP-deficient human neurons and its correction by metformin treatment. Mol. Autism 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bowling, H.; Bhattacharya, A.; Zhang, G.; Alam, D.; Lebowitz, J.Z.; Bohm-Levine, N.; Lin, D.; Singha, P.; Mamcarz, M.; Puckett, R. Altered steady state and activity-dependent de novo protein expression in fragile X syndrome. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med. 2017, 23, 674. [Google Scholar] [CrossRef] [PubMed]
- Monyak, R.E.; Emerson, D.; Schoenfeld, B.P.; Zheng, X.; Chambers, D.B.; Rosenfelt, C.; Langer, S.; Hinchey, P.; Choi, C.H.; McDonald, T.V. Insulin signaling misregulation underlies circadian and cognitive deficits in a Drosophila fragile X model. Mol. Psychiatry 2017, 22, 1140–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dy, A.B.C.; Tassone, F.; Eldeeb, M.; Salcedo-Arellano, M.J.; Tartaglia, N.; Hagerman, R. Metformin as targeted treatment in fragile X syndrome. Clin. Genet. 2018, 93, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Pan, R.; Amiri, K.; Taylor, A.K.; Hagerman, P.J. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J. Mol. Diagn. 2008, 10, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipovic-Sadic, S.; Sah, S.; Chen, L.; Krosting, J.; Sekinger, E.; Zhang, W.; Hagerman, P.J.; Stenzel, T.T.; Hadd, A.G.; Latham, G.J. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin. Chem. 2010, 56, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassone, F.; Hagerman, R.J.; Iklé, D.N.; Dyer, P.N.; Lampe, M.; Willemsen, R.; Oostra, B.A.; Taylor, A.K. FMRP expression as a potential prognostic indicator in fragile X syndrome. Am. J. Med Genet. 1999, 84, 250–261. [Google Scholar] [CrossRef]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000, 66, 6–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busner, J.; Targum, S.D. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry 2007, 4, 28. [Google Scholar] [PubMed]
- Mullard, A. Fragile X drug development flounders. Nat. Rev. Drug Discov. 2016, 15, 77. [Google Scholar] [CrossRef]
- Scharf, S.H.; Jaeschke, G.; Wettstein, J.G.; Lindemann, L. Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr. Opin. Pharmacol. 2015, 20, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, L.B.; Visootsak, J.; Sherman, S.L. Cognitive aspects of fragile X syndrome. Wiley Interdiscip. Rev. Cogn. Sci. 2014, 5, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Keith, K.; Albizua, I.; Malone, T.; Mowrey, J.; Sherman, S.L.; Warren, S.T. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am. J. Hum. Genet. 2009, 85, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, P.J. The fragile X prevalence paradox. J. Med. Genet. 2008, 45, 498–499. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Participants | Age | Gender | Mutation Category | CGG Repeat | % Meth. | Activation Ratio | FMR1 mRNA ± StErr | Metformin Dose | Duration of Treatment (Months) | Other Ongoing Treatments |
|---|---|---|---|---|---|---|---|---|---|---|
| Case 1 | 25 | M | Meth mosaic | >200 (250–480) | 78% | 0.21 ± 0.004 | 1000 mg BID | 28 | Abilify 10 mg QD Effexor 75 mg QD | |
| Case 2 | 10 | M | Full mutation | >200 | 0 | 500 mg BID | 15 | Sertraline 50 mg QD Abilify 5 mg QHS Guanfancine 2 mg BID Minocycline 100 mg QD CBD and THC 2:1 | ||
| Case 3 | 7 | M | Full mutation | >200 | 0.13 ± 0.0001 | 500 mg BID | 6 | Clonidine 0.1 mg TID& 0.2 at QHS Adderall 2.5 mg TID Trileptal 420 mg BID | ||
| Case 4 | 1 | M | Full mutation | >200 | 0.001 ± 0.00 | 150 mg BID | 9 | Melatonin 2 mg Sertraline 4 mg QD | ||
| Case 5 | 2 | M | Full mutation | >200 | 0 | 50 mg BID | 4 | Melatonin 2 mg Sertraline 2.5 mg QAM | ||
| Case 6 | 23 | M | Full mutation | >200 | 0 | 500 mg BID | 16 | Vitamin D Fish Oil Pravastatin 10 mg QD | ||
| Case 7 | 3 | F | Size mosaic | 28, >200 (54) | 93% | 0.38 | 0.95 ± 0.13 | 100 mg BID | 7 | Sertraline 0.2 mL (Zoloft 20 mg/mL) |
| Case 8 | 3 | F | Full mutation | 30, >200 | 0.5 | 0.83 ± 0.03 | 100 mg BID | 1 | Melatonin Multivitamin | |
| Case 9 | 5 | F | Full mutation | 30, 240 | 0.52 | 1.01 ± 0.03 | 200 mg BID | 9 | Sertraline 20 mg QD Melatonin 5 mg QHS | |
| Case 10 | 19 | M | Full mutation | >200 | 0.05 ±0.00 | 500 mg QD | 26 | |||
| Case 11 | 67 | M | Full mutation | >200 | 0.07 ± 0.01 | 1500 mg TID | 28 | Sertraline 50 mg | ||
| Case 12 | 8 | F | Meth. mosaic | 31, >200 (360) | 92% | 0.42 | 0.26 ± 0.01 | 500 mg BID | 1 | |
| Case 13 | 2 | M | Meth. mosaic | >200 (180–300) | 82% | NA | 500 mg BID | 4 | Sertraline 13 mL QD Multivitamins. | |
| Case 14 | 10 | M | Full mutation | >200 | 0 | 500 mg BID | 7 | Sertraline 25 mg Multivitamin Aripiprazole (Abilify) 2 mg Clonidine 01 mg qD + H15:H21 | ||
| Case 15 | 8 | M | Full mutation | >200 | 0 | 500 mg BID | 1 | |||
| Case 16 | 2 | F | Full mutation | 29, >200 | 0.43 | 0.4 ± 0.01 | 50 mg BID | 30 | Melatonin 1 mg/Ml Sertraline HCL 2.5 mg QD Metformin 50 mg BID | |
| Case 17 | 7 | M | Full mutation | >200 | 0 | 500 mg BID | 11 | Guanfacine HCK Aripiprazole (Abilify) 5 mg QD Clonidine 0.1 mg QD |
| Participants | Age | Gender | MMP9/MMP2 Changes | HK1 Changes | Ras Changes | CGI-I Score | BMI Move Towards Normal? |
|---|---|---|---|---|---|---|---|
| Case 1 | 25 | M | 0.46 | 1.08 | 3.92 | 2 | yes |
| Case 2 | 10 | M | 1.09 | 0.65 | - | 2 | NA |
| Case 3 | 7 | M | 0.81 | 0.73 | 1.1 | 1 | stay normal |
| Case 4 | 1 | M | 0.65 | 1.08 | 1.37 | 3 | stay normal |
| Case 5 | 2 | M | 1.64 | 0.65 | - | 2 | yes |
| Case 6 | 23 | M | 0.25 | 6.75 | 1.22 | 1 | yes |
| Case 7 | 3 | F | 1.75 | 0.99 | 1.1 | 2 | yes |
| Case 8 | 3 | F | 0.96 | 1.01 | 2.07 | 4 | NA |
| Case 9 | 5 | F | 0.82 | 1.14 | 2.6 | 4 | NA |
| Case 10 | 19 | M | 1.41 | 1.34 | 1.07 | 3 | yes |
| Case 11 | 67 | M | 5.36 | 0.94 | 0.53 | 2 | no |
| Case 12 | 8 | F | 1.48 | 0.9 | 2.45 | 3 | NA |
| Case 13 | 2 | M | 1.33 | 1.18 | 2.07 | 2 | yes |
| Case 14 | 10 | M | 1.87 | 0.88 | 1.1 | 3 | yes |
| Case 15 | 8 | M | 0.66 | 0.48 | 1.18 | 2 | NA |
| Case 16 | 2 | F | 1.54 | 1.46 | 1.01 | 1 | yes |
| Case 17 | 7 | M | 2.62 | 0.86 | 0.94 | NA | NA |
| Fold Change (95% CI) | p-Value | |
|---|---|---|
| MMP9/MMP2 | 1.32 (0.569, 3.038) | 0.494 |
| HK1 | 0.793 (0.394, 1.595) | 0.488 |
| Ras | 1.72 (0.918, 3.233) | 0.084 |
| Regression Slope (95% CI) | p-Value | |
|---|---|---|
| MMP9/MMP2 | 1.49 (0.235, 2.748) | 0.0271 |
| HK1 | −1.33 (−3.298, 0.634) | 0.148 |
| Ras | −3.17 (−6.490, 0.143) | 0.0572 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jasoliya, M.; Bowling, H.; Petrasic, I.C.; Durbin-Johnson, B.; Klann, E.; Bhattacharya, A.; Hagerman, R.; Tassone, F. Blood-Based Biomarkers Predictive of Metformin Target Engagement in Fragile X Syndrome. Brain Sci. 2020, 10, 361. https://doi.org/10.3390/brainsci10060361
Jasoliya M, Bowling H, Petrasic IC, Durbin-Johnson B, Klann E, Bhattacharya A, Hagerman R, Tassone F. Blood-Based Biomarkers Predictive of Metformin Target Engagement in Fragile X Syndrome. Brain Sciences. 2020; 10(6):361. https://doi.org/10.3390/brainsci10060361
Chicago/Turabian StyleJasoliya, Mittal, Heather Bowling, Ignacio Cortina Petrasic, Blythe Durbin-Johnson, Eric Klann, Aditi Bhattacharya, Randi Hagerman, and Flora Tassone. 2020. "Blood-Based Biomarkers Predictive of Metformin Target Engagement in Fragile X Syndrome" Brain Sciences 10, no. 6: 361. https://doi.org/10.3390/brainsci10060361