
Advances in Recapitulating Alzheimer’s Disease Phenotypes Using Human Induced Pluripotent Stem Cell-Based In Vitro Models


Abstract
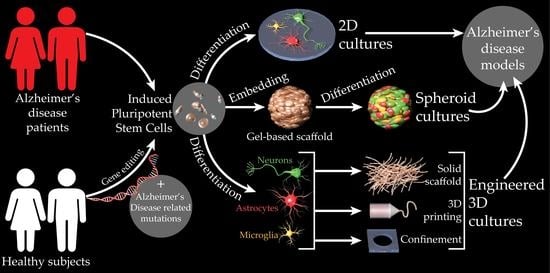
:
1. Introduction
2. HiPSC-Based Models of AD
2.1. HiPSC-Derived 2D AD Models
2.1.1. Neuron-Focused 2D AD Models
2.1.2. Astrocyte-Focused 2D AD Models
2.1.3. Microglia-Focused 2D AD Models
2.2. HiPSC-Derived Organoid Models of AD


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Observed AD Phenotype | Method | AD Source | Experimental Time Point, Days | Reference |
|---|---|---|---|---|
| Neurofibrillary tanglelike inclusions | Dual SMAD inhibition and Matrigel (Corning Life Sciences)—embedded 3D maturation | Application of recombinant human tau (K18) to P301L overexpressed neurons differentiated from NPCs | >28 | [116] |
| Extracellular deposition of Aβ, including Aβ plaques Aggregates of pTau in the soma and neurites and filamentous tau | Matrigel-embedded differentiation [137] | Lentiviral overexpression of FAD-related mutations in APP and PSEN1 of ReNcell VM (ReNeuron Group plc) | 49–100 | [118] |
| Aβ42/Aβ40-correlated increase of pTau and cell death | Matrigel-embedded differentiation [138] | Lentiviral overexpression of FAD-related mutations in APP and PSEN1 of ReNcell VM and FACS purification | 35–84 | [119] |
| Aβ accumulation and elevated pTau | Matrigel-embedded self-organized differentiation | FAD (APP and PSEN1) patient HiPSCs | 60–90 | [121] |
| Aβ oligomers and Aβ aggregation | Hydrogel-embedded dual SMAD-inhibited differentiation [108] | FAD (APP and PSEN1) patient fibroblasts | >14 | [122] |
| Aβ plaques Aggregated and abnormal pTau | Component- and environment-controlled differentiation of cerebral organoids | FAD (PSEN1) and Down syndrome patient HiPSCs | 110 | [123] |
| ↑ Tau fragmentation and mislocalization Impaired axonal transport and functionality that can be improved by microtubule stabilization | Matrigel-embedded self-organized differentiation | Familial frontotemporal dementia patient derived HiPSC with R406W mutation and isogenic control | 60 | [124] |
| Accelerated neuronal differentiation ↑ Synaptic markers ↑ Total tau and pTau | Matrigel-embedded growth factor–directed differentiation of HiPSCs in spinning bioreactor | apoE4+ LOAD patient–derived fibroblasts and gene-edited (apoE4) healthy control–derived fibroblasts | 46 | [128] |
| Early neuronal differentiation Aβ accumulation and elevated pTau | Matrigel-embedded self-organized differentiation | HiPSCs from LOAD patients with apoE4 mutation | >180 | [89] |
| ↑ Secretion of long Aβ peptides (Aβ40, Aβ42, and Aβ43) | Matrigel-embedded growth factor–directed differentiation of HiPSCs in spinning bioreactor | Fibroblasts from FAD patients with FAD-linked mutations in APP or PSEN1 | 100 | [125] |
| Increased Aβ42/Aβ40 peptide ratios and decreased synaptic protein levels | Matrigel-embedded differentiation in suspension | FAD (APP and PSEN1) patient HiPSCs | 35 | [130] |
2.3. Engineered 3D Models of AD

| Observed AD Phenotype | Method | Cell Type | AD Source | Experimental Time Point, Days | Reference |
|---|---|---|---|---|---|
| Decreased cell viability Synaptic dysfunction | Microwell in enclosed PDMS device | NPC-differentiated neurons | Aβ application | 10 | [139] |
| Extracellular Aβ aggregates Elevated intracellular and total pTau | Matrigel (Corning Life Sciences)–scaffolded spheroids in microfabricated microwells | ReNcell VM (ReNeuron Group plc), NPCs | Overexpression of APP variant with FAD mutations in ReNcell VM and FACS | 56 | [140] |
| Aβ aggregation, pTau accumulation, increased neuroinflammatory activity, microglial recruitment, axonal cleavage, and inflammatory damage to AD neurons and astrocytes | Matrigel-based 3D culture in engineered PDMS microfluidic device | ReNcell VM–derived neurons, NPC-derived astrocytes, and immortalized human microglia | Overexpression of APP variant with FAD mutations in ReNcell VM and FACS | 42 | [145] |
| Amyloid plaquelike formations Gliosis Neuroinflammation Decreased functionality | 3D silk sponge ECM [152] | Multiple neuronal and glial subtypes | HSV-1 infection in human NSCs [153] | 32 | [143] |

2.4. HiPSC Xenograft Model
3. Future Directions

4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Giacobini, E.; Gold, G. Alzheimer Disease Therapy—Moving from Amyloid-β to Tau. Nat. Rev. Neurol. 2013, 9, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A Critical Appraisal of Amyloid-β-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Alzheimer Prevention Hopes Continue to Dim. Nat. Rev. Drug Discov. 2020, 19, 226. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain Energy Rescue: An Emerging Therapeutic Concept for Neurodegenerative Disorders of Ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Perlmutter, J.S. Prescribing Aducanumab in the Face of Meager Efficacy and Real Risks. Neurology 2021, 97, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D. Anti-Amyloid Trials Raise Scientific and Ethical Questions. BMJ 2021, 372, n805. [Google Scholar] [CrossRef]
- Salloway, S.; Cummings, J. Aducanumab, Amyloid Lowering, and Slowing of Alzheimer Disease. Neurology 2021, 97, 543–544. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s Disease Drug Development Pipeline: 2021. Alzheimers Dement. Transl. Res. Clin. Interv. 2021, 7, e12179. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s Disease Drug Development Pipeline: 2020. Alzheimers Dement. Transl. Res. Clin. Interv. 2020, 6, e12050. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N. Animal Models of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006320. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, R.J.; Shen, J. Presenilin-1 Mutations and Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef] [Green Version]
- Isik, A.T. Late Onset Alzheimer&Rsquo;s Disease in Older People. Clin. Interv. Aging 2010, 2010, 307–311. [Google Scholar] [CrossRef] [Green Version]
- Oblak, A.L.; Forner, S.; Territo, P.R.; Sasner, M.; Carter, G.W.; Howell, G.R.; Sukoff-Rizzo, S.J.; Logsdon, B.A.; Mangravite, L.M.; Mortazavi, A.; et al. Model Organism Development and Evaluation for Late-onset Alzheimer’s Disease: MODEL-AD. Alzheimers Dement. Transl. Res. Clin. Interv. 2020, 6, e12110. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Eguizabal, C.; Aran, B.; Chuva de Sousa Lopes, S.M.; Geens, M.; Heindryckx, B.; Panula, S.; Popovic, M.; Vassena, R.; Veiga, A. Two Decades of Embryonic Stem Cells: A Historical Overview. Hum. Reprod. Open 2019, 2019, hoy024. [Google Scholar] [CrossRef] [Green Version]
- Lo, B.; Parham, L. Ethical Issues in Stem Cell Research. Endocr. Rev. 2009, 30, 204–213. [Google Scholar] [CrossRef]
- Yuan, S.H.; Martin, J.; Elia, J.; Flippin, J.; Paramban, R.I.; Hefferan, M.P.; Vidal, J.G.; Mu, Y.; Killian, R.L.; Israel, M.A.; et al. Cell-Surface Marker Signatures for the Isolation of Neural Stem Cells, Glia and Neurons Derived from Human Pluripotent Stem Cells. PLoS ONE 2011, 6, e17540. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly Efficient Neural Conversion of Human ES and IPS Cells by Dual Inhibition of SMAD Signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Morizane, A.; Doi, D.; Kikuchi, T.; Nishimura, K.; Takahashi, J. Small-Molecule Inhibitors of Bone Morphogenic Protein and Activin/Nodal Signals Promote Highly Efficient Neural Induction from Human Pluripotent Stem Cells. J. Neurosci. Res. 2011, 89, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.-i.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. A Induces Astrocytic Glutamate Release, Extrasynaptic NMDA Receptor Activation, and Synaptic Loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, C.; Grieder, S.; Yang, N.; Zhang, Y.; Wernig, M.; Sudhof, T. Rapid Generation of Functional and Homogeneous Excitatory Human Forebrain Neurons Using Neurogenin-2 (Ngn2). Protoc. Exch. 2018. [Google Scholar] [CrossRef] [Green Version]
- Canals, I.; Ginisty, A.; Quist, E.; Timmerman, R.; Fritze, J.; Miskinyte, G.; Monni, E.; Hansen, M.G.; Hidalgo, I.; Bryder, D.; et al. Rapid and Efficient Induction of Functional Astrocytes from Human Pluripotent Stem Cells. Nat. Methods 2018, 15, 693–696. [Google Scholar] [CrossRef]
- Konstantinides, N.; Desplan, C. Neuronal Differentiation Strategies: Insights from Single-Cell Sequencing and Machine Learning. Development 2020, 147, dev193631. [Google Scholar] [CrossRef]
- Vieira, M.S.; Santos, A.K.; Vasconcellos, R.; Goulart, V.A.M.; Parreira, R.C.; Kihara, A.H.; Ulrich, H.; Resende, R.R. Neural Stem Cell Differentiation into Mature Neurons: Mechanisms of Regulation and Biotechnological Applications. Biotechnol. Adv. 2018, 36, 1946–1970. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Avci, H.X.; Leist, M.; Kobolák, J.; Dinnyés, A. Astrocyte Differentiation of Human Pluripotent Stem Cells: New Tools for Neurological Disorder Research. Front. Cell. Neurosci. 2016, 10, 215. [Google Scholar] [CrossRef] [Green Version]
- Speicher, A.M.; Wiendl, H.; Meuth, S.G.; Pawlowski, M. Generating Microglia from Human Pluripotent Stem Cells: Novel in Vitro Models for the Study of Neurodegeneration. Mol. Neurodegener. 2019, 14, 46. [Google Scholar] [CrossRef]
- Li, W.; Sun, W.; Zhang, Y.; Wei, W.; Ambasudhan, R.; Xia, P.; Talantova, M.; Lin, T.; Kim, J.; Wang, X.; et al. Rapid Induction and Long-Term Self-Renewal of Primitive Neural Precursors from Human Embryonic Stem Cells by Small Molecule Inhibitors. Proc. Natl. Acad. Sci. USA 2011, 108, 8299–8304. [Google Scholar] [CrossRef] [Green Version]
- Galiakberova, A.A.; Dashinimaev, E.B. Neural Stem Cells and Methods for Their Generation From Induced Pluripotent Stem Cells in Vitro. Front. Cell Dev. Biol. 2020, 8, 815. [Google Scholar] [CrossRef]
- Richner, M.; Victor, M.B.; Liu, Y.; Abernathy, D.; Yoo, A.S. MicroRNA-Based Conversion of Human Fibroblasts into Striatal Medium Spiny Neurons. Nat. Protoc. 2015, 10, 1543–1555. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease: A Practical Approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Moya-Alvarado, G.; Gershoni-Emek, N.; Perlson, E.; Bronfman, F.C. Neurodegeneration and Alzheimer’s Disease (AD). What Can Proteomics Tell Us About the Alzheimer’s Brain? Mol. Cell. Proteom. 2016, 15, 409–425. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s Disease: Synaptic Dysfunction and Aβ. Mol. Neurodegener. 2009, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing Sporadic and Familial Alzheimer’s Disease Using Induced Pluripotent Stem Cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s Disease with IPSCs Reveals Stress Phenotypes Associated with Intracellular Aβ and Differential Drug Responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Téglási, A.; Bock, I.; Giudice, M.L.; Táncos, Z.; Molnár, K.; László, L.; et al. Neurons Derived from Sporadic Alzheimer’s Disease IPSCs Reveal Elevated TAU Hyperphosphorylation, Increased Amyloid Levels, and GSK3B Activation. Alzheimers Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jiao, B.; Shen, L. The Epigenetics of Alzheimer’s Disease: Factors and Therapeutic Implications. Front. Genet. 2018, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. A Large Panel of Isogenic APP and PSEN1 Mutant Human IPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP β-CTFs, Not Aβ. Neuron 2019, 104, 256–270.e5. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An Emerging Therapeutic Target for Alzheimer’s Disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Wadhwani, A.R.; Affaneh, A.; Van Gulden, S.; Kessler, J.A. Neuronal Apolipoprotein E4 Increases Cell Death and Phosphorylated Tau Release in Alzheimer Disease. Ann. Neurol. 2019, 85, 726–739. [Google Scholar] [CrossRef]
- van der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B.; et al. Cholesterol Metabolism Is a Druggable Axis That Independently Regulates Tau and Amyloid-β in IPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 2019, 24, 363–375.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodruff, G.; Reyna, S.M.; Dunlap, M.; Van Der Kant, R.; Callender, J.A.; Young, J.E.; Roberts, E.A.; Goldstein, L.S.B. Defective Transcytosis of APP and Lipoproteins in Human IPSC-Derived Neurons with Familial Alzheimer’s Disease Mutations. Cell Rep. 2016, 17, 759–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, S.; Alvisi, G.; Shahin, F.; Husain, U.; Rabbani, M.; Yaqub, T.; Anjum, A.A.; Sheikh, A.A.; Nawaz, M.; Ali, M.A. Role of Rab GTPases in HSV-1 Infection: Molecular Understanding of Viral Maturation and Egress. Microb. Pathog. 2018, 118, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Trushin, S.; Hasan, M.F. Mitochondrial Complex I as a Therapeutic Target for Alzheimer’s Disease. Acta Pharm. Sin. B 2021, 12, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial Dysfunction Is a Trigger of Alzheimer’s Disease Pathophysiology. Biochim. Biophys. Acta (BBA) -Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Hauptmann, S.; Scherping, I.; Dröse, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Müller, W.E. Mitochondrial Dysfunction: An Early Event in Alzheimer Pathology Accumulates with Age in AD Transgenic Mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kündig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative Stress and Altered Mitochondrial Protein Expression in the Absence of Amyloid-β and Tau Pathology in IPSC-Derived Neurons from Sporadic Alzheimer’s Disease Patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, M.; Ren, Q.; Liu, G.; Meng, S.; Xiahou, K.; Zhang, Y.; Jiang, N.; Zhou, W. Human Induced Pluripotent Stem Cell-Derived Neural Cells from Alzheimer’s Disease Patients Exhibited Different Susceptibility to Oxidative Stress. Stem Cells Dev. 2020, 29, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy Inhibits Amyloid-β and Tau Pathology and Reverses Cognitive Deficits in Models of Alzheimer’s Disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; Sproul, A.A.; García, E.; Antón, L.C.; Noggle, S.; Arancio, O.; Avila, J.; García-Escudero, V. Mitophagy Failure in Fibroblasts and IPSC-Derived Neurons of Alzheimer’s Disease-Associated Presenilin 1 Mutation. Front. Mol. Neurosci. 2017, 10, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojakovic, A.; Trushin, S.; Sheu, A.; Khalili, L.; Chang, S.-Y.; Li, X.; Christensen, T.; Salisbury, J.L.; Geroux, R.E.; Gateno, B.; et al. Partial Inhibition of Mitochondrial Complex I Ameliorates Alzheimer’s Disease Pathology and Cognition in APP/PS1 Female Mice. Commun. Biol. 2021, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Stojakovic, A.; Chang, S.-Y.; Nesbitt, J.; Pichurin, N.P.; Ostroot, M.A.; Aikawa, T.; Kanekiyo, T.; Trushina, E. Partial Inhibition of Mitochondrial Complex I Reduces Tau Pathology and Improves Energy Homeostasis and Synaptic Function in 3xTg-AD Mice. J. Alzheimers Dis. 2021, 79, 335–353. [Google Scholar] [CrossRef]
- Selkoe, D.J. Early Network Dysfunction in Alzheimer’s Disease. Science 2019, 365, 540–541. [Google Scholar] [CrossRef]
- Ghatak, S.; Dolatabadi, N.; Trudler, D.; Zhang, X.; Wu, Y.; Mohata, M.; Ambasudhan, R.; Talantova, M.; Lipton, S.A. Mechanisms of Hyperexcitability in Alzheimer’s Disease HiPSC-Derived Neurons and Cerebral Organoids vs Isogenic Controls. Elife 2019, 8, e50333. [Google Scholar] [CrossRef]
- Nicholas, C.R.; Chen, J.; Tang, Y.; Southwell, D.G.; Chalmers, N.; Vogt, D.; Arnold, C.M.; Chen, Y.-J.J.; Stanley, E.G.; Elefanty, A.G.; et al. Functional Maturation of HPSC-Derived Forebrain Interneurons Requires an Extended Timeline and Mimics Human Neural Development. Cell Stem Cell 2013, 12, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of Toxic Apolipoprotein E4 Effects in Human IPSC-Derived Neurons Is Ameliorated by a Small-Molecule Structure Corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef]
- García-León, J.A.; Cabrera-Socorro, A.; Eggermont, K.; Swijsen, A.; Terryn, J.; Fazal, R.; Nami, F.; Ordovás, L.; Quiles, A.; Lluis, F.; et al. Generation of a Human Induced Pluripotent Stem Cell-Based Model for Tauopathies Combining Three Microtubule-Associated Protein TAU Mutations Which Displays Several Phenotypes Linked to Neurodegeneration. Alzheimers Dement. 2018, 14, 1261–1280. [Google Scholar] [CrossRef]
- Duan, L.; Bhattacharyya, B.J.; Belmadani, A.; Pan, L.; Miller, R.J.; Kessler, J.A. Stem Cell Derived Basal Forebrain Cholinergic Neurons from Alzheimer’s Disease Patients Are More Susceptible to Cell Death. Mol. Neurodegener. 2014, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.L.; Della Guardia, L.; Shnyder, V.; Ortiz-Virumbrales, M.; Kruglikov, I.; Zhang, B.; Schadt, E.E.; Tanzi, R.E.; Noggle, S.; Buettner, C.; et al. IPSC-Derived Familial Alzheimer’s PSEN2 N141I Cholinergic Neurons Exhibit Mutation-Dependent Molecular Pathology Corrected by Insulin Signaling. Mol. Neurodegener. 2018, 13, 33. [Google Scholar] [CrossRef]
- Klapper, S.D.; Garg, P.; Dagar, S.; Lenk, K.; Gottmann, K.; Nieweg, K. Astrocyte Lineage Cells Are Essential for Functional Neuronal Differentiation and Synapse Maturation in Human IPSC-Derived Neural Networks. Glia 2019, 67, 1893–1909. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-Associated Astrocytes in Alzheimer’s Disease and Aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Olabarria, M.; Noristani, H.N.; Yeh, C.-Y.; Rodriguez, J.J. Astrocytes in Alzheimer’s Disease. Neurotherapeutics 2010, 7, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune Attack: The Role of Inflammation in Alzheimer Disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Stipursky, J.; Gomes, F.C.A. TGF-Β1/SMAD Signaling Induces Astrocyte Fate Commitmentin Vitro: Implications for Radial Glia Development. Glia 2007, 55, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhang, S.-C. Neural Subtype Specification from Human Pluripotent Stem Cells. Cell Stem Cell 2016, 19, 573–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perriot, S.; Mathias, A.; Perriard, G.; Canales, M.; Jonkmans, N.; Merienne, N.; Meunier, C.; El Kassar, L.; Perrier, A.L.; Laplaud, D.-A.; et al. Human Induced Pluripotent Stem Cell-Derived Astrocytes Are Differentially Activated by Multiple Sclerosis-Associated Cytokines. Stem Cell Rep. 2018, 11, 1199–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Gubert Olivé, M.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant IPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Leon, J.A.; Caceres-Palomo, L.; Sanchez-Mejias, E.; Mejias-Ortega, M.; Nuñez-Diaz, C.; Fernandez-Valenzuela, J.J.; Sanchez-Varo, R.; Davila, J.C.; Vitorica, J.; Gutierrez, A. Human Pluripotent Stem Cell-Derived Neural Cells as a Relevant Platform for Drug Screening in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6867. [Google Scholar] [CrossRef]
- Wilkins, J.M.; Trushina, E. Application of Metabolomics in Alzheimer’s Disease. Front. Neurol. 2018, 8, 719. [Google Scholar] [CrossRef] [Green Version]
- Konttinen, H.; Gureviciene, I.; Oksanen, M.; Grubman, A.; Loppi, S.; Huuskonen, M.T.; Korhonen, P.; Lampinen, R.; Keuters, M.; Belaya, I.; et al. PPARβ/δ-Agonist GW0742 Ameliorates Dysfunction in Fatty Acid Oxidation in PSEN1ΔE9 Astrocytes. Glia 2019, 67, 146–159. [Google Scholar] [CrossRef] [Green Version]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant IPSC-Derived Human Astrocytes in Alzheimer’s Disease. Cell Death Dis. 2017, 8, e2696. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Davis, M.D.; Martens, Y.A.; Shinohara, M.; Graff-Radford, N.R.; Younkin, S.G.; Wszolek, Z.K.; Kanekiyo, T.; Bu, G. APOE Ε4/Ε4 Diminishes Neurotrophic Function of Human IPSC-Derived Astrocytes. Hum. Mol. Genet. 2017, 26, 2690–2700. [Google Scholar] [CrossRef]
- Ryu, W.-I.; Bormann, M.K.; Shen, M.; Kim, D.; Forester, B.; Park, Y.; So, J.; Seo, H.; Sonntag, K.-C.; Cohen, B.M. Brain Cells Derived from Alzheimer’s Disease Patients Have Multiple Specific Innate Abnormalities in Energy Metabolism. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
- Lee, S.-I.; Jeong, W.; Lim, H.; Cho, S.; Lee, H.; Jang, Y.; Cho, J.; Bae, S.; Lin, Y.-T.; Tsai, L.-H.; et al. APOE4-Carrying Human Astrocytes Oversupply Cholesterol to Promote Neuronal Lipid Raft Expansion and Aβ Generation. Stem Cell Rep. 2021, 16, 2128–2137. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s Disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Mizee, M.R.; Miedema, S.S.M.; van der Poel, M.; Adelia; Schuurman, K.G.; van Strien, M.E.; Melief, J.; Smolders, J.; Hendrickx, D.A.; Heutinck, K.M.; et al. Isolation of Primary Microglia from the Human Post-Mortem Brain: Effects of Ante- and Post-Mortem Variables. Acta Neuropathol. Commun. 2017, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Melief, J.; Sneeboer, M.A.M.; Litjens, M.; Ormel, P.R.; Palmen, S.J.M.C.; Huitinga, I.; Kahn, R.S.; Hol, E.M.; de Witte, L.D. Characterizing Primary Human Microglia: A Comparative Study with Myeloid Subsets and Culture Models. Glia 2016, 64, 1857–1868. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-Resident Macrophages Originate from Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.-H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient Derivation of Microglia-like Cells from Human Pluripotent Stem Cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Jiang, P.; Xue, H.; Peterson, S.E.; Tran, H.T.; McCann, A.E.; Parast, M.M.; Li, S.; Pleasure, D.E.; Laurent, L.C.; et al. Role of Astroglia in Down’s Syndrome Revealed by Patient-Derived Human-Induced Pluripotent Stem Cells. Nat. Commun. 2014, 5, 4430. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-T.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human IPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef] [Green Version]
- Guttikonda, S.R.; Sikkema, L.; Tchieu, J.; Saurat, N.; Walsh, R.M.; Harschnitz, O.; Ciceri, G.; Sneeboer, M.; Mazutis, L.; Setty, M.; et al. Fully Defined Human Pluripotent Stem Cell-Derived Microglia and Tri-Culture System Model C3 Production in Alzheimer’s Disease. Nat. Neurosci. 2021, 24, 343–354. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, X.-J.; Renier, N.; Wu, Z.; Atkin, T.; Sun, Z.; Ozair, M.Z.; Tchieu, J.; Zimmer, B.; Fattahi, F.; et al. Combined Small-Molecule Inhibition Accelerates the Derivation of Functional Cortical Neurons from Human Pluripotent Stem Cells. Nat. Biotechnol. 2017, 35, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, H.; Cabral-da-Silva, M.e.C.; Ohtonen, S.; Wojciechowski, S.; Shakirzyanova, A.; Caligola, S.; Giugno, R.; Ishchenko, Y.; Hernández, D.; Fazaludeen, M.F.; et al. PSEN1ΔE9, APPswe, and APOE4 Confer Disparate Phenotypes in Human IPSC-Derived Microglia. Stem Cell Rep. 2019, 13, 669–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassil, R.; Shields, K.; Granger, K.; Zein, I.; Ng, S.; Chih, B. Improved Modeling of Human AD with an Automated Culturing Platform for IPSC Neurons, Astrocytes and Microglia. Nat. Commun. 2021, 12, 5220. [Google Scholar] [CrossRef] [PubMed]
- Le Pichon, C.E.; Meilandt, W.J.; Dominguez, S.; Solanoy, H.; Lin, H.; Ngu, H.; Gogineni, A.; Sengupta Ghosh, A.; Jiang, Z.; Lee, S.-H.; et al. Loss of Dual Leucine Zipper Kinase Signaling Is Protective in Animal Models of Neurodegenerative Disease. Sci. Transl. Med. 2017, 9, eaag0394. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.-Z.; Mandelkow, E.-M.; et al. Indirubins Inhibit Glycogen Synthase Kinase-3β and CDK5/P25, Two Protein Kinases Involved in Abnormal Tau Phosphorylation in Alzheimer’s Disease. J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, A.C.; Salazar, S.V.; Haas, L.T.; Yang, J.; Kostylev, M.A.; Jeng, A.T.; Robinson, S.A.; Gunther, E.C.; van Dyck, C.H.; Nygaard, H.B.; et al. Fyn Inhibition Rescues Established Memory and Synapse Loss in Alzheimer Mice. Ann. Neurol. 2015, 77, 953–971. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.X.; Lertvorachon, J.; Hou, S.T.; Konishi, Y.; Webster, J.; Mealing, G.; Brunette, E.; Tauskela, J.; Preston, E. Chlortetracycline and Demeclocycline Inhibit Calpains and Protect Mouse Neurons against Glutamate Toxicity and Cerebral Ischemia. J. Biol. Chem. 2005, 280, 33811–33818. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y. Luteolin as a Potential Preventive and Therapeutic Candidate for Alzheimer’s Disease. Exp. Gerontol. 2017, 95, 39–43. [Google Scholar] [CrossRef]
- Yu, T.-X.; Zhang, P.; Guan, Y.; Wang, M.; Zhen, M.-Q. Protective Effects of Luteolin against Cognitive Impairment Induced by Infusion of Aβ Peptide in Rats. Int. J. Clin. Exp. Pathol. 2015, 8, 6740–6747. [Google Scholar]
- Prior, M.; Dargusch, R.; Ehren, J.L.; Chiruta, C.; Schubert, D. The Neurotrophic Compound J147 Reverses Cognitive Impairment in Aged Alzheimer’s Disease Mice. Alzheimers Res. Ther. 2013, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Huang, G.; Sun, X.; Guo, Z.; Zhang, J.H. Tozasertib Attenuates Neuronal Apoptosis via DLK/JIP3/MA2K7/JNK Pathway in Early Brain Injury after SAH in Rats. Neuropharmacology 2016, 108, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Ndubaku, C.O.; Crawford, T.D.; Chen, H.; Boggs, J.W.; Drobnick, J.; Harris, S.F.; Jesudason, R.; McNamara, E.; Nonomiya, J.; Sambrone, A.; et al. Structure-Based Design of GNE-495, a Potent and Selective MAP4K4 Inhibitor with Efficacy in Retinal Angiogenesis. ACS Med. Chem. Lett. 2015, 6, 913–918. [Google Scholar] [CrossRef] [Green Version]
- Logan, S.; Arzua, T.; Canfield, S.G.; Seminary, E.R.; Sison, S.L.; Ebert, A.D.; Bai, X. Studying Human Neurological Disorders Using Induced Pluripotent Stem Cells: From 2D Monolayer to 3D Organoid and Blood Brain Barrier Models. Compr. Physiol. 2019, 9, 565–611. [Google Scholar] [CrossRef]
- Centeno, E.G.Z.; Cimarosti, H.; Bithell, A. 2D versus 3D Human Induced Pluripotent Stem Cell-Derived Cultures for Neurodegenerative Disease Modelling. Mol. Neurodegener. 2018, 13, 27. [Google Scholar] [CrossRef]
- Liu, C.; Oikonomopoulos, A.; Sayed, N.; Wu, J.C. Modeling Human Diseases with Induced Pluripotent Stem Cells: From 2D to 3D and Beyond. Development 2018, 145, dev156166. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Pekkanen-Mattila, M.; Shahsavani, M.; Falk, A.; Teixeira, A.I.; Herland, A. A 3D Alzheimer’s Disease Culture Model and the Induction of P21-Activated Kinase Mediated Sensing in IPSC Derived Neurons. Biomaterials 2014, 35, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Martín-Maestro, P.; Sproul, A.; Martinez, H.; Paquet, D.; Gerges, M.; Noggle, S.; Starkov, A.A. Autophagy Induction by Bexarotene Promotes Mitophagy in Presenilin 1 Familial Alzheimer’s Disease IPSC-Derived Neural Stem Cells. Mol. Neurobiol. 2019, 56, 8220–8236. [Google Scholar] [CrossRef]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed Differentiation of Human Pluripotent Stem Cells to Cerebral Cortex Neurons and Neural Networks. Nat. Protoc. 2012, 7, 1836–1846. [Google Scholar] [CrossRef]
- Landry, J.; Bernier, D.; Ouellet, C.; Goyette, R.; Marceau, N. Spheroidal Aggregate Culture of Rat Liver Cells: Histotypic Reorganization, Biomatrix Deposition, and Maintenance of Functional Activities. J. Cell Biol. 1985, 101, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, G.A.; Westmoreland, C.; George, E. Effects of Medium Composition on the Morphology and Function of Rat Hepatocytes Cultured as Spheroids and Monolayers. Vitr. Cell. Dev. Biol. -Anim. 2001, 37, 656. [Google Scholar] [CrossRef]
- Choi, Y.J.; Park, J.; Lee, S.-H. Size-Controllable Networked Neurospheres as a 3D Neuronal Tissue Model for Alzheimer’s Disease Studies. Biomaterials 2013, 34, 2938–2946. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Song, H.; Ming, G. Brain Organoids: Advances, Applications and Challenges. Development 2019, 146, dev166074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, M.; Techy, G.B.; Saroufeem, R.; Yazan, O.; Narayan, K.S.; Goodwin, T.J.; Spaulding, G.F. Three-Dimensional Growth Patterns of Various Human Tumor Cell Lines in Simulated Microgravity of a NASA Bioreactor. Vitr. Cell. Dev. Biol. -Anim. 1997, 33, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Khaoustov, V.I.; Darlington, G.J.; Soriano, H.E.; Krishnan, B.; Risin, D.; Pellis, N.R.; Yoffe, B. Induction of Three-Dimensional Assembly of Human Liver Cells by Simulated Microgravity. Vitr. Cell. Dev. Biol. -Anim. 1999, 35, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Knoblich, J.A.; Lutolf, M.P.; Martinez-Arias, A. The Hope and the Hype of Organoid Research. Development 2017, 144, 938–941. [Google Scholar] [CrossRef] [Green Version]
- Medda, X.; Mertens, L.; Versweyveld, S.; Diels, A.; Barnham, L.; Bretteville, A.; Buist, A.; Verheyen, A.; Royaux, I.; Ebneth, A.; et al. Development of a Scalable, High-Throughput-Compatible Assay to Detect Tau Aggregates Using IPSC-Derived Cortical Neurons Maintained in a Three-Dimensional Culture Format. J. Biomol. Screen. 2016, 21, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Mirra, S.S.; Murrell, J.R.; Gearing, M.; Spillantini, M.G.; Goedert, M.; Crowther, R.A.; Levey, A.I.; Jones, R.; Green, J.; Shoffner, J.M.; et al. Tau Pathology in a Family with Dementia and a P301L Mutation in Tau. J. Neuropathol. Exp. Neurol. 1999, 58, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A Three-Dimensional Human Neural Cell Culture Model of Alzheimer’s Disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Kwak, S.S.; Washicosky, K.J.; Brand, E.; von Maydell, D.; Aronson, J.; Kim, S.; Capen, D.E.; Cetinbas, M.; Sadreyev, R.; Ning, S.; et al. Amyloid-Β42/40 Ratio Drives Tau Pathology in 3D Human Neural Cell Culture Models of Alzheimer’s Disease. Nat. Commun. 2020, 11, 1377. [Google Scholar] [CrossRef]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-Organization of Axial Polarity, inside-out Layer Pattern, and Species-Specific Progenitor Dynamics in Human ES Cell-Derived Neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [Green Version]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.-T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.-H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Sapiéns, M.A.; Reza-Zaldívar, E.E.; Cevallos, R.R.; Márquez-Aguirre, A.L.; Gazarian, K.; Canales-Aguirre, A.A. A Three-Dimensional Alzheimer’s Disease Cell Culture Model Using IPSC-Derived Neurons Carrying A246E Mutation in PSEN1. Front. Cell. Neurosci. 2020, 14, 151. [Google Scholar] [CrossRef]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling Amyloid Beta and Tau Pathology in Human Cerebral Organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef]
- Nakamura, M.; Shiozawa, S.; Tsuboi, D.; Amano, M.; Watanabe, H.; Maeda, S.; Kimura, T.; Yoshimatsu, S.; Kisa, F.; Karch, C.M.; et al. Pathological Progression Induced by the Frontotemporal Dementia-Associated R406W Tau Mutation in Patient-Derived IPSCs. Stem Cell Rep. 2019, 13, 684–699. [Google Scholar] [CrossRef] [Green Version]
- Arber, C.; Toombs, J.; Lovejoy, C.; Ryan, N.S.; Paterson, R.W.; Willumsen, N.; Gkanatsiou, E.; Portelius, E.; Blennow, K.; Heslegrave, A.; et al. Familial Alzheimer’s Disease Patient-Derived Neurons Reveal Distinct Mutation-Specific Effects on Amyloid Beta. Mol. Psychiatry 2020, 25, 2919–2931. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-X.; Tan, L.; Yu, J.-T. Axonal Transport Defects in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 1309–1321. [Google Scholar] [CrossRef]
- Ygland, E.; van Westen, D.; Englund, E.; Rademakers, R.; Wszolek, Z.K.; Nilsson, K.; Nilsson, C.; Landqvist Waldö, M.; Alafuzoff, I.; Hansson, O.; et al. Slowly Progressive Dementia Caused by MAPT R406W Mutations: Longitudinal Report on a New Kindred and Systematic Review. Alzheimers Res. Ther. 2018, 10, 2. [Google Scholar] [CrossRef]
- Meyer, K.; Feldman, H.M.; Lu, T.; Drake, D.; Lim, E.T.; Ling, K.-H.; Bishop, N.A.; Pan, Y.; Seo, J.; Lin, Y.-T.; et al. REST and Neural Gene Network Dysregulation in IPSC Models of Alzheimer’s Disease. Cell Rep. 2019, 26, 1112–1127.e9. [Google Scholar] [CrossRef] [Green Version]
- Ashton, N.J.; Hye, A.; Leckey, C.A.; Jones, A.R.; Gardner, A.; Elliott, C.; Wetherell, J.L.; Lenze, E.J.; Killick, R.; Marchant, N.L. Plasma REST: A Novel Candidate Biomarker of Alzheimer’s Disease Is Modified by Psychological Intervention in an at-Risk Population. Transl. Psychiatry 2017, 7, e1148. [Google Scholar] [CrossRef] [Green Version]
- Pomeshchik, Y.; Klementieva, O.; Gil, J.; Martinsson, I.; Hansen, M.G.; de Vries, T.; Sancho-Balsells, A.; Russ, K.; Savchenko, E.; Collin, A.; et al. Human IPSC-Derived Hippocampal Spheroids: An Innovative Tool for Stratifying Alzheimer Disease Patient-Specific Cellular Phenotypes and Developing Therapies. Stem Cell Rep. 2020, 15, 256–273. [Google Scholar] [CrossRef]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.-Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional Cortical Neurons and Astrocytes from Human Pluripotent Stem Cells in 3D Culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.J.; Elahi, L.S.; Pașca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of Human Cortical Organoid Generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef]
- Pas, S.P. The Rise of Three-Dimensional Human Brain Cultures. Nature 2018, 553, 437–445. [Google Scholar] [CrossRef]
- Chen, H.I.; Song, H.; Ming, G. li Applications of Human Brain Organoids to Clinical Problems. Dev. Dyn. 2019, 248, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Giandomenico, S.L.; Sutcliffe, M.; Lancaster, M.A. Generation and Long-Term Culture of Advanced Cerebral Organoids for Studying Later Stages of Neural Development. Nat. Protoc. 2021, 16, 579–602. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of Cerebral Organoids from Human Pluripotent Stem Cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Donato, R.; Miljan, E.A.; Hines, S.J.; Aouabdi, S.; Pollock, K.; Patel, S.; Edwards, F.A.; Sinden, J.D. Differential Development of Neuronal Physiological Responsiveness in Two Human Neural Stem Cell Lines. BMC Neurosci. 2007, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Choi, S.H.; D’Avanzo, C.; Hebisch, M.; Sliwinski, C.; Bylykbashi, E.; Washicosky, K.J.; Klee, J.B.; Brüstle, O.; Tanzi, R.E.; et al. A 3D Human Neural Cell Culture System for Modeling Alzheimer’s Disease. Nat. Protoc. 2015, 10, 985–1006. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, B.K.; Jeong, G.S.; Hyun, J.K.; Lee, C.J.; Lee, S.-H. Three-Dimensional Brain-on-a-Chip with an Interstitial Level of Flow and Its Application as an in Vitro Model of Alzheimer’s Disease. Lab Chip 2015, 15, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, M.; D’Avanzo, C.; Tanzi, R.E.; Kim, D.Y.; Irimia, D. Human Neurospheroid Arrays for In Vitro Studies of Alzheimer’s Disease. Sci. Rep. 2018, 8, 2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, A.R.; Laslett, A.; O’Brien, C.M.; Cameron, N.R. Scaffolds for 3D in Vitro Culture of Neural Lineage Cells. Acta Biomater. 2017, 54, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorfi, M.; D’Avanzo, C.; Kim, D.Y.; Irimia, D. Three-Dimensional Models of the Human Brain Development and Diseases. Adv. Healthc. Mater. 2018, 7, 1700723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, D.M.; Chwalek, K.; Moore, Y.E.; Kelley, M.R.; Abbott, R.D.; Moss, S.; Kaplan, D.L. Expandable and Rapidly Differentiating Human Induced Neural Stem Cell Lines for Multiple Tissue Engineering Applications. Stem Cell Rep. 2016, 7, 557–570. [Google Scholar] [CrossRef] [Green Version]
- Cairns, D.M.; Rouleau, N.; Parker, R.N.; Walsh, K.G.; Gehrke, L.; Kaplan, D.L. A 3D Human Brain–like Tissue Model of Herpes-Induced Alzheimer’s Disease. Sci. Adv. 2020, 6, eaay8828. [Google Scholar] [CrossRef]
- Chwalek, K.; Tang-Schomer, M.D.; Omenetto, F.G.; Kaplan, D.L. In Vitro Bioengineered Model of Cortical Brain Tissue. Nature Protocols 2015, 10, 1362–1373. [Google Scholar] [CrossRef]
- Rizzo, R. Controversial Role of Herpesviruses in Alzheimer’s Disease. PLoS Pathog. 2020, 16, e1008575. [Google Scholar] [CrossRef]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D Human Triculture System Modeling Neurodegeneration and Neuroinflammation in Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Hasan, M.F.; Ghiasvand, S.; Wang, H.; Miwa, J.M.; Berdichevsky, Y. Neural Layer Self-Assembly in Geometrically Confined Rat and Human 3D Cultures. Biofabrication 2019, 11, 045011. [Google Scholar] [CrossRef]
- Hasan, M.F.; Berdichevsky, Y. Neuron and Astrocyte Aggregation and Sorting in Three-Dimensional Neuronal Constructs. Commun. Biol. 2021, 4, 587. [Google Scholar] [CrossRef]
- Hasan, M.F. Mdfayadhasan/Cellular-Aggregation-Model: Cellular Aggregation Model (Version V1.0). Zenodo. GitHub 2021, 4, 587. [Google Scholar] [CrossRef]
- Hasan, M.F.; Berdichevsky, Y. Designing and Manipulating Interconnectivity between Cortical and Striatal 3D Cultures. Proc. Int. IEEE/EMBS Conf. Neural Eng. NER 2019, 2019, 163–166. [Google Scholar]
- Ming, Y.; Hasan, M.F.; Tatic-Lucic, S.; Berdichevsky, Y. Micro Three-Dimensional Neuronal Cultures Generate Developing Cortex-Like Activity Patterns. Front. Neurosci. 2020, 14, 563905. [Google Scholar] [CrossRef]
- Espuny-Camacho, I.; Michelsen, K.A.; Gall, D.; Linaro, D.; Hasche, A.; Bonnefont, J.; Bali, C.; Orduz, D.; Bilheu, A.; Herpoel, A.; et al. Pyramidal Neurons Derived from Human Pluripotent Stem Cells Integrate Efficiently into Mouse Brain Circuits In Vivo. Neuron 2013, 77, 440–456. [Google Scholar] [CrossRef] [Green Version]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. IPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [Green Version]
- Hasselmann, J.; Coburn, M.A.; England, W.; Figueroa Velez, D.X.; Kiani Shabestari, S.; Tu, C.H.; McQuade, A.; Kolahdouzan, M.; Echeverria, K.; Claes, C.; et al. Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron 2019, 103, 1016–1033.e10. [Google Scholar] [CrossRef] [Green Version]
- Gleichmann, M.; Mattson, M.P. Alzheimer’s Disease and Neuronal Network Activity. NeuroMol. Med. 2010, 12, 44–47. [Google Scholar] [CrossRef] [Green Version]
- de Leeuw, S.M.; Davaz, S.; Wanner, D.; Milleret, V.; Ehrbar, M.; Gietl, A.; Tackenberg, C. Increased Maturation of IPSC-Derived Neurons in a Hydrogel-Based 3D Culture. J. Neurosci. Methods 2021, 360, 109254. [Google Scholar] [CrossRef]
- Dolega, M.E.; Abeille, F.; Picollet-D’hahan, N.; Gidrol, X. Controlled 3D Culture in Matrigel Microbeads to Analyze Clonal Acinar Development. Biomaterials 2015, 52, 347–357. [Google Scholar] [CrossRef]
- SODUNKE, T.; TURNER, K.; CALDWELL, S.; MCBRIDE, K.; REGINATO, M.; NOH, H. Micropatterns of Matrigel for Three-Dimensional Epithelial Cultures. Biomaterials 2007, 28, 4006–4016. [Google Scholar] [CrossRef]
- Dattola, E.; Parrotta, E.I.; Scalise, S.; Perozziello, G.; Limongi, T.; Candeloro, P.; Coluccio, M.L.; Maletta, C.; Bruno, L.; De Angelis, M.T.; et al. Development of 3D PVA Scaffolds for Cardiac Tissue Engineering and Cell Screening Applications. RSC Adv. 2019, 9, 4246–4257. [Google Scholar] [CrossRef] [Green Version]
- Puig-Sanvicens, V.A.C.; Semino, C.E.; zur Nieden, N.I. Cardiac Differentiation Potential of Human Induced Pluripotent Stem Cells in a 3D Self-Assembling Peptide Scaffold. Differentiation 2015, 90, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Tomaskovic-Crook, E.; Wallace, G.G.; Crook, J.M. 3D Bioprinting Human Induced Pluripotent Stem Cell Constructs for In Situ Cell Proliferation and Successive Multilineage Differentiation. Adv. Healthc. Mater. 2017, 6, 1700175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Lim, C.H.J.; Low, M.J.; Tham, N.; Murukeshan, V.M.; Kim, Y.-J. Lasers in Additive Manufacturing: A Review. Int. J. Precis. Eng. Manuf. Green Technol. 2017, 4, 307–322. [Google Scholar] [CrossRef]
- Koroleva, A.; Deiwick, A.; El-Tamer, A.; Koch, L.; Shi, Y.; Estévez-Priego, E.; Ludl, A.-A.; Soriano, J.; Guseva, D.; Ponimaskin, E.; et al. In Vitro Development of Human IPSC-Derived Functional Neuronal Networks on Laser-Fabricated 3D Scaffolds. ACS Appl. Mater. Interfaces 2021, 13, 7839–7853. [Google Scholar] [CrossRef]
- Sibbett, W.; Lagatsky, A.A.; Brown, C.T.A. The Development and Application of Femtosecond Laser Systems. Opt. Express 2012, 20, 6989. [Google Scholar] [CrossRef]
- Jakobsson, A.; Ottosson, M.; Zalis, M.C.; O’Carroll, D.; Johansson, U.E.; Johansson, F. Three-Dimensional Functional Human Neuronal Networks in Uncompressed Low-Density Electrospun Fiber Scaffolds. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1563–1573. [Google Scholar] [CrossRef]
- Askari, M.; Afzali Naniz, M.; Kouhi, M.; Saberi, A.; Zolfagharian, A.; Bodaghi, M. Recent Progress in Extrusion 3D Bioprinting of Hydrogel Biomaterials for Tissue Regeneration: A Comprehensive Review with Focus on Advanced Fabrication Techniques. Biomater. Sci. 2021, 9, 535–573. [Google Scholar] [CrossRef]
- Soliman, E.; Bianchi, F.; Sleigh, J.N.; George, J.H.; Cader, M.Z.; Cui, Z.; Ye, H. Aligned Electrospun Fibers for Neural Patterning. Biotechnol. Lett. 2018, 40, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Rengarajan, V.; Kjar, A.; Huang, Y. A Matrigel-Free Method to Generate Matured Human Cerebral Organoids Using 3D-Printed Microwell Arrays. Bioact. Mater. 2021, 6, 1130–1139. [Google Scholar] [CrossRef]
- Frega, M.; Tedesco, M.; Massobrio, P.; Pesce, M.; Martinoia, S. Network Dynamics of 3D Engineered Neuronal Cultures: A New Experimental Model for in-Vitro Electrophysiology. Sci. Rep. 2015, 4, 5489. [Google Scholar] [CrossRef]
- Lozano, R.; Stevens, L.; Thompson, B.C.; Gilmore, K.J.; Gorkin, R.; Stewart, E.M.; in het Panhuis, M.; Romero-Ortega, M.; Wallace, G.G. 3D Printing of Layered Brain-like Structures Using Peptide Modified Gellan Gum Substrates. Biomaterials 2015, 67, 264–273. [Google Scholar] [CrossRef]
- Frimat, J.-P.; Xie, S.; Bastiaens, A.; Schurink, B.; Wolbers, F.; den Toonder, J.; Luttge, R. Advances in 3D Neuronal Cell Culture. J. Vac. Sci. Technol. B Nanotechnol. Microelectron. Mater. Processing Meas. Phenom. 2015, 33, 06F902. [Google Scholar] [CrossRef] [Green Version]

| Observed Key AD Phenotype | Differentiation Method | Cell Type | AD Source | Experimental Timeline, Days | Reference |
|---|---|---|---|---|---|
| ↑ Aβ, pTau levels ↑ GSK3β activation Neuronal endosomal accumulation | Growth factor–guided differentiation of FACS-purified NPCs | Cortical neurons and normal astrocytes | FAD (APP) and LOAD patient fibroblasts | 21 | [35] |
| ↑ Aβ42/Aβ40 ratio Intracellular Aβ oligomers ER and cell stress | Small molecule–guided differentiation of EB | Cortical neurons and normal astrocytes | FAD (APP) and LOAD patient fibroblasts | 180 | [36] a |
| β-CTF but not Aβ-mediated endosomal abnormality ↓ Endocytosis and transcytosis of APP and lipoproteins | FACS purification of NPCs and neuronal differentiation | Cortical neurons | Gene-edited (PSEN1 ∆E9, APP V717F, or APP SWE) HiPSCs | >21 | [45] b |
| β-CTF but not Aβ-mediated endosomal abnormality | Dual SMAD inhibition and neuronal maturation | Cortical neurons | Multiple FAD-related gene knock-in HiPSCs | 80 | [40] b |
| ↑ Aβ, pTau levels ↑ GSK3β activation ↑ Sensitivity to Aβ | Differentiated from NPCs, obtained by dual SMAD inhibition of HiPSCs | Cortical neurons and glia | FAD (PSEN1) and LOAD patient fibroblasts | 70 | [37] |
| Aberrant cholesterol metabolism–correlated pTau accumulation | Neurons: dual SMAD inhibition and FACS Astrocytes: extended culture of neutrospheres | Neurons and astrocytes | FAD and LOAD patient fibroblasts and gene-edited lines | >35 | [44] |
| ↓ Resistance to H2O2 injury | Serum-free induction of NSCs from HiPSCs and neuronal differentiation | Cortical neurons | HiPSCs from LOAD patients | 35 | [53] |
| ↑ Aβ and pTau levels GABAergic neuron degeneration | Differentiated from NPCs, obtained by dual SMAD inhibition of HiPSCs | Cortical neurons and glia | Fibroblasts of LOAD patients with apoE4 mutation | >56 | [61] c |
| Lysosomal dysfunction–mediated impaired mitophagy | Dual SMAD inhibition and neuronal maturation | Cortical neurons | FAD patients with PSEN1 A246E mutation–derived fibroblasts | >40 | [55] |
| Several mitochondrial respiratory chain defects Aberrant mitophagy | PSC Neural Induction Medium (Gibco) | NSCs | PSEN1 M146L knock-in HiPSCs | >7 | [107] d |
| Impaired mitophagy | Differentiated from NPCs, obtained by dual SMAD inhibition of HiPSCs [108] | Cortical neurons and glia | HiPSCs from LOAD patients with apoE4 mutation | 28 | [54] |
| ↑ Aβ42/Aβ40 ratio ↑ Total Aβ level ↑ Frequency of spontaneous action potentials and evoked activity ↑ Action potential height ↓ Action potential half-width ↓ Neuritic processes length Altered neuronal sodium channel activity ↓ Inhibitory GABA- and PV-positive neurons | Small molecule cocktail | Cortical neurons | CRISPR/CAS9 gene–edited PSEN1 and APP HiPSCs | 35 | [59] |
| ↑ Vulnerability to glutamate-mediated cell death | Overexpression of transcription factors in NPCs | Cholinergic neurons | LOAD patient fibroblasts | 14 | [63] |
| ↑ Aβ42/Aβ40 ratio Altered Ca2+ flux | Dual SMAD inhibition with ventralizing agents and maturation in BrainPhys (STEMCELL Technologies Inc.) medium | Cholinergic neurons | FAD with PSEN2 N141I mutation patient–derived HiPSCs | 30 | [64] e |
| ↑ pTau ↑ ERK1/2 phosphoactivation ↑ Extracellular pTau release | Overexpression of transcription factor in HiPSCs | Cortical neurons | HiPSCs from LOAD patients with apoE4 mutation | 38 | [43] |
| Aberrant Aβ or pTau uncorrelated, DNA damage correlated ROS production Altered levels of OXPHOS complexes | Overexpression of transcription factor in HiPSCs | Cortical neurons | LOAD patient fibroblast–derived HiPSCs | 21–23 | [52] |
| ↑ 4R tau, pTau ↑ Tau aggregation ↑ Neuronal activity ↓ Neurite outgrowth Altered GABAergic gene expression Aberrant differentiation Activation of stress pathways Upregulation of neurodegenerative pathways | Dual SMAD inhibition | Cortical neurons | N279K, P301L, and E10 + 16 mutations in HiPSCs from healthy patients | >70 | [62] |
| ↑ Synapse number ↑ Neuronal Aβ42 secretion Impaired astrocytic Aβ uptake and cholesterol accumulation Altered microglia morphologies Reduced microglial Aβ phagocytosis | Neurons: overexpression of transcription factor in HiPSCs Astrocytes: differentiated from HiPSC-derived NPCs Microglia: defined serum-free differentiation from HiPSCs | Neurons, astrocytes, and microglia | HiPSCs from LOAD patients with apoE4 mutation | 28 | [89] |
| Altered astrocytic mitochondrial metabolism ↑ Oxidative stress Disturbed Ca2+ signaling in the astrocytic ER Astrocyte-mediated reduction of neuronal calcium signaling | Differentiated from NPCs, obtained by dual SMAD inhibition of HiPSCs and chemical differentiation | Astrocytes | Early-onset FAD (PSEN1) patient fibroblasts | 210 | [74] |
| Impairment in astrocytic fatty acid oxidation | Differentiated from NPCs, obtained by dual SMAD inhibition of HiPSCs and chemical differentiation | Astrocytes | Early-onset FAD (PSEN1) patient fibroblasts | 210 | [77] f |
| ↓ Morphologic complexity Abnormal localization of key functional astroglial markers Altered nonstimulated release of soluble inflammatory mediators | Chemically defined differentiation method from cortical NPCs | Astrocytes | FAD (PSEN1) and LOAD (apoE4) patient HiPSCs | 30 | [78] |
| Less supportive in neuronal survival and synaptogenesis than apoE3 astrocytes | Differentiated from HiPSC-derived NPCs | Neurons and astrocytes | HiPSCs from LOAD patients with apoE4 mutation | 45 | [79] |
| ↓ Glucose uptake ↓ IGF-1 or insulin responses Altered bioenergetic metabolites and metabolic transcriptomes | Differentiated from HiPSC-derived NPCs | Neurons and astrocytes | LOAD patient fibroblasts and peripheral blood mononucleocytes | 60–90 | [80] |
| ↑ Inflammatory response ↓ Metabolism ↓ Phagocytosis ↓ Migration | Small molecule–directed differentiation of HiPSCs under defined oxygen conditions | Microglia | FAD (PSEN1 and APP) and LOAD (apoE4) patient HiPSCs | >24 | [92] |
| Mutual activation of microglia and astrocytes | Neurons: small molecule–directed dual SMAD inhibition Astrocytes: lentiviral overexpression of transcriptome factor Microglia: defined chemical differentiation | Neurons, astrocytes, and microglia | FAD (APP) patient HiPSCs | 80 | [90] |
| Neuronal synaptic loss, dendrite reduction, axon fragmentation, pTau, Aβ plaque formation, dystrophic neurite around plaque, microglial migration | Aβ oligomer application to triculture with: Neurons: overexpression of transcription factor in HiPSCs Astrocytes: commercially available primary Microglia: defined chemical differentiation | Neurons, astrocytes, and microglia | Neurons: apoE3 or apoE4 Astrocytes and microglia: apoE3 | <30 | [93] g |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, M.F.; Trushina, E. Advances in Recapitulating Alzheimer’s Disease Phenotypes Using Human Induced Pluripotent Stem Cell-Based In Vitro Models. Brain Sci. 2022, 12, 552. https://doi.org/10.3390/brainsci12050552
Hasan MF, Trushina E. Advances in Recapitulating Alzheimer’s Disease Phenotypes Using Human Induced Pluripotent Stem Cell-Based In Vitro Models. Brain Sciences. 2022; 12(5):552. https://doi.org/10.3390/brainsci12050552
Chicago/Turabian StyleHasan, Md Fayad, and Eugenia Trushina. 2022. "Advances in Recapitulating Alzheimer’s Disease Phenotypes Using Human Induced Pluripotent Stem Cell-Based In Vitro Models" Brain Sciences 12, no. 5: 552. https://doi.org/10.3390/brainsci12050552
APA StyleHasan, M. F., & Trushina, E. (2022). Advances in Recapitulating Alzheimer’s Disease Phenotypes Using Human Induced Pluripotent Stem Cell-Based In Vitro Models. Brain Sciences, 12(5), 552. https://doi.org/10.3390/brainsci12050552