
Identification and Validation of Novel Potential Pathogenesis and Biomarkers to Predict the Neurological Outcome after Cardiac Arrest

 , and
, and
Abstract
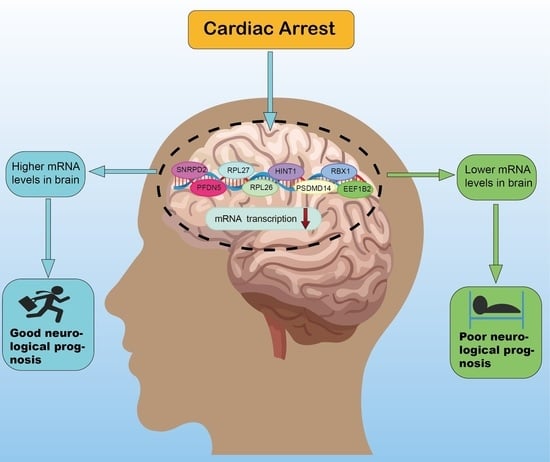
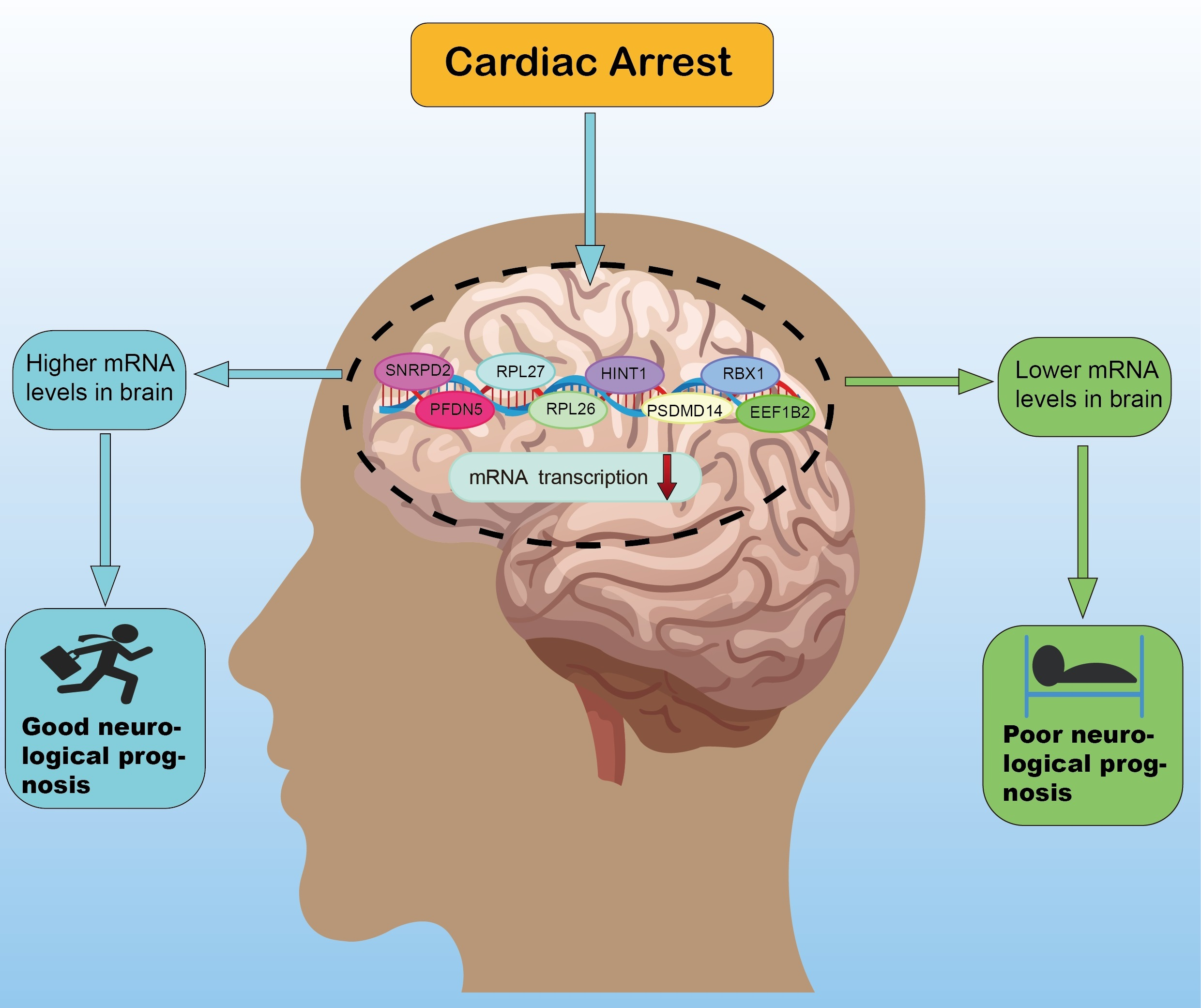
:
1. Introduction
2. Materials and Methods
2.1. Datasets Collection
2.2. Data Normalization and Differential Expression Analysis
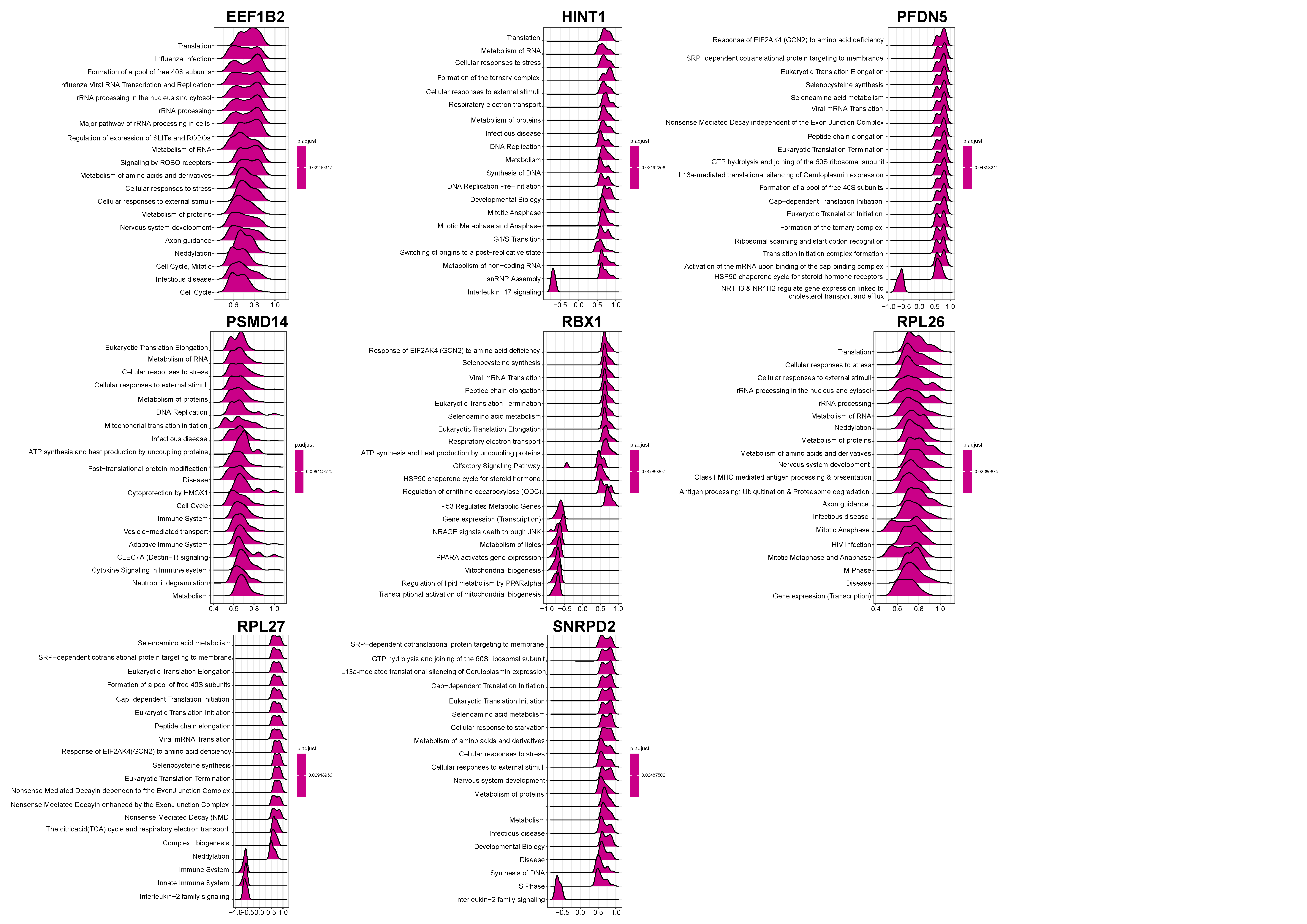
2.3. Functional Enrichment Analysis
2.4. WGCNA Analysis
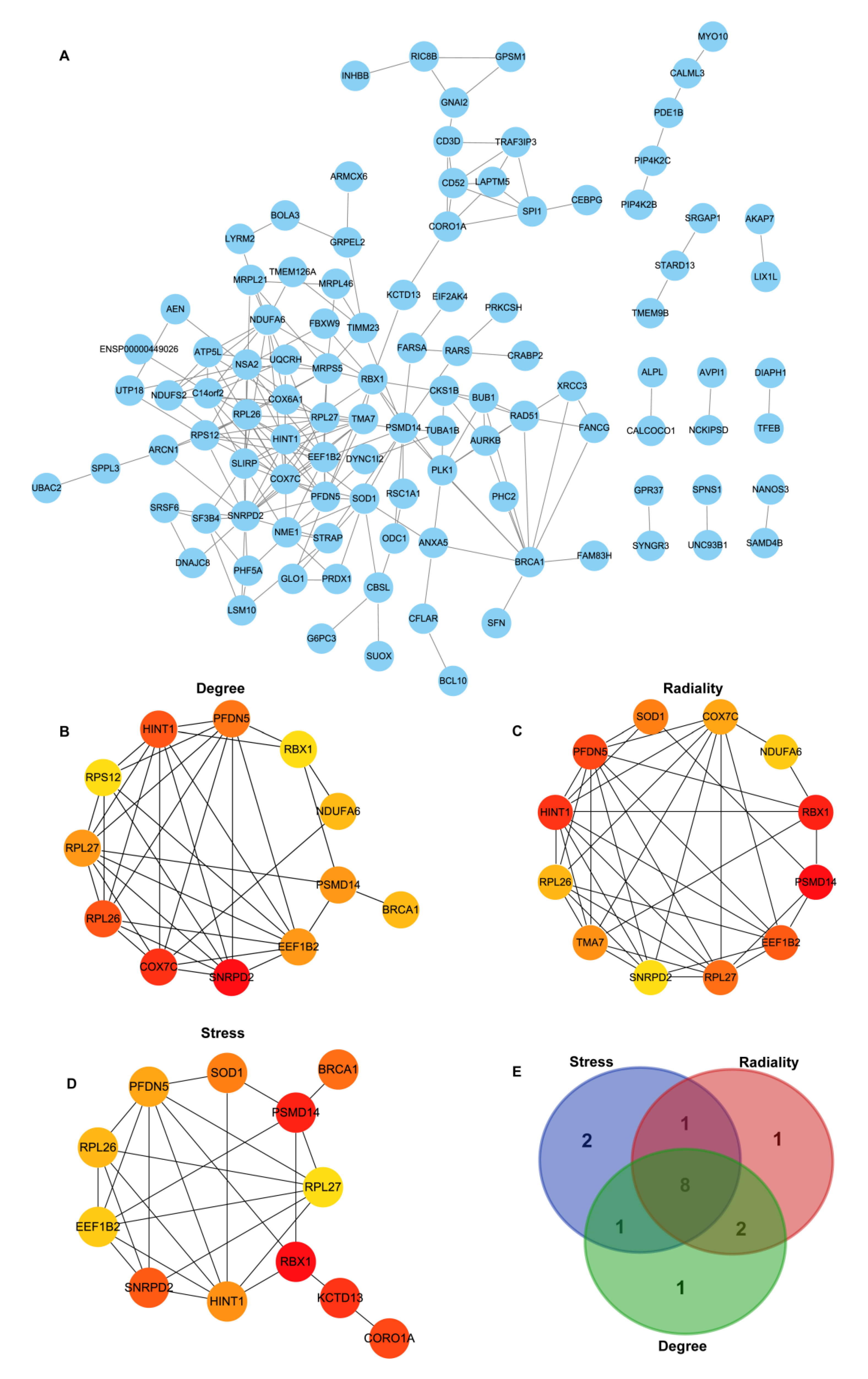
2.5. Protein–Protein Interaction (PPI) Analysis and Hub Genes Recognition
2.6. Immune Cell Infiltration
2.7. Construction of the Animal Model
2.8. TUNEL Staining
2.9. Hematoxylin–Eosin (H&E) Staining and Nissl Staining
2.10. Quantitative Real-Time Reverse Transcription PCR (qRT-PCR)
2.11. Western Blotting Assay
2.12. Statistics
3. Results
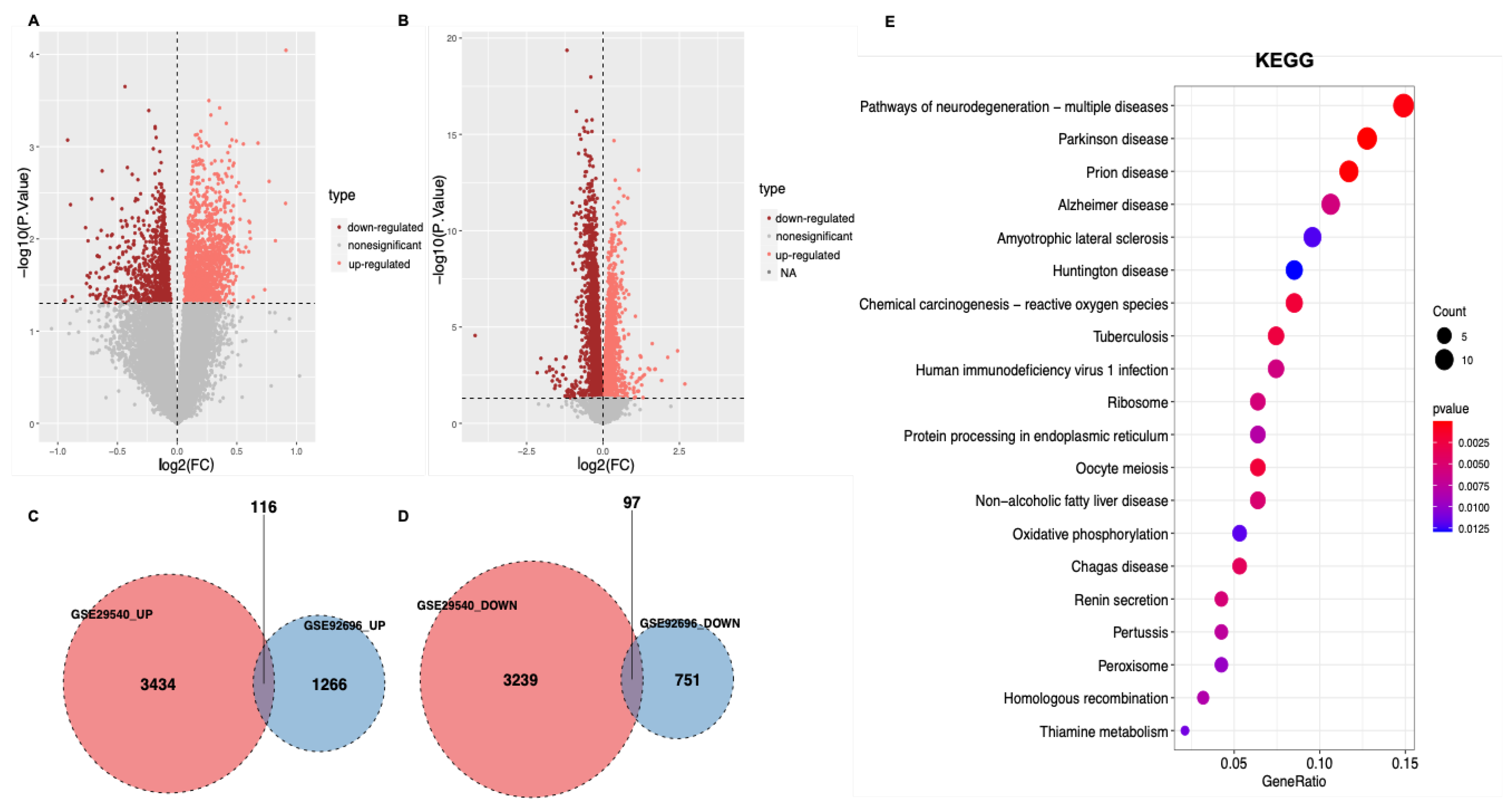
3.1. Identification of DEGs
3.2. Functional Enrichment Analysis
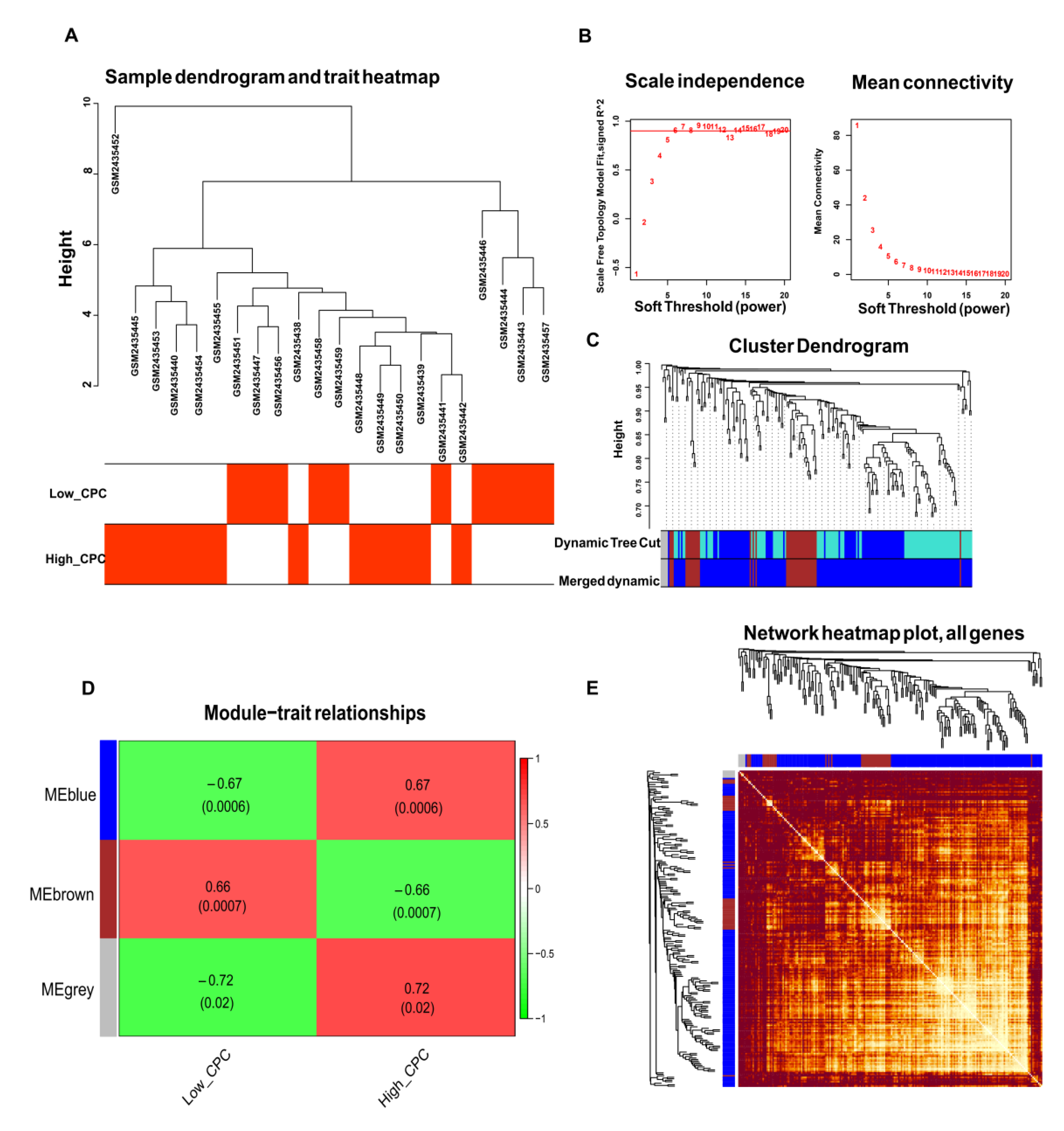
3.3. WGCNA and Identification of Key Modules
3.4. Construction of PPI Network Construction and Identification of Hub Genes
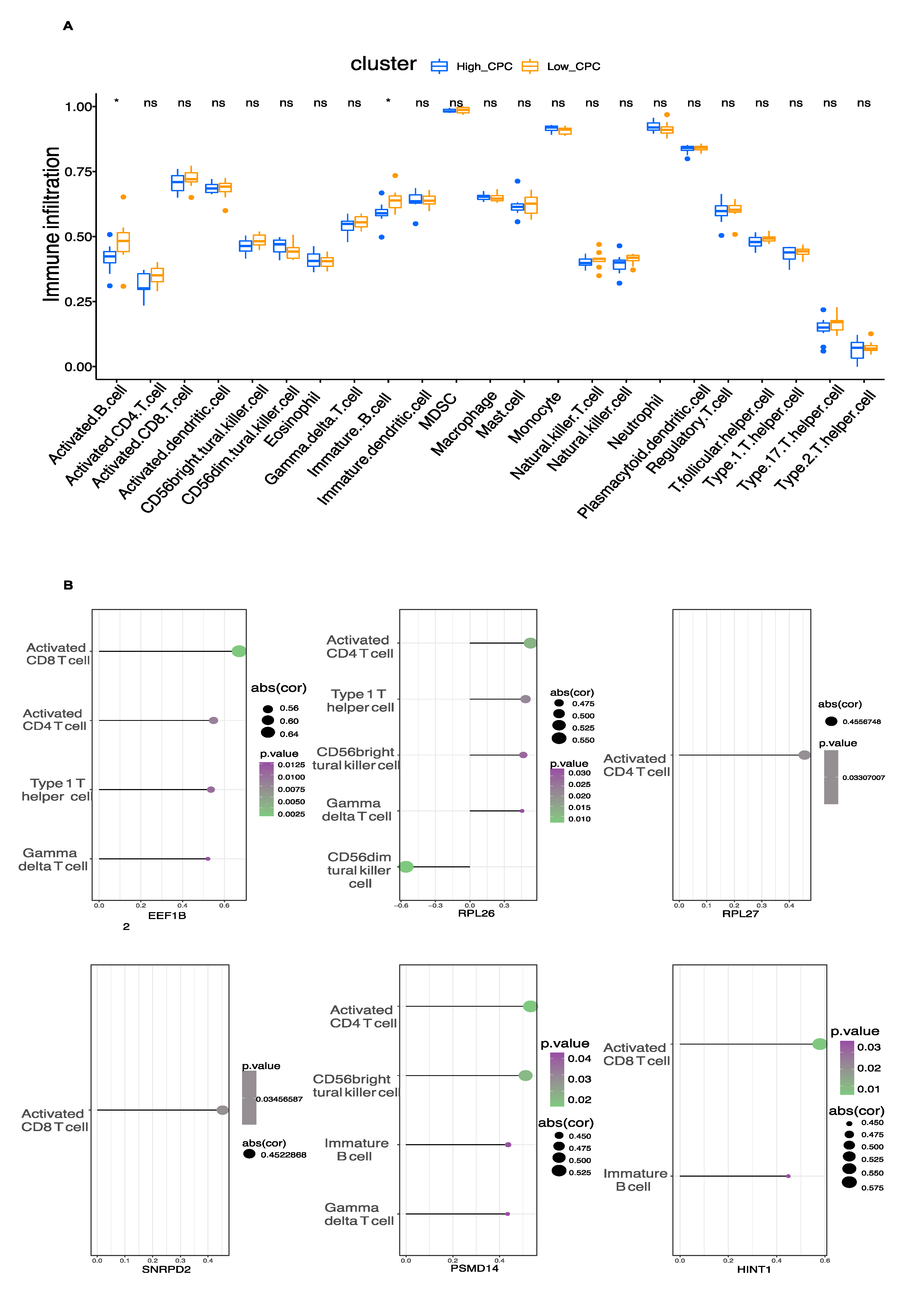
3.5. Immune Cell Infiltration
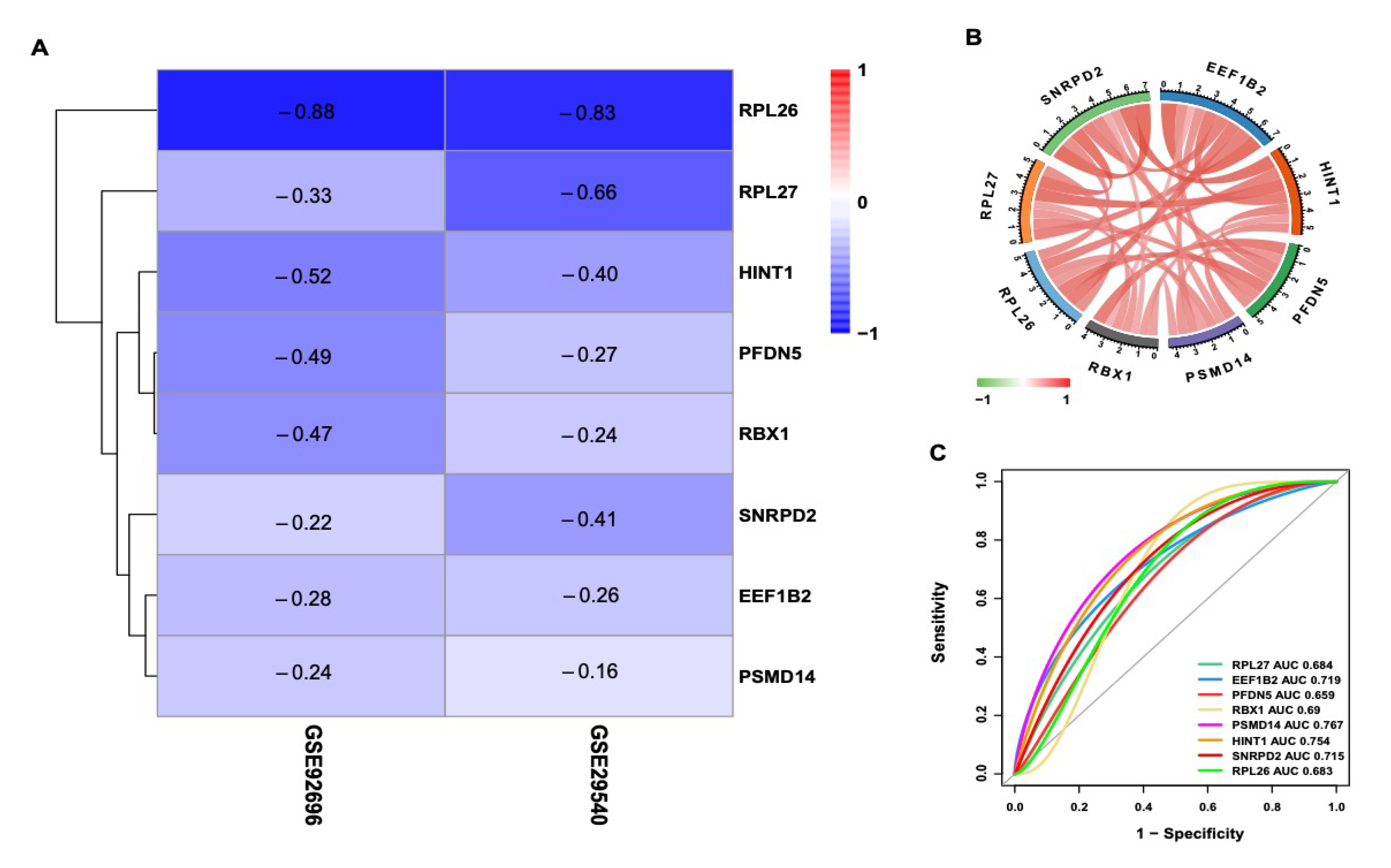
3.6. Exploration of Hub Genes
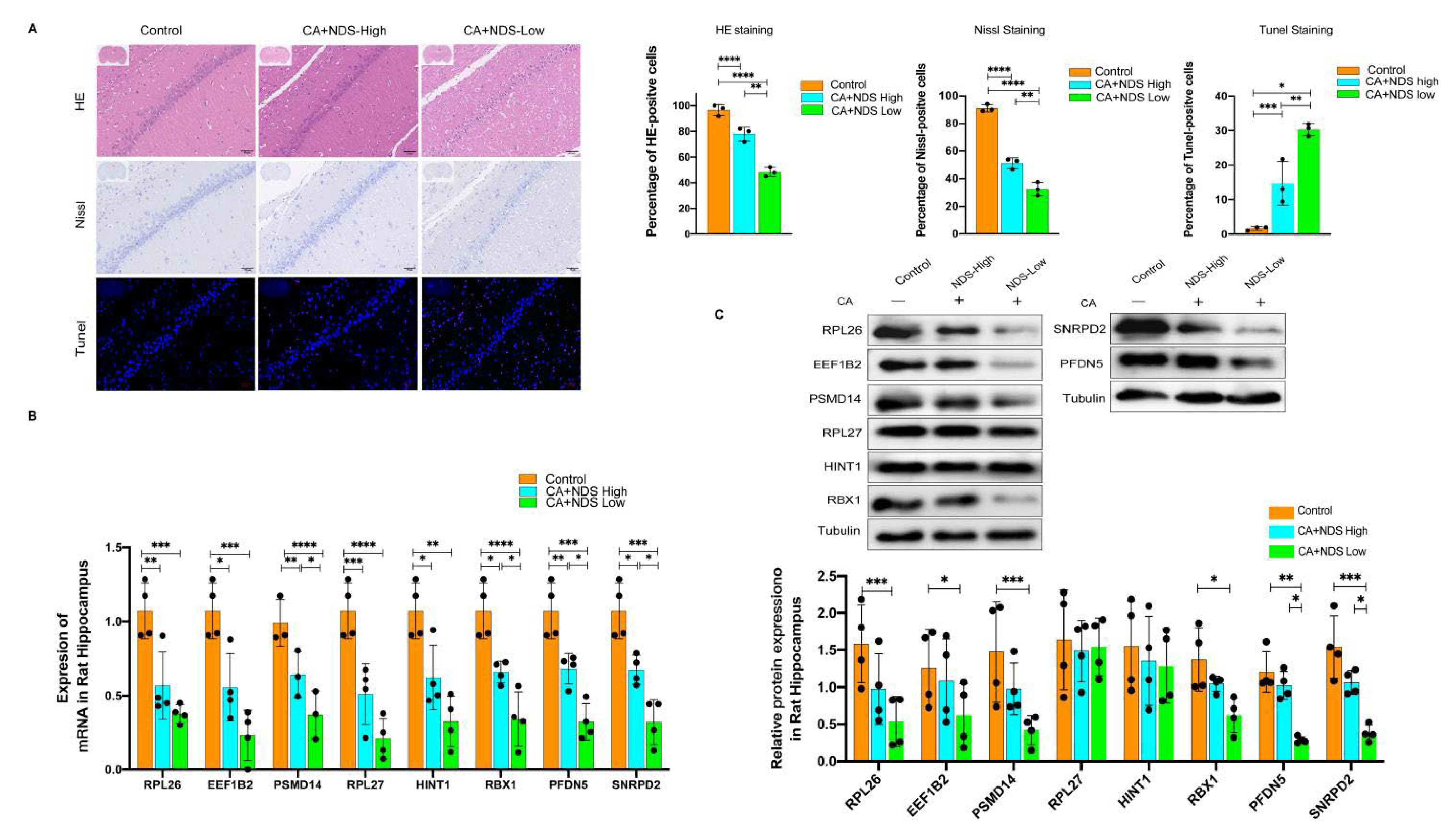
3.7. Validation of Hub Genes
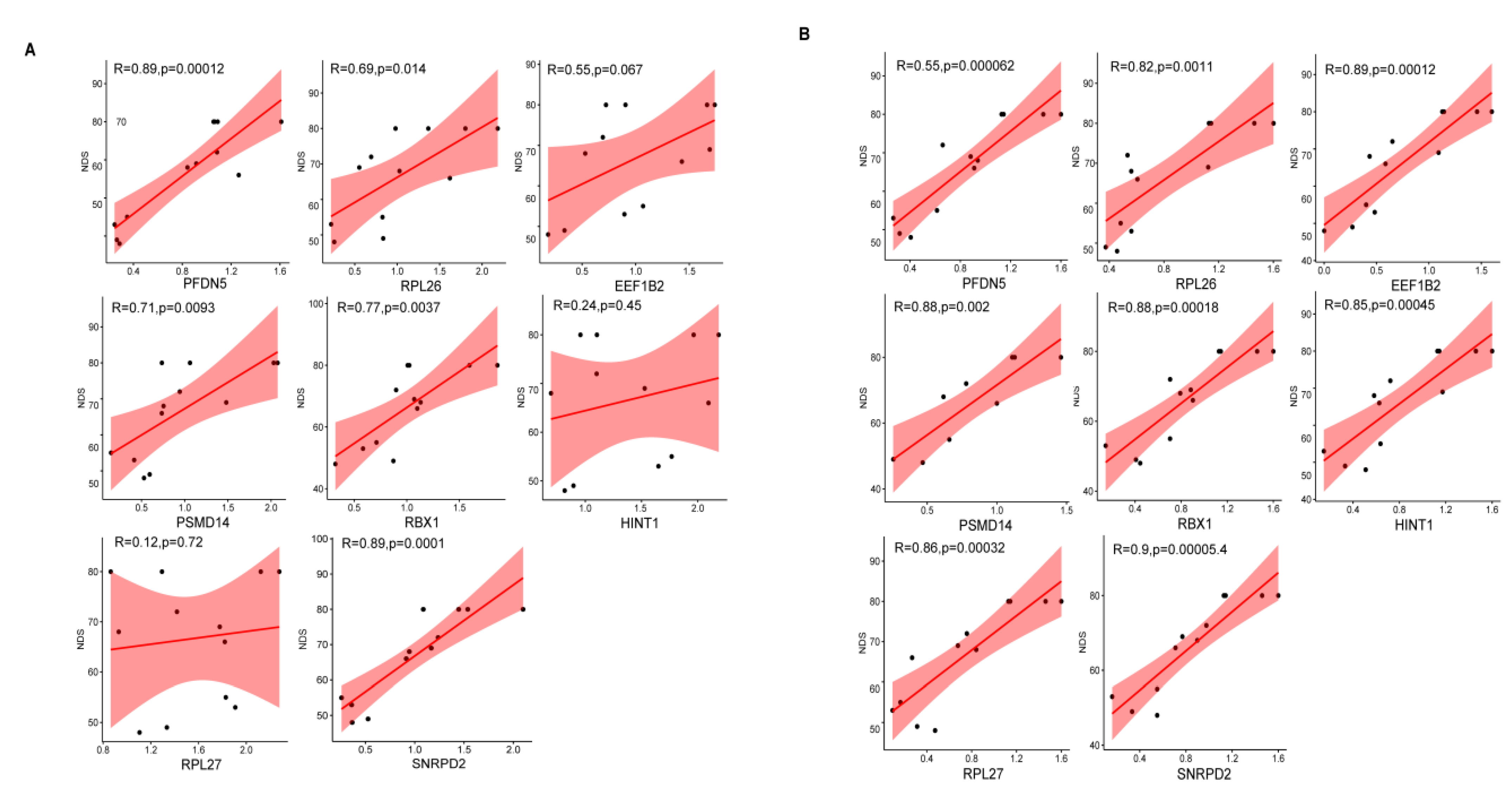
3.8. Correlation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Porzer, M.; Mrazkova, E.; Homza, M.; Janout, V. Out-of-hospital cardiac arrest. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czechoslov. 2017, 161, 348–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herlitz, J.; Engdahl, J.; Svensson, L.; Angquist, K.A.; Young, M.; Holmberg, S. Factors associated with an increased chance of survival among patients suffering from an out-of-hospital cardiac arrest in a national perspective in Sweden. Am. Heart J. 2005, 149, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, M.J.; Granfeldt, A.; Stankovic, N.; Andersen, L.W. Intra-cardiac arrest transport and survival from out-of-hospital cardiac arrest: A nationwide observational study. Resuscitation 2022, 175, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Hirunpattarasilp, C.; Shiina, H.; Na-Ek, N.; Attwell, D. The Effect of Hyperoxemia on Neurological Outcomes of Adult Patients: A Systematic Review and Meta-Analysis. Neurocrit Care 2022, 36, 1027–1043. [Google Scholar] [CrossRef]
- Sanfilippo, F.; La Via, L.; Lanzafame, B.; Dezio, V.; Busalacchi, D.; Messina, A.; Ristagno, G.; Pelosi, P.; Astuto, M. Targeted Temperature Management after Cardiac Arrest: A Systematic Review and Meta-Analysis with Trial Sequential Analysis. J. Clin. Med. 2021, 10, 3943. [Google Scholar] [CrossRef]
- Fernando, S.M.; Di Santo, P.; Sadeghirad, B.; Lascarrou, J.-B.; Rochwerg, B.; Mathew, R.; Sekhon, M.S.; Munshi, L.; Fan, E.; Brodie, D.; et al. Targeted temperature management following out-of-hospital cardiac arrest: A systematic review and network meta-analysis of temperature targets. Intensiv. Care Med. 2021, 47, 1078–1088. [Google Scholar] [CrossRef]
- Laver, S.; Farrow, C.; Turner, D.; Nolan, J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensiv. Care Med. 2004, 30, 2126–2128. [Google Scholar] [CrossRef]
- Dragancea, I.; Wise, M.P.; Al-Subaie, N.; Cranshaw, J.; Friberg, H.; Glover, G.; Pellis, T.; Rylance, R.; Walden, A.; Nielsen, N.; et al. Protocol-driven neurological prognostication and withdrawal of life-sustaining therapy after cardiac arrest and targeted temperature management. Resuscitation 2017, 117, 50–57. [Google Scholar] [CrossRef]
- Rossetti, A.O.; Rabinstein, A.A.; Oddo, M. Neurological prognostication of outcome in patients in coma after cardiac arrest. Lancet Neurol. 2016, 15, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Sandroni, C.; D’Arrigo, S.; Cacciola, S.; Hoedemaekers, C.W.E.; Kamps, M.J.A.; Oddo, M.; Taccone, F.S.; Di Rocco, A.; Meijer, F.J.A.; Westhall, E.; et al. Prediction of poor neurological outcome in comatose survivors of cardiac arrest: A systematic review. Intensiv. Care Med. 2020, 46, 1803–1851. [Google Scholar] [CrossRef]
- Cronberg, T.; Brizzi, M.; Liedholm, L.J.; Rosén, I.; Rubertsson, S.; Rylander, C.; Friberg, H. Neurological prognostication after cardiac arrest—Recommendations from the Swedish Resuscitation Council. Resuscitation 2013, 84, 867–872. [Google Scholar] [CrossRef]
- Sandroni, C.; Cronberg, T.; Sekhon, M. Brain injury after cardiac arrest: Pathophysiology, treatment, and prognosis. Intensiv. Care Med. 2021, 47, 1393–1414. [Google Scholar] [CrossRef]
- Rundgren, M.; Karlsson, T.; Nielsen, N.; Cronberg, T.; Johnsson, P.; Friberg, H. Neuron specific enolase and S-100B as predictors of outcome after cardiac arrest and induced hypothermia. Resuscitation 2009, 80, 784–789. [Google Scholar] [CrossRef]
- Oksanen, T.; Tiainen, M.; Skrifvars, M.B.; Varpula, T.; Kuitunen, A.; Castrén, M.; Pettilä, V. Predictive power of serum NSE and OHCA score regarding 6-month neurologic outcome after out-of-hospital ventricular fibrillation and therapeutic hypothermia. Resuscitation 2009, 80, 165–170. [Google Scholar] [CrossRef]
- Ok, G.; Aydin, D.; Erbüyün, K.; Gürsoy, C.; Taneli, F.; Bilge, S.; Horasan, G.D. Neurological outcome after cardiac arrest: A prospective study of the predictive ability of prognostic biomarkers neuron-specific enolase, glial fibrillary acidic protein, S-100B, and procalcitonin. Turk. J. Med. Sci. 2016, 46, 1459–1468. [Google Scholar] [CrossRef]
- Qi, Z.; Zhang, Q.; Liu, B.; Shao, F.; Li, C. Presepsin as a Biomarker for Evaluating Prognosis and Early Innate Immune Response of Out-of-Hospital Cardiac Arrest Patients After Return of Spontaneous Circulation. Crit. Care Med. 2019, 47, e538–e546. [Google Scholar] [CrossRef]
- Iwahana, T.; Okada, S.; Kanda, M.; Oshima, M.; Iwama, A.; Matsumiya, G.; Kobayashi, Y. Novel myocardial markers GADD45G and NDUFS5 identified by RNA-sequencing predicts left ventricular reverse remodeling in advanced non-ischemic heart failure: A retrospective cohort study. BMC Cardiovasc. Disord. 2020, 20, 116. [Google Scholar] [CrossRef]
- Chen, C.; Liu, C.; Niu, Z.; Li, M.; Zhang, Y.; Gao, R.; Chen, H.; Wang, Q.; Zhang, S.; Zhou, R.; et al. RNA-seq analysis of the key long noncoding RNAs and mRNAs related to cognitive impairment after cardiac arrest and cardiopulmonary resuscitation. Aging 2020, 12, 14490–14505. [Google Scholar] [CrossRef]
- Barr, T.L.; Alexander, S.; Conley, Y. Gene Expression Profiling for Discovery of Novel Targets in Human Traumatic Brain Injury. Biol. Res. Nurs. 2011, 13, 140–153. [Google Scholar] [CrossRef]
- Oh, J.S.; Tulasi, J.; Xiaodan, R.; Stacey, W.C.; Neumar, R.W. Valproic Acid Combined with Postcardiac Arrest Hypothermic-Targeted Temperature Management Prevents Delayed Seizures and Improves Survival in a Rat Cardiac Arrest Model. Crit. Care Med. 2017, 45, e1149–e1156. [Google Scholar] [CrossRef]
- Huang, L.; Applegate, R.L., 2nd; Applegate, P.M.; Boling, W.; Zhang, J.H. Inhalation of high concentration hydrogen gas improves short-term outcomes in a rat model of asphyxia induced-cardiac arrest. Med. Gas. Res. 2018, 8, 73–78. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y. Curative efficacy of penehyclidine combined with edaravone on acute cerebral infarction and their effects on serum TNF-α and NDS score in rats. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 223–228. [Google Scholar]
- Geocadin, R.G.; Sherman, D.L.; Hansen, H.C.; Kimura, T.; Niedermeyer, E.; Thakor, N.V.; Hanley, D.F. Neurological recovery by EEG bursting after resuscitation from cardiac arrest in rats. Resuscitation 2002, 55, 193–200. [Google Scholar] [CrossRef]
- Myat, A.; Song, K.-J.; Rea, T. Out-of-hospital cardiac arrest: Current concepts. Lancet 2018, 391, 970–979. [Google Scholar] [CrossRef]
- Andersen, L.W.; Holmberg, M.J.; Berg, K.M.; Donnino, M.W.; Granfeldt, A. In-Hospital Cardiac Arrest: A Review. JAMA 2019, 321, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Presciutti, A.; Perman, S.M. The evolution of hypothermia for neuroprotection after cardiac arrest: A history in the making. Ann. N. Y. Acad. Sci. 2022, 1507, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Zhuang, Y.; Ying, Z.; Agrawal, R.; Yang, X.; Gomez-Pinilla, F. Traumatic Brain Injury Induces Genome-Wide Transcriptomic, Methylomic, and Network Perturbations in Brain and Blood Predicting Neurological Disorders. eBioMedicine 2017, 16, 184–194. [Google Scholar] [CrossRef] [Green Version]
- Ebner, F.; Moseby-Knappe, M.; Mattsson-Carlgren, N.; Lilja, G.; Dragancea, I.; Undén, J.; Friberg, H.; Erlinge, D.; Kjaergaard, J.; Hassager, C.; et al. Serum GFAP and UCH-L1 for the prediction of neurological outcome in comatose cardiac arrest patients. Resuscitation 2020, 154, 61–68. [Google Scholar] [CrossRef]
- Stammet, P.; Collignon, O.; Hassager, C.; Wise, M.; Hovdenes, J.; Aneman, A.; Horn, J.; Devaux, Y.; Erlinge, D.; Kjaergaard, J.; et al. Neuron-Specific Enolase as a Predictor of Death or Poor Neurological Outcome After Out-of-Hospital Cardiac Arrest and Targeted Temperature Management at 33 °C and 36 °C. J. Am. Coll. Cardiol. 2015, 65, 2104–2114. [Google Scholar] [CrossRef]
- Henson, T.; Rawanduzy, C.; Salazar, M.; Sebastian, A.; Weber, H.; Al-Mufti, F.; Mayer, S.A. Outcome and prognostication after cardiac arrest. Ann. N. Y. Acad. Sci. 2021, 1508, 23–34. [Google Scholar] [CrossRef]
- Martínez-Losas, P.; De Sá, E.L.; Armada, E.; Rosillo, S.; Monedero, M.C.; Rey, J.R.; Caro-Codón, J.; Soto, A.B.; Sendón, J.L.L. Neuron-specific enolase kinetics: An additional tool for neurological prognostication after cardiac arrest. Rev. Esp. Cardiol. 2020, 73, 123–130. [Google Scholar] [CrossRef]
- Osier, N.D.; Conley, Y.P.; Okonkwo, D.O.; Puccio, A.M. Variation in Candidate Traumatic Brain Injury Biomarker Genes Are Associated with Gross Neurological Outcomes after Severe Traumatic Brain Injury. J. Neurotrauma 2018, 35, 2684–2690. [Google Scholar] [CrossRef]
- Dergunova, L.V.; Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Yuzhakov, V.V.; Mozerov, S.A.; Gubsky, L.V.; Limborska, S.A. Genome-wide transcriptome analysis using RNA-Seq reveals a large number of differentially expressed genes in a transient MCAO rat model. BMC Genom. 2018, 19, 655. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, Q.; Cui, Y.; Li, X.; Yang, H. In-depth transcriptomic and proteomic analyses of the hippocampus and cortex in a rat model after cerebral ischemic injury and repair by Shuxuetong (SXT) injection. J. Ethnopharmacol. 2020, 249, 112362. [Google Scholar] [CrossRef]
- Zhou, R.; Guo, F.; Xiang, C.; Zhang, Y.; Yang, H.; Zhang, J. Systematic Study of Crucial Transcription Factors of Coptidis rhizoma Alkaloids against Cerebral Ischemia-Reperfusion Injury. ACS Chem. Neurosci. 2021, 12, 2308–2319. [Google Scholar] [CrossRef]
- Gong, P.; Liu, J.; Jiao, X.; Niu, Y.; Wang, J.; Wang, X.; Yang, Z. Novel biallelic loss of EEF1B2 function links to autosomal recessive intellectual disability. Hum. Mutat. 2022, 43, 299–304. [Google Scholar] [CrossRef]
- Wang, H.; Han, X.; Gao, S. Identification of potential biomarkers for pathogenesis of Alzheimer’s disease. Hereditas 2021, 158, 23. [Google Scholar] [CrossRef]
- Yokoyama, S.; Iwakami, Y.; Hang, Z.; Kin, R.; Zhou, Y.; Yasuta, Y.; Takahashi, A.; Hayakawa, Y.; Sakurai, H. Targeting PSMD14 inhibits melanoma growth through SMAD3 stabilization. Sci. Rep. 2020, 10, 19214. [Google Scholar] [CrossRef]
- Wang, S.; Jun, J.; Cong, L.; Du, L.; Wang, C. miR-328-3p, a Predictor of Stroke, Aggravates the Cerebral Ischemia-Reperfusion Injury. Int. J. Gen. Med. 2021, 14, 2367–2376. [Google Scholar] [CrossRef]
- Boissady, E.; Kohlhauer, M.; Lidouren, F.; Hocini, H.; Lefebvre, C.; Chateau-Jouber, S.; Mongardon, N.; Deye, N.; Cariou, A.; Micheau, P.; et al. Ultrafast Hypothermia Selectively Mitigates the Early Humoral Response After Cardiac Arrest. J. Am. Heart Assoc. 2020, 9, e017413. [Google Scholar] [CrossRef]
- Kreimann, K.; Jang, M.-S.; Rong, S.; Greite, R.; Von Vietinghoff, S.; Schmitt, R.; Bräsen, J.H.; Schiffer, L.; Gerstenberg, J.; Vijayan, V.; et al. Ischemia Reperfusion Injury Triggers CXCL13 Release and B-Cell Recruitment After Allogenic Kidney Transplantation. Front. Immunol. 2020, 11, 1204. [Google Scholar] [CrossRef]
- Stammet, P. Blood Biomarkers of Hypoxic-Ischemic Brain Injury after Cardiac Arrest. Skull Base 2017, 37, 75–80. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequence |
|---|---|
| RPL26 | F: TCACCCCAGCAAGGTGGTTAT |
| R: TGCCCTTCTCCTTTCCTACTTG | |
| PSMD14 | F: AAACAGGAAGGCCCGAGATG |
| R: CACACCAGAAAGCCAACAGC | |
| HINT1 | F: GCAGATGATGATGATGAAAGTCTTC |
| R: CCCGTCTGCACCTTCATTCA | |
| RBX1 | F: TTGGGGAGTCTGTAACCACG |
| R: CTCTGTTGTCCAAGGGGCAC | |
| EEF1B2 | F: AGCTACATTGAGGGGTACGTG |
| R: TACCAACGCAGAGCATGACA | |
| RPL27 | F: CCTCATGCCCACAAGGTACT |
| R: AAACTTGACCTTGGCCTCCC | |
| SNRPD2 | F: CTCTCGGTGCTCACACAGTC |
| R: ACCATGTTGCAGTGCCTGTC | |
| PFDN5 | F: AGACAGCTGAGGATGCCAAG |
| R: TCATTTCCACGACGGCTTGT | |
| β-actin | F: CACCCGCGAGTACAACCTTC |
| R: CCCATACCCACCCATCACACC |
| Name | Corporation | Co. Number | Dilution Ratio |
|---|---|---|---|
| Anti-RPL26 | Proteintech | 16487-1-AP | 1:800 |
| Anti-PSMD14 | Abcam | Ab109130 | 1:1000 |
| Anti-HINT1 | Proteintech | 10717-1-AP | 1:500 |
| Anti-RBX1 | Abcam | Ab133565 | 1:1000 |
| Anti-EEF1B2 | Proteintech | 10483-1-AP | 1:500 |
| Anti-RPL27 | Proteintech | 14980-1-AP | 1:500 |
| Anti-SNRPD2 | Abcam | Ab198296 | 1:500 |
| Anti-PFDN5 | Proteintech | 15078-1-AP | 1:500 |
| Anti-Tubulin | Beyotime | AF1216 | 1:1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Zhang, C.; Liu, C.; Zhan, H.; Li, B.; Lu, Y.; Wei, H.; Cheng, J.; Li, S.; Wang, C.; et al. Identification and Validation of Novel Potential Pathogenesis and Biomarkers to Predict the Neurological Outcome after Cardiac Arrest. Brain Sci. 2022, 12, 928. https://doi.org/10.3390/brainsci12070928
Zhang Q, Zhang C, Liu C, Zhan H, Li B, Lu Y, Wei H, Cheng J, Li S, Wang C, et al. Identification and Validation of Novel Potential Pathogenesis and Biomarkers to Predict the Neurological Outcome after Cardiac Arrest. Brain Sciences. 2022; 12(7):928. https://doi.org/10.3390/brainsci12070928
Chicago/Turabian StyleZhang, Qiang, Chenyu Zhang, Cong Liu, Haohong Zhan, Bo Li, Yuanzhen Lu, Hongyan Wei, Jingge Cheng, Shuhao Li, Chuyue Wang, and et al. 2022. "Identification and Validation of Novel Potential Pathogenesis and Biomarkers to Predict the Neurological Outcome after Cardiac Arrest" Brain Sciences 12, no. 7: 928. https://doi.org/10.3390/brainsci12070928
APA StyleZhang, Q., Zhang, C., Liu, C., Zhan, H., Li, B., Lu, Y., Wei, H., Cheng, J., Li, S., Wang, C., Hu, C., & Liao, X. (2022). Identification and Validation of Novel Potential Pathogenesis and Biomarkers to Predict the Neurological Outcome after Cardiac Arrest. Brain Sciences, 12(7), 928. https://doi.org/10.3390/brainsci12070928