Precision Medicine Care in ADHD: The Case for Neural Excitation and Inhibition


{kind=link}
Abstract
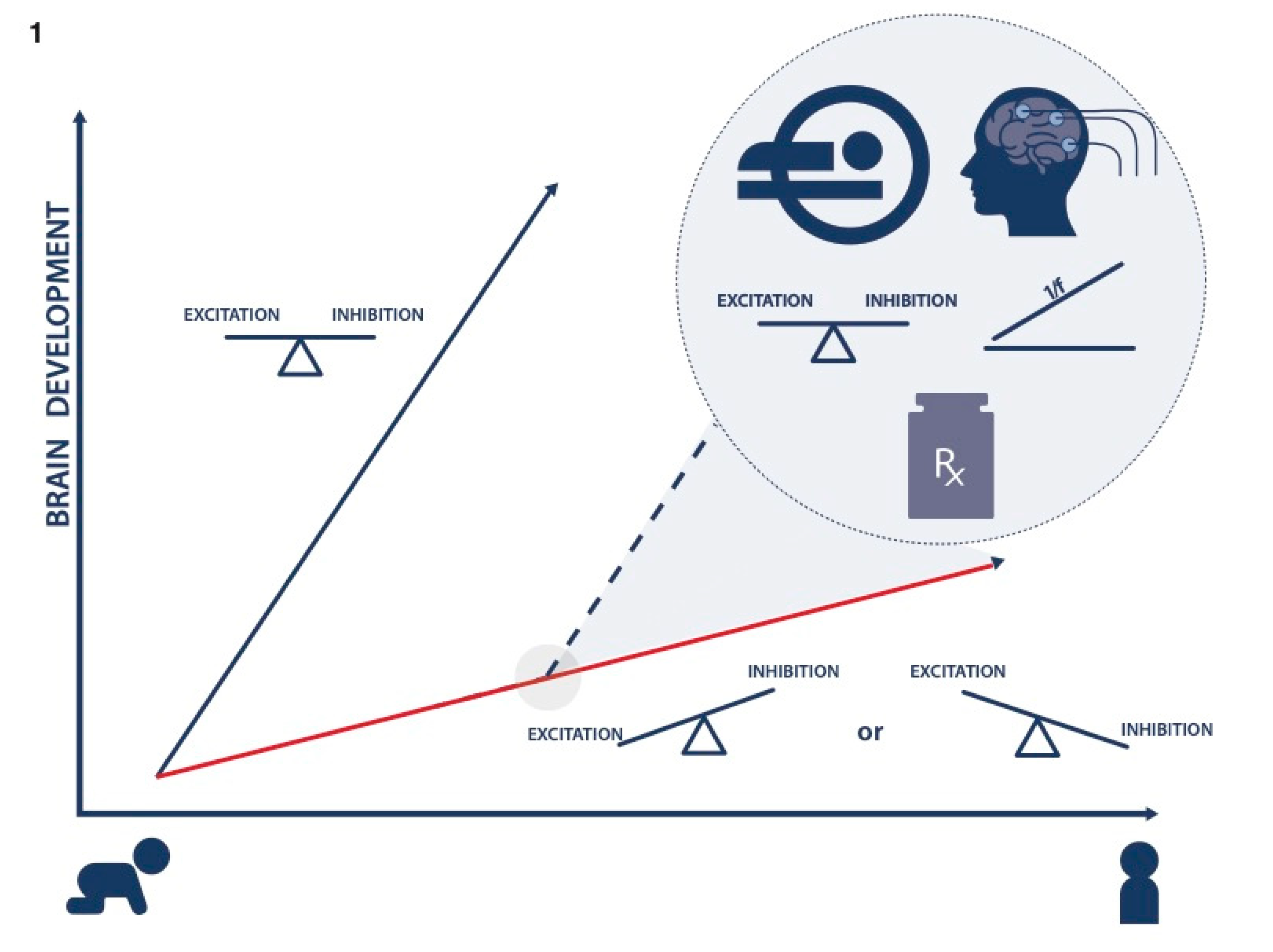
:1. Introduction: Need for Precision Medicine Care in ADHD
2. Imbalance of E/I in ADHD
3. Developmental Sequelae of E/I Imbalance: Altered Brain Connectivity
4. GABA and Glutamate Modulating Treatments in ADHD and Related Neurodevelopmental Disorders
5. Conclusions and Future Directions: Implications for Precision Medicine Care
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sudre, G.; Mangalmurti, A.; Shaw, P. Growing out of attention deficit hyperactivity disorder: Insights from the ‘remitted’brain. Neurosci. Biobehav. Rev. 2018, 94, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Barbaresi, W.J.; Colligan, R.C.; Weaver, A.L.; Voigt, R.G.; Killian, J.M.; Katusic, S.K.; Chawla, A.; Sprinz, P.G.; Welch, J.; Heeney, M.; et al. Mortality, ADHD, and Psychosocial Adversity in Adults with Childhood ADHD: A Prospective Study. Pediatrics 2013, 131, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkley, R.A.; Fischer, M. Hyperactive Child Syndrome and Estimated Life Expectancy at Young Adult Follow-Up: The Role of ADHD Persistence and Other Potential Predictors. J. Atten. Disord. 2018, 23, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Page, T.F.; Altszuler, A.R.; Pelham, W.E.; Kipp, H.; Gnagy, E.M.; Coxe, S.; Schatz, N.K.; Merrill, B.M.; Macphee, F.L. Family Burden of Raising a Child with ADHD. J. Abnorm. Child Psychol. 2019, 47, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Weibman, D.; Halperin, J.M.; Li, X. A Review of Heterogeneity in Attention Deficit/Hyperactivity Disorder (ADHD). Front. Hum. Neurosci. 2019, 13, 42. [Google Scholar] [CrossRef] [Green Version]
- Cortese, S. Pharmacologic Treatment of Attention Deficit–Hyperactivity Disorder. New Engl. J. Med. 2020, 383, 1050–1056. [Google Scholar] [CrossRef]
- Arnett, A.; Stein, M.A. Refining treatment choices for ADHD. Lancet Psychiatry 2018, 5, 691–692. [Google Scholar] [CrossRef] [Green Version]
- Sikirica, V.; Lü, M.; Greven, P.; Zhong, Y.; Qin, P.; Xie, J.; Gajria, K. Adherence, persistence, and medication discontinuation in patients with attention-deficit/hyperactivity disorder – a systematic literature review. Neuropsychiatr. Dis. Treat. 2014, 10, 1543–1569. [Google Scholar] [CrossRef] [Green Version]
- Barbaresi, W.J.; Katusic, S.K.; Colligan, R.C.; Weaver, A.L.; Leibson, C.L.; Jacobsen, S.J. Long-Term Stimulant Medication Treatment of Attention-Deficit/Hyperactivity Disorder. J. Dev. Behav. Pediatr. 2014, 35, 448–457. [Google Scholar] [CrossRef]
- Solanto, M.V.; Newcorn, J.H.; Vail, L.; Gilbert, S.; Ivanov, I.; Lara, R. Stimulant Drug Response in the Predominantly Inattentive and Combined Subtypes of Attention-Deficit/Hyperactivity Disorder. J. Child Adolesc. Psychopharmacol. 2009, 19, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Lahey, B.B.; Pelham, W.E.; Loney, J.; Lee, S.S.; Willcutt, E. Instability of the DSM-IV Subtypes of ADHD From Preschool Through Elementary School. Arch. Gen. Psychiatry 2005, 62, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Peadon, E. Distinguishing between attention-deficit hyperactivity and fetal alcohol spectrum disorders in children: Clinical guidelines. Neuropsychiatr. Dis. Treat. 2010, 6, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, J.F.; Griffiths, K.R.; Korgaonkar, M.S. A Systematic Review of Imaging Studies in the Combined and Inattentive Subtypes of Attention Deficit Hyperactivity Disorder. Front. Integr. Neurosci. 2020, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Kasparek, T.; Theiner, P.; Filova, A. Neurobiology of ADHD from childhood to adulthood: Findings of imaging methods. J. Atten. Disord. 2015, 19, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Bush, G. Cingulate, Frontal, and Parietal Cortical Dysfunction in Attention-Deficit/Hyperactivity Disorder. Biol. Psychiatry 2011, 69, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Todd, R.D.; Huang, H.; Smalley, S.L.; Nelson, S.F.; Willcutt, E.G.; Pennington, B.F.; Smith, S.D.; Faraone, S.V.; Neuman, R.J. Collaborative analysis of DRD4 and DAT genotypes in population-defined ADHD subtypes. J. Child Psychol. Psychiatry 2005, 46, 1067–1073. [Google Scholar] [CrossRef]
- Cook, E.H.; A Stein, M.; Krasowski, M.D.; Cox, N.J.; Olkon, D.M.; E Kieffer, J.; Leventhal, B.L. Association of attention-deficit disorder and the dopamine transporter gene. Am. J. Hum. Genet. 1995, 56, 993–998. [Google Scholar]
- Li, D.; Sham, P.C.; Owen, M.J.; He, L. Meta-analysis shows significant association between dopamine system genes and attention deficit hyperactivity disorder (ADHD). Hum. Mol. Genet. 2006, 15, 2276–2284. [Google Scholar] [CrossRef]
- Schrantee, A.; Tamminga, H.G.; Bouziane, C.; Bottelier, M.A.; Bron, E.E.; Mutsaerts, H.-J.M.; Zwinderman, A.H.; Groote, I.R.; Rombouts, S.A.; Lindauer, R.J. Age-dependent effects of methylphenidate on the human dopaminergic system in young vs adult patients with attention-deficit/hyperactivity disorder: A randomized clinical trial. JAMA Psychiatry 2016, 73, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Rosa-Neto, P.; Lou, H.C.; Cumming, P.; Pryds, O.; Karrebaek, H.; Lunding, J.; Gjedde, A. Methylphenidate-evoked changes in striatal dopamine correlate with inattention and impulsivity in adolescents with attention deficit hyperactivity disorder. NeuroImage 2005, 25, 868–876. [Google Scholar] [CrossRef]
- Shoham, R.; Sonuga-Barke, E.; Yaniv, I.; Pollak, Y. ADHD Is Associated with a Widespread Pattern of Risky Behavior Across Activity Domains. J. Atten. Disord. 2019, 1087054719875786. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Ghahremani, D.G.; Morales, A.M.; Robertson, C.L.; Ishibashi, K.; Morgan, A.T.; Mandelkern, M.A.; London, E.D. Risk-Taking Behavior: Dopamine D2/D3 Receptors, Feedback, and Frontolimbic Activity. Cereb. Cortex 2013, 25, 236–245. [Google Scholar] [CrossRef]
- Rosch, K.S.; Mostofsky, S.H.; Nebel, M.B. ADHD-related sex differences in fronto-subcortical intrinsic functional connectivity and associations with delay discounting. J. Neurodev. Disord. 2018, 10, 34. [Google Scholar]
- Tedford, S.E.; Persons, A.L.; Napier, T.C. Dopaminergic Lesions of the Dorsolateral Striatum in Rats Increase Delay Discounting in an Impulsive Choice Task. PLOS ONE 2015, 10, e0122063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielson, M.L.; Bitsko, R.H.; Ghandour, R.M.; Holbrook, J.R.; Kogan, M.D.; Blumberg, S.J. Prevalence of Parent-Reported ADHD Diagnosis and Associated Treatment Among U.S. Children and Adolescents, 2016. J. Clin. Child Adolesc. Psychol. 2018, 47, 199–212. [Google Scholar] [CrossRef]
- Faraone, S.V. The pharmacology of amphetamine and methylphenidate: Relevance to the neurobiology of attention-deficit/hyperactivity disorder and other psychiatric comorbidities. Neurosci. Biobehav. Rev. 2018, 87, 255–270. [Google Scholar] [CrossRef]
- Puts, N.A.; Ryand, M.; Oeltzschner, G.; Horska, A.; Edden, R.A.E.; Mahone, E.M. Reduced striatal GABA in unmedicated children with ADHD at 7T. Psychiatry Res. Neuroimaging 2020, 301, 111082. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Portera-Cailliau, C. Autism in the Balance: Elevated E-I Ratio as a Homeostatic Stabilization of Synaptic Drive. Neuron 2019, 101, 543–545. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Regal, M.P.; Luengas-Escuza, I.; Bayón-Cordero, L.; Ibarra-Aizpurua, N.; Alberdi, E.; Pérez-Samartín, A.; Matute, C.; Sánchez-Gómez, M.V. Oligodendrocyte Differentiation and Myelination Is Potentiated via GABAB Receptor Activation. Neuroscience 2020, 439, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.J.; Paul, A. The diversity of GABAergic neurons and neural communication elements. Nat. Rev. Neurosci. 2019, 20, 563–572. [Google Scholar] [CrossRef]
- Gu, Y.; Tran, T.; Murase, S.; Borrell, A.; Kirkwood, A.; Quinlan, E.M. Neuregulin-Dependent Regulation of Fast-Spiking Interneuron Excitability Controls the Timing of the Critical Period. J. Neurosci. 2016, 36, 10285–10295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Huang, S.; Chang, M.C.; Worley, P.; Kirkwood, A.; Quinlan, E.M. Obligatory Role for the Immediate Early Gene NARP in Critical Period Plasticity. Neuron 2013, 79, 335–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, S.; Di Nardo, A.A.; Aizawa, S.; Matsuo, I.; Volovitch, M.; Prochiantz, A.; Hensch, T.K. Experience-Dependent Transfer of Otx2 Homeoprotein into the Visual Cortex Activates Postnatal Plasticity. Cell 2008, 134, 508–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Kirkwood, A.; Pizzorusso, T.; Porciatti, V.; Morales, B.; Bear, M.F.; Maffei, L.; Tonegawa, S. BDNF Regulates the Maturation of Inhibition and the Critical Period of Plasticity in Mouse Visual Cortex. Cell 1999, 98, 739–755. [Google Scholar] [CrossRef] [Green Version]
- Deco, G.; Hugues, E.; Sporns, O.; Deco, G. Role of local network oscillations in resting-state functional connectivity. NeuroImage 2011, 57, 130–139. [Google Scholar] [CrossRef]
- Edden, R.A.E.; Crocetti, D.; Zhu, H.; Gilbert, D.L.; Mostofsky, S.H. Reduced GABA Concentration in Attention-Deficit/Hyperactivity Disorder. Arch. Gen. Psychiatry 2012, 69, 750–753. [Google Scholar] [CrossRef]
- Courvoisie, H.; Hooper, S.R.; Fine, C.; Kwock, L.; Castillo, M. Neurometabolic Functioning and Neuropsychological Correlates in Children With ADHD-H: Preliminary Findings. J. Neuropsychiatry Clin. Neurosci. 2004, 16, 63–69. [Google Scholar] [CrossRef]
- MacMaster, F.P.; Carrey, N.; Sparkes, S.; Kusumakar, V. Proton spectroscopy in medication-free pediatric attention-deficit/hyperactivity disorder. Biol. Psychiatry 2003, 53, 184–187. [Google Scholar] [CrossRef]
- Bollmann, S.; Ghisleni, C.; Poil, S.-S.; Martin, E.L.; Ball, J.S.; Eich-Höchli, D.; A E Edden, R.; Klaver, P.; Michels, L.; Brandeis, D.; et al. Developmental changes in gamma-aminobutyric acid levels in attention-deficit/hyperactivity disorder. Transl. Psychiatry 2015, 5, e589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dramsdahl, M.; Ersland, L.; Plessen, K.; Haavik, J.; Hugdahl, K.; Specht, K. Adults with Attention-Deficit/Hyperactivity Disorder? A Brain Magnetic Resonance Spectroscopy Study. Front. Psychiatry 2011, 2, 65. [Google Scholar] [CrossRef] [Green Version]
- Ende, G.; Cackowski, S.; Van Eijk, J.; Sack, M.; Demirakca, T.; Kleindienst, N.; Bohus, M.; Sobanski, E.; Krause-Utz, A.; Schmahl, C. Impulsivity and Aggression in Female BPD and ADHD Patients: Association with ACC Glutamate and GABA Concentrations. Neuropsychopharmacology 2015, 41, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Perlov, E.; Philipsen, A.; Hesslinger, B.; Büchert, M.; Ahrendts, J.; Feige, B.; Bubl, E.; Hennig, J.; Ebert, D.; Van Elst, L.T. Reduced cingulate glutamate/glutamine-to-creatine ratios in adult patients with attention deficit/hyperactivity disorder – A magnet resonance spectroscopy study. J. Psychiatr. Res. 2007, 41, 934–941. [Google Scholar] [CrossRef]
- Endres, D.; Perlov, E.; Maier, S.; Feige, B.; Nickel, K.; Goll, P.; Bubl, E.; Lange, T.; Glauche, V.; Graf, E.; et al. Normal Neurochemistry in the Prefrontal and Cerebellar Brain of Adults with Attention-Deficit Hyperactivity Disorder. Front. Behav. Neurosci. 2015, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridi, M.C.; Zong, F.-J.; Min, X.; Luo, N.; Tran, T.; Qiu, J.; Severin, D.; Zhang, X.-T.; Wang, G.; Zhu, Z.-J.; et al. Daily Oscillation of the Excitation-Inhibition Balance in Visual Cortical Circuits. Neuron 2020, 105, 621–629.e4. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Bridi, M.; Koh, M.T.; Gallagher, M.; Kirkwood, A. Reduced cognitive performance in aged rats correlates with increased excitation/inhibition ratio in the dentate gyrus in response to lateral entorhinal input. Neurobiol. Aging 2019, 82, 120–127. [Google Scholar] [CrossRef]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nat. Cell Biol. 2011, 477, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, S.L.; Gaggioni, G.; Ly, J.Q.M.; Papachilleos, S.; Borsu, C.; Brzozowski, A.; Rosanova, M.; Sarasso, S.; Luxen, A.; Middleton, B.; et al. Circadian dynamics in measures of cortical excitation and inhibition balance. Sci. Rep. 2016, 6, 33661. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.L.; Jeitner, T. Central Role of Glutamate Metabolism in the Maintenance of Nitrogen Homeostasis in Normal and Hyperammonemic Brain. Biomolecules 2016, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.B.; De Graaf, R.A.; Mason, G.F.; Rothman, D.L.; Shulman, R.G.; Behar, K.L. The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 5588–5593. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Atallah, B.V.; Scanziani, M. Equalizing excitation–inhibition ratios across visual cortical neurons. Nat. Cell Biol. 2014, 511, 596–600. [Google Scholar] [CrossRef]
- Vincent, S.R.; Hokfelt, T.; Skirboll, L.R.; Wu, J.Y. Hypothalamic gamma-aminobutyric acid neurons project to the neocortex. Science 1983, 220, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Freund, T.F.; Meskenaite, V. gamma-Aminobutyric acid-containing basal forebrain neurons innervate inhibitory interneurons in the neocortex. Proc. Natl. Acad. Sci. USA 1992, 89, 738–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- Mamiya, P.C.; Richards, T.L.; Lee, A.K.C.; Edden, R.A.E.; Stein, A.M.; Kuhl, P.K. Reduced gamma-aminobutyric acid levels during Flanker tasks are associated with impaired attention control deficits in adults with Attention-Deficit/Hyperactivity Disorder. In Proceedings of the 5th International Symposium on GABA and advanced MRS, Park City, UT, USA, 18–20 November 2019. [Google Scholar]
- Muthukumaraswamy, S.D.; Edden, R.A.E.; Jones, D.K.; Swettenham, J.B.; Singh, K. Resting GABA concentration predicts peak gamma frequency and fMRI amplitude in response to visual stimulation in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8356–8361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grent, T.; Gross, J.; Goense, J.; Wibral, M.; Gajwani, R.; Gumley, A.I.; Lawrie, S.M.; Schwannauer, M.; Schultze-Lutter, F.; Schröder, T.N.; et al. Resting-state gamma-band power alterations in schizophrenia reveal E/I-balance abnormalities across illness-stages. eLife 2018, 7. [Google Scholar] [CrossRef]
- Buzsáki, G. Rhythms of the Brain; Oxford University Press (OUP): Oxford, UK, 2006. [Google Scholar]
- He, B.J.; Zempel, J.M.; Snyder, A.Z.; Raichle, M.E. The Temporal Structures and Functional Significance of Scale-free Brain Activity. Neuron 2010, 66, 353–369. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Peterson, E.J.; Voytek, B. Inferring synaptic excitation/inhibition balance from field potentials. NeuroImage 2017, 158, 70–78. [Google Scholar] [CrossRef]
- Arns, M.; Conners, C.K.; Kraemer, H.C. A decade of EEG theta/beta ratio research in ADHD: A meta-analysis. J. Atten. Disord. 2013, 17, 374–383. [Google Scholar]
- Lenartowicz, A.; Loo, S.K. Use of EEG to Diagnose ADHD. Curr. Psychiatry Rep. 2014, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Robertson, M.M.; Furlong, S.; Voytek, B.; Donoghue, T.; Boettiger, C.A.; Sheridan, M.A. EEG power spectral slope differs by ADHD status and stimulant medication exposure in early childhood. J. Neurophysiol. 2019, 122, 2427–2437. [Google Scholar] [CrossRef]
- Pertermann, M.; Bluschke, A.; Roessner, V.; Beste, C. The Modulation of Neural Noise Underlies the Effectiveness of Methylphenidate Treatment in Attention-Deficit/Hyperactivity Disorder. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2019, 4, 743–750. [Google Scholar] [CrossRef]
- Demontis, D.; Walters, R.K.; Martin, J.; Mattheisen, M.; Als, T.D.; Agerbo, E.; Baldursson, G.; Belliveau, R.; Bybjerg-Grauholm, J.; Bækvad-Hansen, M.; et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 2019, 51, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Fair, D.A.; Bathula, D.; Nikolas, M.A.; Nigg, J.T. Distinct neuropsychological subgroups in typically developing youth inform heterogeneity in children with ADHD. Proc. Natl. Acad. Sci. USA 2012, 109, 6769–6774. [Google Scholar] [CrossRef] [Green Version]
- Mescher, M.; Merkle, H.; Kirsch, J.; Garwood, M.; Gruetter, R. Simultaneous in vivo spectral editing and water suppression. NMR Biomed. 1998, 11, 266–272. [Google Scholar] [CrossRef]
- Edden, R.A.E.; Barker, P.B. Spatial effects in the detection of γ-aminobutyric acid: Improved sensitivity at high fields using inner volume saturation. Magn. Reson. Med. 2007, 58, 1276–1282. [Google Scholar] [CrossRef]
- Luck, S.J. An Introduction to the Event-Related Potential Technique; MIT Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Ben-Ari, Y. Excitatory actions of gaba during development: The nature of the nurture. Nat. Rev. Neurosci. 2002, 3, 728–739. [Google Scholar] [CrossRef]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Marín, O. Developmental timing and critical windows for the treatment of psychiatric disorders. Nat. Med. 2016, 22, 1229–1238. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, A.; Holton, K.F. Measuring Treatment Response in Pharmacological and Lifestyle Interventions Using Electroencephalography in ADHD: A Review. Clin. EEG Neurosci. 2019, 50, 256–266. [Google Scholar] [CrossRef]
- Arns, M.; Gunkelman, J.; Breteler, M.; Spronk, D. EEG phenotypes predict treatment outcome to stimulants in children with ADHD. J. Integr. Neurosci. 2008, 7, 421–438. [Google Scholar] [CrossRef]
- Battel, L.; Kieling, R.R.; Kieling, C.; Anés, M.; Aurich, N.K.; Da Costa, J.C.; Rohde, L.A.; Franco, A.R. Intrinsic Brain Connectivity Following Long-Term Treatment with Methylphenidate in Children with Attention-Deficit/Hyperactivity Disorder. J. Child Adolesc. Psychopharmacol. 2016, 26, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Ogrim, G.; A Hestad, K.; Kropotov, J.; Sandvik, L.; Candrian, G.; Brunner, J.F. Predicting the clinical outcome of stimulant medication in pediatric attention-deficit/hyperactivity disorder: Data from quantitative electroencephalography, event-related potentials, and a go/no-go test. Neuropsychiatr. Dis. Treat. 2014, 10, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresnahan, S.M.; Barry, R.J.; Clarke, A.R.; Johnstone, S.J. Quantitative EEG analysis in dexamphetamine-responsive adults with attention-deficit/hyperactivity disorder. Psychiatry Res. 2006, 141, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Schweren, L.J.S.; de Zeeuw, P.; Durston, S. MR imaging of the effects of methylphenidate on brain structure and function in Attention-Deficit/Hyperactivity Disorder. Eur. Neuropsychopharmacol. 2013, 23, 1151–1164. [Google Scholar] [CrossRef]
- Rubia, K.; Alegria, A.A.; Cubillo, A.I.; Smith, A.B.; Brammer, M.J.; Radua, J. Effects of Stimulants on Brain Function in Attention-Deficit/Hyperactivity Disorder: A Systematic Review and Meta-Analysis. Biol. Psychiatry 2014, 76, 616–628. [Google Scholar] [CrossRef] [Green Version]
- Rubia, K.; Halari, R.; Mohammad, A.-M.; Taylor, E.; Brammer, M. Methylphenidate Normalizes Frontocingulate Underactivation During Error Processing in Attention-Deficit/Hyperactivity Disorder. Biol. Psychiatry 2011, 70, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Hart, H.; Radua, J.; Nakao, T.; Mataix-Cols, D.; Rubia, K. Meta-analysis of Functional Magnetic Resonance Imaging Studies of Inhibition and Attention in Attention-deficit/Hyperactivity Disorder. JAMA Psychiatry 2013, 70, 185–198. [Google Scholar] [CrossRef] [Green Version]
- Schweren, L.J.; Hartman, C.; Zwiers, M.; Heslenfeld, D.; Franke, L.; Oosterlaan, J.; Buitelaar, J.; Hoekstra, P. Stimulant treatment history predicts frontal-striatal structural connectivity in adolescents with attention-deficit/hyperactivity disorder. Eur. Neuropsychopharmacol. 2016, 26, 674–683. [Google Scholar] [CrossRef]
- Frodl, T.; Skokauskas, N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr. Scand. 2012, 125, 114–126. [Google Scholar] [CrossRef]
- Samea, F.; Soluki, S.; Nejati, V.; Zarei, M.; Cortese, S.; Eickhoff, S.B.; Tahmasian, M.; Eickhoff, C.R. Brain alterations in children/adolescents with ADHD revisited: A neuroimaging meta-analysis of 96 structural and functional studies. Neurosci. Biobehav. Rev. 2019, 100, 1–8. [Google Scholar] [CrossRef]
- Hoogman, M.; Van Rooij, D.; Klein, M.; Boedhoe, P.; Ilioska, I.; Li, T.; Patel, Y.; Postema, M.C.; Zhang-James, Y.; Anagnostou, E.; et al. Consortium neuroscience of attention deficit/hyperactivity disorder and autism spectrum disorder: The ENIGMA adventure. Hum. Brain Mapp. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bouziane, C.; Caan, M.W.; Tamminga, H.G.; Schrantee, A.; Bottelier, M.A.; De Ruiter, M.B.; Kooij, S.J.; Reneman, L. ADHD and maturation of brain white matter: A DTI study in medication naive children and adults. NeuroImage: Clin. 2018, 17, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Saad, J.F.; Griffiths, K.R.; Kohn, M.R.; Clarke, S.; Williams, L.M.; Korgaonkar, M.S. Regional brain network organization distinguishes the combined and inattentive subtypes of Attention Deficit Hyperactivity Disorder. NeuroImage Clin. 2017, 15, 383–390. [Google Scholar] [CrossRef]
- Greven, C.U.; Bralten, J.; Mennes, M.; O’Dwyer, L.; Van Hulzen, K.J.E.; Rommelse, N.; Schweren, L.J.S.; Hoekstra, P.J.; Hartman, C.A.; Heslenfeld, D.; et al. Developmentally Stable Whole-Brain Volume Reductions and Developmentally Sensitive Caudate and Putamen Volume Alterations in Those with Attention-Deficit/Hyperactivity Disorder and Their Unaffected Siblings. JAMA Psychiatry 2015, 72, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Nakao, T.; Radua, J.; Rubia, K.; Mataix-Cols, D. Gray Matter Volume Abnormalities in ADHD: Voxel-Based Meta-Analysis Exploring the Effects of Age and Stimulant Medication. Am. J. Psychiatry 2011, 168, 1154–1163. [Google Scholar] [CrossRef]
- Murias, M.; Swanson, J.M.; Srinivasan, R. Functional Connectivity of Frontal Cortex in Healthy and ADHD Children Reflected in EEG Coherence. Cereb. Cortex 2006, 17, 1788–1799. [Google Scholar] [CrossRef] [Green Version]
- Furlong, S.; Cohen, J.R.; Hopfinger, J.; Snyder, J.; Robertson, M.M.; Sheridan, M. Resting-state EEG Connectivity in Young Children with ADHD. J. Clin. Child Adolesc. Psychol. 2020, 1–17. [Google Scholar] [CrossRef]
- Kessler, D.; Angstadt, M.; Sripada, C. Growth Charting of Brain Connectivity Networks and the Identification of Attention Impairment in Youth. JAMA Psychiatry 2016, 73, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Mattfeld, A.T.; Gabrieli, J.D.E.; Biederman, J.; Spencer, T.; Brown, A.; Kotte, A.; Kagan, E.; Whitfield-Gabrieli, S. Brain differences between persistent and remitted attention deficit hyperactivity disorder. Brain 2014, 137, 2423–2428. [Google Scholar] [CrossRef] [Green Version]
- Michelini, G.; Jurgiel, J.; Bakolis, I.; Cheung, C.H.M.; Asherson, P.; Loo, S.K.; Kuntsi, J.; Mohammad-Rezazadeh, I. Atypical functional connectivity in adolescents and adults with persistent and remitted ADHD during a cognitive control task. Transl. Psychiatry 2019, 9, 137. [Google Scholar] [CrossRef]
- Pfeffer, T.; Avramiea, A.-E.; Nolte, G.; Engel, A.K.; Linkenkaer-Hansen, K.; Donner, T.H. Catecholamines alter the intrinsic variability of cortical population activity and perception. PLoS Biol. 2018, 16, e2003453. [Google Scholar] [CrossRef] [Green Version]
- Purkayastha, P.; Malapati, A.; Yogeeswari, P.; Sriram, D. A Review on GABA/Glutamate Pathway for Therapeutic Intervention of ASD and ADHD. Curr. Med. Chem. 2015, 22, 1850–1859. [Google Scholar] [CrossRef]
- Cohen, R.; Senecky, Y.; Shuper, A.; Inbar, D.; Chodick, G.; Shalev, V.; Raz, R. Prevalence of epilepsy and attention-deficit hyperactivity (ADHD) disorder: A population-based study. J. Child Neurol. 2013, 28, 120–123. [Google Scholar] [CrossRef]
- Leitner, Y. The co-occurrence of autism and attention deficit hyperactivity disorder in children—What do we know? Front. Hum. Neurosci. 2014, 8, 268. [Google Scholar] [CrossRef]
- Hagerman, R.; Protic, D.; Rajaratnam, A.; Salcedo-Arellano, M.J.; Aydin, E.Y.; Schneider, A. Fragile X-Associated Neuropsychiatric Disorders (FXAND). Front. Psychiatry 2018, 9, 564. [Google Scholar] [CrossRef] [Green Version]
- Lo-Castro, A.; Curatolo, P. Epilepsy associated with autism and attention deficit hyperactivity disorder: Is there a genetic link? Brain Dev. 2014, 36, 185–193. [Google Scholar] [CrossRef]
- Clark, T.; Feehan, C.; Tinline, C.; Vostanis, P. Autistic symptoms in children with attention deficit-hyperactivity disorder. Eur. Child Adolesc. Psychiatry 1999, 8, 50–55. [Google Scholar] [CrossRef]
- Treiman, D.M. GABAergic Mechanisms in Epilepsy. Epilepsia 2001, 42, 8–12. [Google Scholar] [CrossRef]
- Stoppel, L.J.; Kazdoba, T.M.; Schaffler, M.D.; Preza, A.R.; Heynen, A.; Crawley, J.N.; Bear, M.F. R-Baclofen Reverses Cognitive Deficits and Improves Social Interactions in Two Lines of 16p11.2 Deletion Mice. Neuropsychopharmacology 2018, 43, 513–524. [Google Scholar] [CrossRef]
- Dölen, G.; Carpenter, R.L.; Ocain, T.D.; Bear, M.F. Mechanism-based approaches to treating fragile X. Pharmacol. Ther. 2010, 127, 78–93. [Google Scholar] [CrossRef]
- Anagnostou, E. Clinical trials in autism spectrum disorder: Evidence, challenges and future directions. Curr. Opin. Neurol. 2018, 31, 119–125. [Google Scholar] [CrossRef]
- Lemonnier, E.; Villeneuve, N.; Sonie, S.; Serret, S.; Rosier, A.; Roue, M.; Brosset, P.; Viellard, M.; Bernoux, D.; Rondeau, S.; et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. Psychiatry 2017, 7, e1056. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, C.-C.; Dai, Y.; Luo, Q.; Ji, Y.; Wang, K.; Deng, S.; Yu, J.; Xu, M.; Du, X.; et al. Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl. Psychiatry 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Berry-Kravis, E.; Hagerman, R.; Visootsak, J.; Budimirovic, D.; Kaufmann, W.E.; Cherubini, M.; Zarevics, P.; Walton-Bowen, K.; Wang, P.; Bear, M.F.; et al. Arbaclofen in fragile X syndrome: Results of phase 3 trials. J. Neurodev. Disord. 2017, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Veenstra-VanderWeele, J.; Cook, E.H.; King, B.H.; Zarevics, P.; Cherubini, M.; Walton-Bowen, K.; Bear, M.F.; Wang, P.P.; Carpenter, R.L. Arbaclofen in Children and Adolescents with Autism Spectrum Disorder: A Randomized, Controlled, Phase 2 Trial. Neuropsychopharmacology 2017, 42, 1390–1398. [Google Scholar] [CrossRef]
- Novell-Alsina, R.; Esteba-Castillo, S.; Rodriguez, E. Efficacy and safety of a GABAergic drug (Gamalate® B6): Effects on behavior and cognition in young adults with borderline-to-mild intellectual developmental disabilities and ADHD. Drugs Context 2020, 9, 212601–212612. [Google Scholar] [CrossRef]
- Adler, L.A.; Kroon, R.A.; Stein, M.; Shahid, M.; Tarazi, F.I.; Szegedi, A.; Schipper, J.; Cazorla, P. A Translational Approach to Evaluate the Efficacy and Safety of the Novel AMPA Receptor Positive Allosteric Modulator Org 26576 in Adult Attention-Deficit/Hyperactivity Disorder. Biol. Psychiatry 2012, 72, 971–977. [Google Scholar] [CrossRef]
- Elia, J.; Ungal, G.; Kao, C.; Ambrosini, A.; De Jesus-Rosario, N.; Larsen, L.; Chiavacci, R.; Wang, T.; Kurian, C.; Titchen, K.; et al. Fasoracetam in adolescents with ADHD and glutamatergic gene network variants disrupting mGluR neurotransmitter signaling. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Di Miceli, M.; Gronier, B. Psychostimulants and atomoxetine alter the electrophysiological activity of prefrontal cortex neurons, interaction with catecholamine and glutamate NMDA receptors. Psychopharmacology 2015, 232, 2191–2205. [Google Scholar] [CrossRef]
- Tritsch, N.X.; Ding, J.B.; Sabatini, B.L. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nat. Cell Biol. 2012, 490, 262–266. [Google Scholar] [CrossRef] [Green Version]
- Tritsch, N.X.; Oh, W.-J.; Gu, C.; Sabatini, B.L. Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. eLife 2014, 3, e01936. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.C.; Rosenstand, A.; Brooks, D.J.; Bender, D.; Jakobsen, S.; Blicher, J.U.; Hansen, K.V.; Møller, A. Exogenous dopamine reduces GABA receptor availability in the human brain. Brain Behav. 2016, 6, e00484. [Google Scholar] [CrossRef] [Green Version]
- Solleveld, M.M.; Schrantee, A.; Puts, N.A.; Reneman, L.; Lucassen, P.J. Age-dependent, lasting effects of methylphenidate on the GABAergic system of ADHD patients. NeuroImage Clin. 2017, 15, 812–818. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamiya, P.C.; Arnett, A.B.; Stein, M.A. Precision Medicine Care in ADHD: The Case for Neural Excitation and Inhibition. Brain Sci. 2021, 11, 91. https://doi.org/10.3390/brainsci11010091
Mamiya PC, Arnett AB, Stein MA. Precision Medicine Care in ADHD: The Case for Neural Excitation and Inhibition. Brain Sciences. 2021; 11(1):91. https://doi.org/10.3390/brainsci11010091
Chicago/Turabian StyleMamiya, Ping C., Anne B. Arnett, and Mark A. Stein. 2021. "Precision Medicine Care in ADHD: The Case for Neural Excitation and Inhibition" Brain Sciences 11, no. 1: 91. https://doi.org/10.3390/brainsci11010091
APA StyleMamiya, P. C., Arnett, A. B., & Stein, M. A. (2021). Precision Medicine Care in ADHD: The Case for Neural Excitation and Inhibition. Brain Sciences, 11(1), 91. https://doi.org/10.3390/brainsci11010091