Insulin Resistance in Skeletal Muscle of Chronic Stroke




Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. VO2peak and Body Composition
2.3. Oral Glucose Tolerance Test (OGTT)
2.4. Hyperinsulinemic-Euglycemic Clamps with Skeletal Muscle Biopsies and Substrate Oxidation
2.5. Skeletal Muscle Analysis
2.6. Statistical Analyses
3. Results
3.1. Physical Characteristics
3.2. Glucose Metabolism
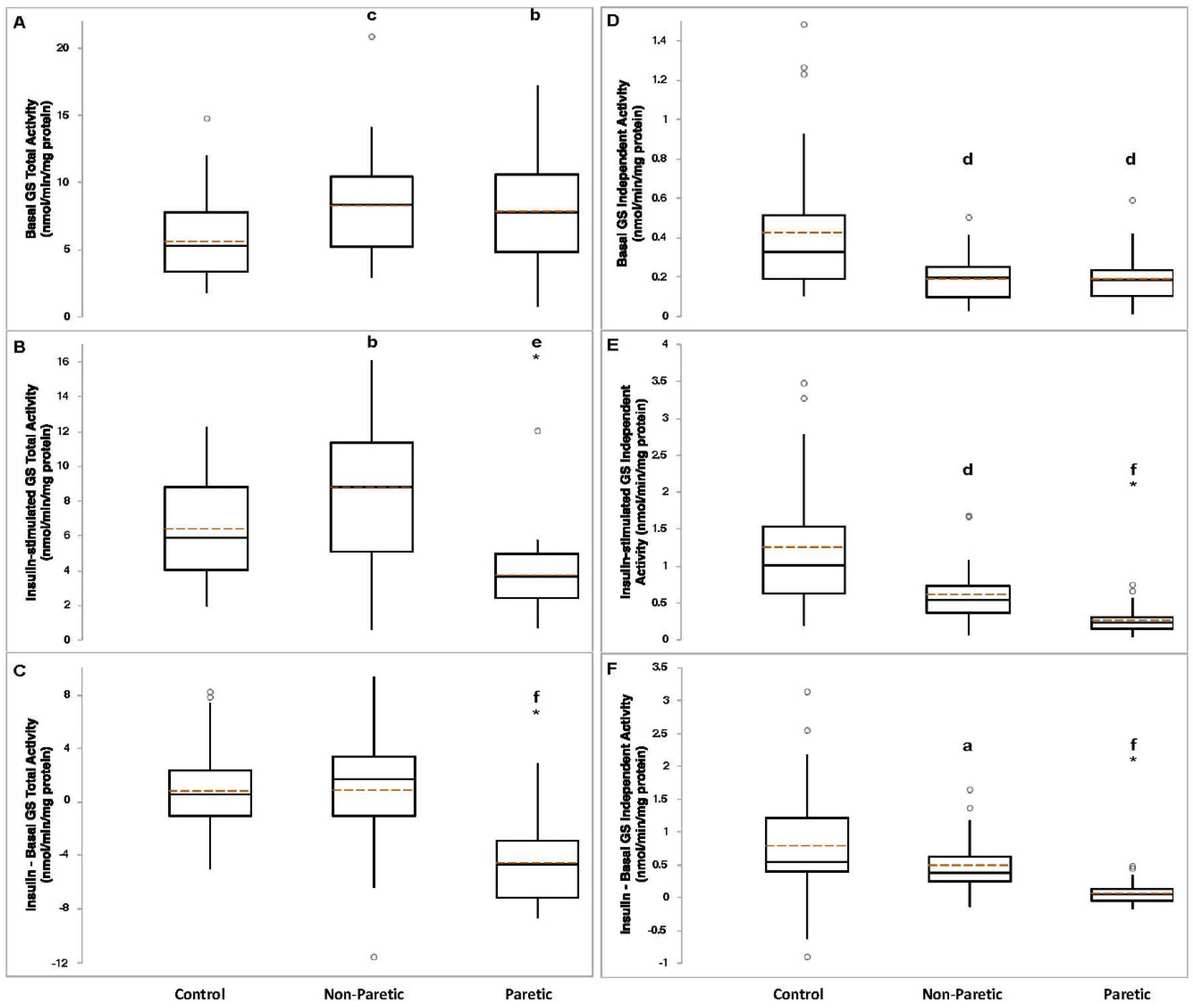
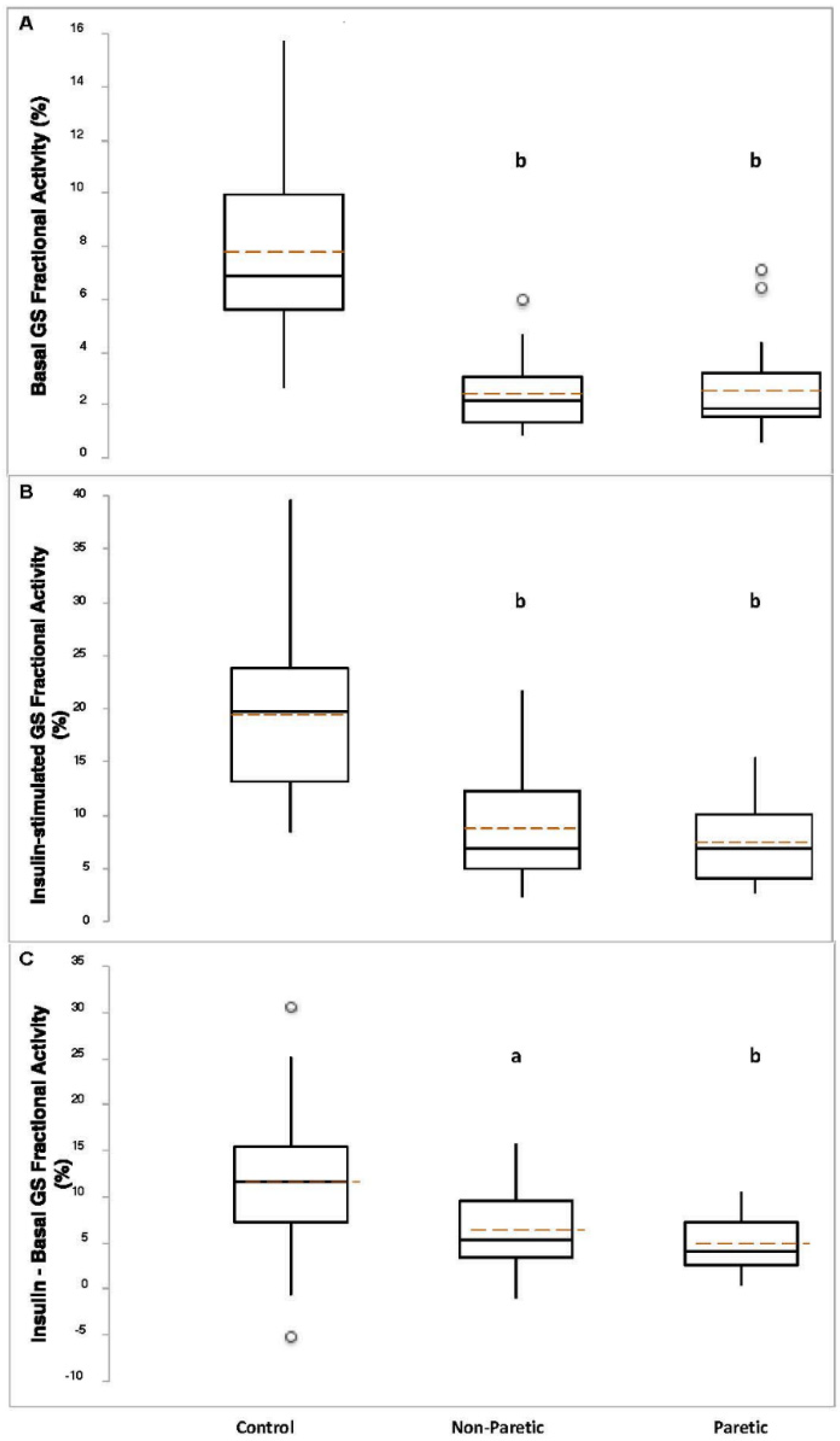
3.3. Skeletal Muscle Glycogen Synthase Activity in Stroke Survivors (Paired-t)
3.4. Skeletal Muscle Glycogen Synthase Activity in Stroke Survivors vs. Control (ANOVA)
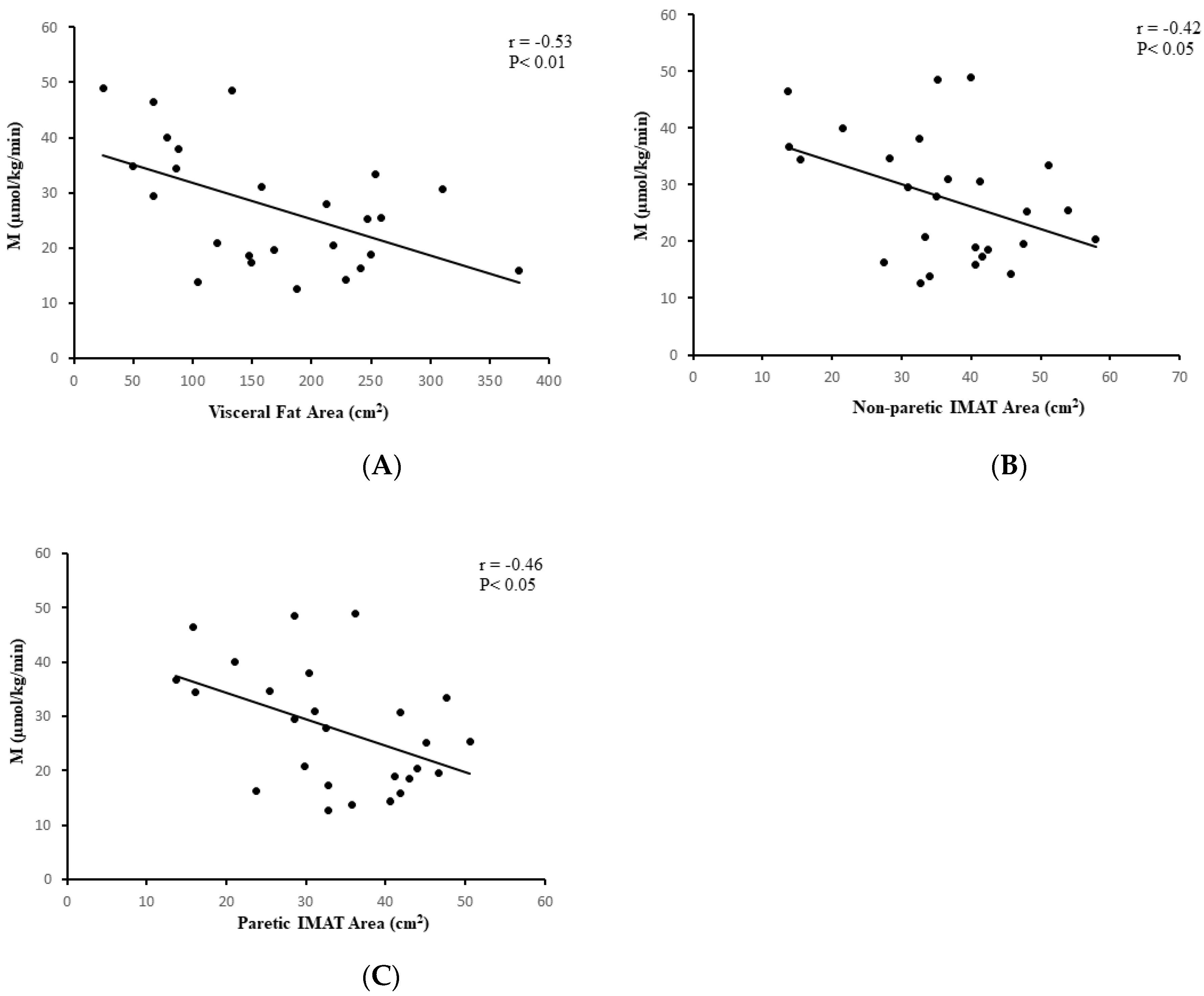
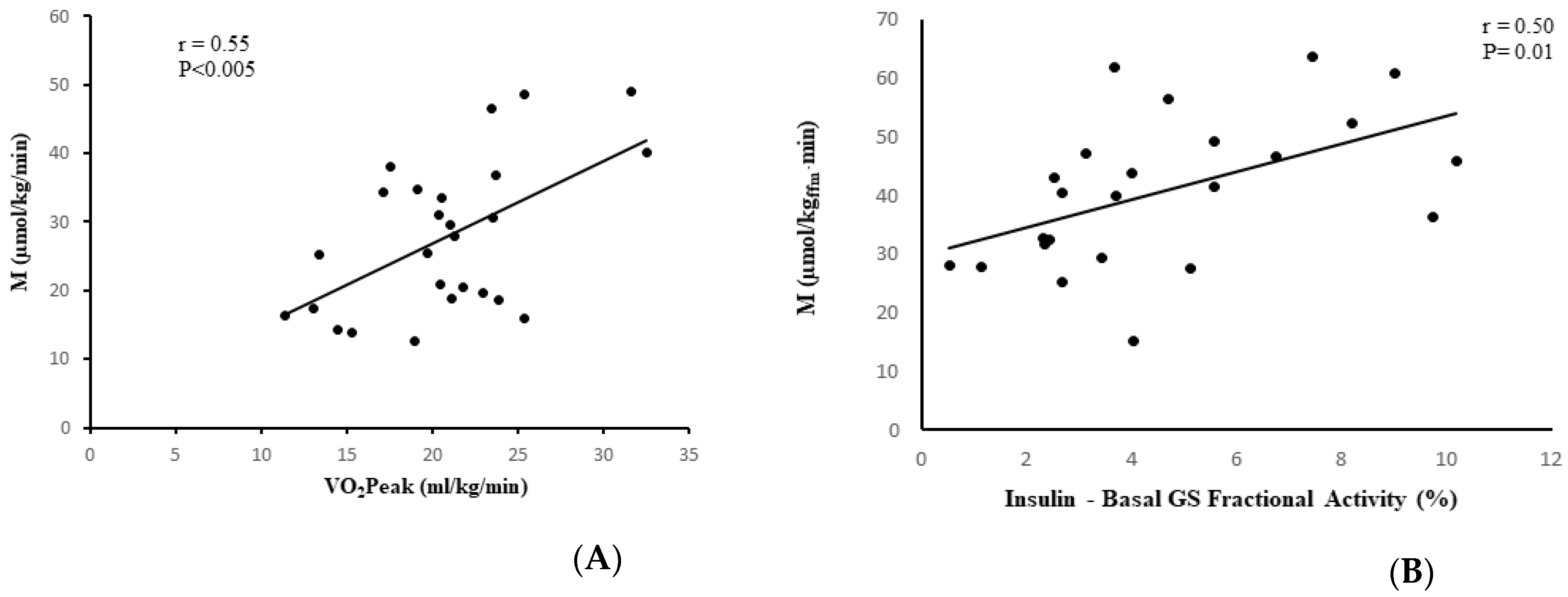
3.5. Relationships between Glucose Metabolism and Body Fat and Fitness
3.6. Skeletal Muscle Glycogen Synthase Activity versus Insulin Sensitivity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ovbiagele, B.; Goldstein, L.B.; Higashida, R.T.; Howard, V.J.; Johnston, S.C.; Khavjou, O.A.; Lackland, D.T.; Lichtman, J.H.; Mohl, S.; Sacco, R.L.; et al. Forecasting the future of stroke in the united states: A policy statement from the American Heart Association and American Stroke Association. Stroke 2013, 44, 2361–2375. [Google Scholar] [CrossRef] [Green Version]
- McGrath, R.; Al Snih, S.; Markides, K.; Hall, O.; Peterson, M. The burden of health conditions for middle-aged and older adults in the united states: Disability-adjusted life years. BMC Geriatr. 2019, 19, 100. [Google Scholar] [CrossRef]
- Ryan, A.S.; Dobrovolny, C.L.; Smith, G.V.; Silver, K.H.; Macko, R.F. Hemiparetic muscle atrophy and increased intramuscular fat in stroke patients. Arch. Phys. Med. Rehabil. 2002, 83, 1703–1707. [Google Scholar] [CrossRef]
- Ryan, A.S.; Buscemi, A.; Forrester, L.; Hafer-Macko, C.E.; Ivey, F.M. Atrophy and intramuscular fat in specific muscles of the thigh: Associated weakness and hyperinsulinemia in stroke survivors. Neurorehabilit. Neural Repair 2011, 25, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.S.; Ivey, F.M.; Serra, M.C.; Hartsetin, J.; Hafer-Macko, C.E. Sarcopenia and physical function in middle-aged and older stroke survivors. Arch Phys Med. Rahbil. 2017, 98, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, A.S.; Nicklas, B.J. Age-related changes in fat deposition in mid-thigh muscle in women: Relationships with metabolic cardiovascular disease risk factors. Int. J. Obes. 1999, 23, 126–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodpaster, B.H.; Thaete, F.L.; Simoneau, J.A.; Kelley, D.E. Subcutaneous abdominal fat and thigh muscle composition predict insulin sensitivity independently of visceral fat. Diabetes 1997, 46, 1579–1585. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Wolf, D. Skeletal muscle lipid accumulation in obesity, insulin resistance, and type 2 diabetes. Pediatr. Diabetes 2004, 5, 219–226. [Google Scholar] [CrossRef]
- De Deyne, P.G.; Hafer-Macko, C.E.; Ivey, F.M.; Ryan, A.S.; Macko, R.F. Muscle molecular phenotype after stroke is associated with gait speed. Muscle Nerve 2004, 30, 209–215. [Google Scholar] [CrossRef]
- Hafer-Macko, C.E.; Yu, S.; Ryan, A.S.; Ivey, F.M.; Macko, R.F. Elevated tumor necrosis factor-α in skeletal muscle after stroke. Stroke 2005, 36, 2021–2023. [Google Scholar] [CrossRef] [Green Version]
- Wiberg, B.; Sundstrom, J.; Zethelius, B.; Lind, L. Insulin sensitivity measured by the euglycaemic insulin clamp and proinsulin levels as predictors of stroke in elderly men. Diabetologia 2009, 52, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folstein, M.F.; Folstein, S.E.; McHugh, P. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Radloff, L.S.; Rae, D.S. Susceptibility and precipitating factors in depression: Sex differences and similarities. J. Abnorm. Psychol. 1979, 88, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.S.; Ortmeyer, H.K.; Sorkin, J.D. Exercise with calorie restriction improves insulin sensitivity and glycogen synthase activity in obese postmenopausal women with impaired glucose tolerance. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E145–E152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macko, R.F.; DeSouza, C.A.; Tretter, L.D.; Silver, K.H.; Smith, G.V.; Anderson, P.A.; Tomoyasu, N.; Gorman, P.; Dengel, D. Treadmill aerobic exercise training reduces the energy expenditure and cardiovascular demands of hemiparetic gait in chronic stroke patients. A preliminary report. Stroke 1997, 28, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.S.; Nicklas, B.J.; Berman, D.M. Aerobic exercise is necessary to improve glucose utilization with moderate weight loss in women. Obesity 2006, 14, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tobin, J.D.; Andres, R. Glucose clamp technique: A method for quantifying insulin secretion and resistance. Am. J. Physiol. Endocrinol. Metab. 1979, 237, E214–E223. [Google Scholar] [CrossRef]
- McGuire, E.A.; Helderman, J.H.; Tobin, J.D.; Andres, R.; Berman, M. Effects of arterial versus venous sampling on analysis of glucose kinetics in man. J. Appl. Physiol. 1976, 41, 565–573. [Google Scholar] [CrossRef]
- Ryan, A.S.; Nicklas, B.J.; Berman, D.M. Racial differences in insulin resistance and mid-thigh fat deposition in postmenopausal women. Obes. Res. 2002, 10, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Ovbiagele, B.; Feng, W. Diabetes and stroke: Epidemiology, pathophysiology, pharmaceuticals and outcomes. Am. J. Med. Sci. 2016, 351, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Ivey, F.M.; Ryan, A.S. Resistive training improves insulin sensitivity after stroke. J. Stroke Cerebrovasc. Dis. 2014, 23, 225–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamide, K.; Rakugi, H.; Nakano, N.; Ohishi, M.; Nakata, Y.; Takami, S.; Katsuya, T.; Higaki, J.; Ogihara, T. Insulin resistance is related to silent cerebral infarction in patients with essential hypertension. Am. J. Hypertens. 1997, 10, 1245–1249. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Y.; Gao, Z. Correlations of c-reactive protein (CRP), interleukin-6 (il-6), and insulin resistance with cerebral infarction in hypertensive patients. Med. Sci. Monit. 2019, 25, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yin, C.; Zhao, W.; Zhu, H.; Xu, D.; Xu, Q.; Jiao, Y.; Wang, X.; Qiao, H. Homeostasis model assessment of insulin resistance in relation to the poor functional outcomes in nondiabetic patients with ischemic stroke. Biosci. Rep. 2018, 38, BSR20180330. [Google Scholar] [CrossRef] [Green Version]
- Ryan, A.S.; Nicklas, B.J.; Elahi, D. A cross-sectional study on body composition and energy expenditure in women athletes during aging. Am. J. Physiol. Endocrinol. Metab. 1996, 271, E916–E921. [Google Scholar] [CrossRef]
- Huth, C.; Pigeon, E.; Riou, M.E.; St-Onge, J.; Arguin, H.; Couillard, E.; Dubois, M.-J.; Marette, A.; Tremblat, A.; Weisnagel, S.J.; et al. Fitness, adiposopathy, and adiposity are independent predictors of insulin sensitivity in middle-aged men without diabetes. J. Physiol. Biochem. 2016, 72, 435–444. [Google Scholar] [CrossRef]
- Sachs, S.; Zarini, S.; Kahn, D.E.; Harrison, K.A.; Perreault, L.; Phang, T.; Newsom, S.A.; Strauss, A.; Kerege, A.; Schoen, J.A.; et al. Intermuscular adipose tissue directly modulates skeletal muscle insulin sensitivity in humans. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E866–E879. [Google Scholar] [CrossRef] [Green Version]
- Ryan, A.S.; Macko, R.F.; Peters, M.N.; Ivey, F.M.; Prior, S.J.; Joseph, L.J.; Hafer-Macko, C.E. Plasma adiponectin levels are associated with insulin sensitivity in stroke survivors. J. Stroke Cerebrovasc. Dis. 2009, 18, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Henry, R.R.; Ciaraldi, T.P.; Abrams-Carter, L.; Mudaliar, S.; Park, K.S.; Nikoulina, S.E. Glycogen synthase activity is reduced in cultured skeletal muscle cells of non-insulin-dependent diabetes mellitus subjects. Biochemical and molecular mechanisms. J. Clin. Investig. 1996, 98, 1231–1236. [Google Scholar] [CrossRef]
- Thorburn, A.W.; Gumbiner, B.; Bulacan, F.; Brechtel, G.; Henry, R.R. Multiple defects in muscle glycogen synthase activity contribute to reduced glycogen synthesis in non-insulin dependent diabetes mellitus. J. Clin. Investig. 1991, 87, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Kida, Y.; Puente, A.E.-D.; Bogardus, C.; Mott, D.M. Insulin resistance is associated with reduced fasting and insulin-stimulated glycogen synthase phosphatase activity in human skeletal muscle. J. Clin. Investig. 1990, 85, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, H.; Bjorbaek, C.; Andersen, P.H.; Bak, J.F.; Pedersen, O. Impaired expression of glycogen synthase mRNA in skeletal muscle of NIDDM patients. Diabetes 1991, 40, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.S.; Ivey, F.M.; Prior, S.; Li, G.; Hafer-Macko, C. Skeletal muscle hypertrophy and muscle myostatin reduction after resistive training in stroke survivors. Stroke 2011, 42, 416–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck-Nielsen, H. The role of glycogen synthase in the development of hyperglycemia in type 2 diabetes—‘To store or not to store glucose, that’s the question’. Diabetes/Metab. Res. Rev. 2012, 28, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Bogardus, C.; Lillioja, S.; Stone, K.; Mott, D. Correlation between muscle glycogen synthase activity and in vivo insulin action in man. J. Clin. Investig. 1984, 73, 1185–1190. [Google Scholar] [CrossRef]
- Azpiazu, I.; Manchester, J.; Skurat, A.V.; Roach, P.J.; Lawrence, J.C., Jr. Control of glycogen synthesis is shared between glucose transport and glycogen synthase in skeletal muscle fibers. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E234–E243. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; De Ferranti, S.; Després, J.-P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Popa-Wagner, A.; Dumitrascu, D.I.; Capitanescu, B.; Petcu, E.B.; Surugiu, R.; Fang, W.H.; Dumbrava, D.A. Dietary habits, lifestyle factors and neurodegenerative diseases. Neural Regen. Res. 2020, 15, 394–400. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stroke (n = 30) | Controls (n = 35) | |
|---|---|---|
| Age (yr) | 60 ± 8 | 62 ± 8 |
| Weight (kg) | 93.2 ± 21.5 | 96.2 ± 16.9 |
| BMI (kg/m2) | 31.6 ± 7.5 | 33.5 ± 5.0 |
| % Body fat | 37.6 ± 8.6 | 41.8 ± 8.2 * |
| Fat mass (kg) | 36.1 ± 13.8 | 40.5 ± 10.8 |
| Fat-free mass (kg) | 58.3 ± 11.4 | 55.9 ± 11.6 |
| VO2max (mL/kg/min) | 20.1 ± 5.1 | 23.7 ± 5.2 † |
| VO2max (L/min) | 1.82 ± 0.55 | 2.29 ± 0.59 ‡ |
| Visceral fat area L2-L3 (cm2) | 226.8 ± 109.7 | 229.7 ± 109.9 |
| Visceral fat area L4-L5(cm2) | 169.7 ± 85.5 | 182.3 ± 71.4 |
| Subcutaneous abdominal fat L2-L3 (cm2) | 308.6 ± 151.6 | 317.3 ± 121.8 |
| Subcutaneous abdominal fat L4-L5 (cm2) | 364.2 ± 177.1 | 423.6 ± 140.6 |
| Sagittal diameter L2-L3 (cm) | 27.3 ± 5.3 | 26.8 ± 3.5 |
| Sagittal diameter L4-L5 (cm) | 26.7 ± 5.8 | 27.6 ± 3.9 |
| Mid-thigh NP muscle area (cm2) Mid-thigh NP muscle (%) | 91.2 ± 17.1 57.2 ± 1.1 | 88.0 ± 26.8 ** |
| Mid-thigh P muscle area (cm2) Mid-thigh P muscle (%) | 74.4 ± 17.2 ‡‡ 52.3 ± 1.0 ‡‡‡ | |
| Mid-thigh NP subcutaneous fat (cm2) | 119.6 ± 64.9 | 142.5 ± 71.7 |
| Mid-thigh P subcutaneous fat (cm2) | 121.9 ± 63.3 | |
| Mid-thigh NP IMAT (cm2) Mid-thigh NP IMAT (%) | 35.7 ± 11.3 21.4 ± 3.2 | 34.8 ± 10.4 |
| Mid-thigh P IMAT (cm2) Mid-thigh P IMAT (%) | 33.3 ± 10.1 ‡‡ 22.6 ± 2.6 ‡‡ | |
| Mid-thigh NP muscle attenuation (HU) | 37.8 ± 4.8 | 36.2 ± 6.9 |
| Mid-thigh P muscle attenuation (HU) | 33.8 ± 6.1 ‡‡ |
| Stroke (n = 30) a | Controls (n = 35) a | |
|---|---|---|
| Fasting plasma glucose (mmol/L) | 5.40 ± 0.99 | 5.46 ± 0.66 |
| Fasting plasma insulin (pmol/L) | 105 ± 80 | 106 ± 49 |
| Glucose at 120 min | 7.74 ± 2.17 | 7.90 ± 2.19 |
| Insulin at 120 min | 622 ± 552 | 661 ± 397 |
| Glucose AUC (mmol/L·120 min) | 1014 ± 202 | 989 ± 213 |
| Insulin AUC (pmol/L·120 min) | 68,928 ± 58,589 | 68,823 ± 34,992 |
| M (µmol·kg−1·min−1) | 27.7 ± 11.0 | 27.2 ± 10.5 |
| M (µmol·kgFFM−1·min−1) | 42.1 ± 15.9 | 45.7 ± 18.1 |
| CHO oxidative metabolism (µmol·kgFFM−1·min−1) | 6.67 ± 7.05 | 7.91 ± 8.41 |
| Nonoxidative metabolism (µmol·kgFFM−1·min−1) | 35.98 ± 14.96 | 41.94 ± 18.30 |
| Glycogen Synthase Activity | |||
|---|---|---|---|
| Nonparetic | Paretic | p-Value | |
| Basal GSI (nmol/min/mg protein) | 0.19 ± 0.12 | 0.20 ± 0.13 | 0.80 |
| Insulin GSI (nmol/min/mg protein) | 0.66 ± 0.39 * | 0.21 ± 0.13 | 0.00003 |
| Insulin-Basal GSI (nmol/min/mg protein) | 0.53 ± 0.42 | 0.008 ± 0.12 | 0.00004 |
| % above Basal GSI | 413 ± 338 | 18 ± 65 | 0.000009 |
| Basal GST (nmol/min/mg protein) | 8.5 ± 4.1 | 7.9 ± 3.3 | 0.43 |
| Insulin GST (nmol/min/mg protein) | 9.5 ± 3.6 | 3.3 ± 1.5 ‡ | 0.000000003 |
| Insulin-Basal GST (nmol/min/mg protein) | 1.0 ± 4.3 | −4.8 ± 2.6 | 0.000004 |
| % above Basal GST | 24 ± 47 | −61 ± 14 | 0.0000001 |
| Basal GSFV (%) | 2.4 ± 1.4 | 2.6 ± 1.6 | 0.51 |
| Insulin GSFV (%) | 8.6 ± 5.1 ‡ | 6.9 ± 3.5 † | 0.048 |
| Insulin-Basal GSFV (%) | 6.2 ± 4.3 | 4.3 ± 2.5 | 0.02 |
| % above Basal GSFV | 296 ± 156 | 205 ± 134 | 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryan, A.S.; Hafer-Macko, C.; Ortmeyer, H.K. Insulin Resistance in Skeletal Muscle of Chronic Stroke. Brain Sci. 2021, 11, 20. https://doi.org/10.3390/brainsci11010020
Ryan AS, Hafer-Macko C, Ortmeyer HK. Insulin Resistance in Skeletal Muscle of Chronic Stroke. Brain Sciences. 2021; 11(1):20. https://doi.org/10.3390/brainsci11010020
Chicago/Turabian StyleRyan, Alice S., Charlene Hafer-Macko, and Heidi K. Ortmeyer. 2021. "Insulin Resistance in Skeletal Muscle of Chronic Stroke" Brain Sciences 11, no. 1: 20. https://doi.org/10.3390/brainsci11010020
APA StyleRyan, A. S., Hafer-Macko, C., & Ortmeyer, H. K. (2021). Insulin Resistance in Skeletal Muscle of Chronic Stroke. Brain Sciences, 11(1), 20. https://doi.org/10.3390/brainsci11010020