

The Absence of Gasdermin D Reduces Nuclear Autophagy in a Cecal Ligation and Puncture-Induced Sepsis-Associated Encephalopathy Mouse Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals
2.3. CLP Model of Sepsis
2.4. Open Field Test (OFT)
2.5. Novel Object Rrecognition Test (NORT)
2.6. Immunofluorescence
2.7. Western Blotting
2.8. Transmission Electron Microscopy (TEM)
2.9. Measurements of Nuclear Distortions and Circularity
2.10. Statistical Analysis
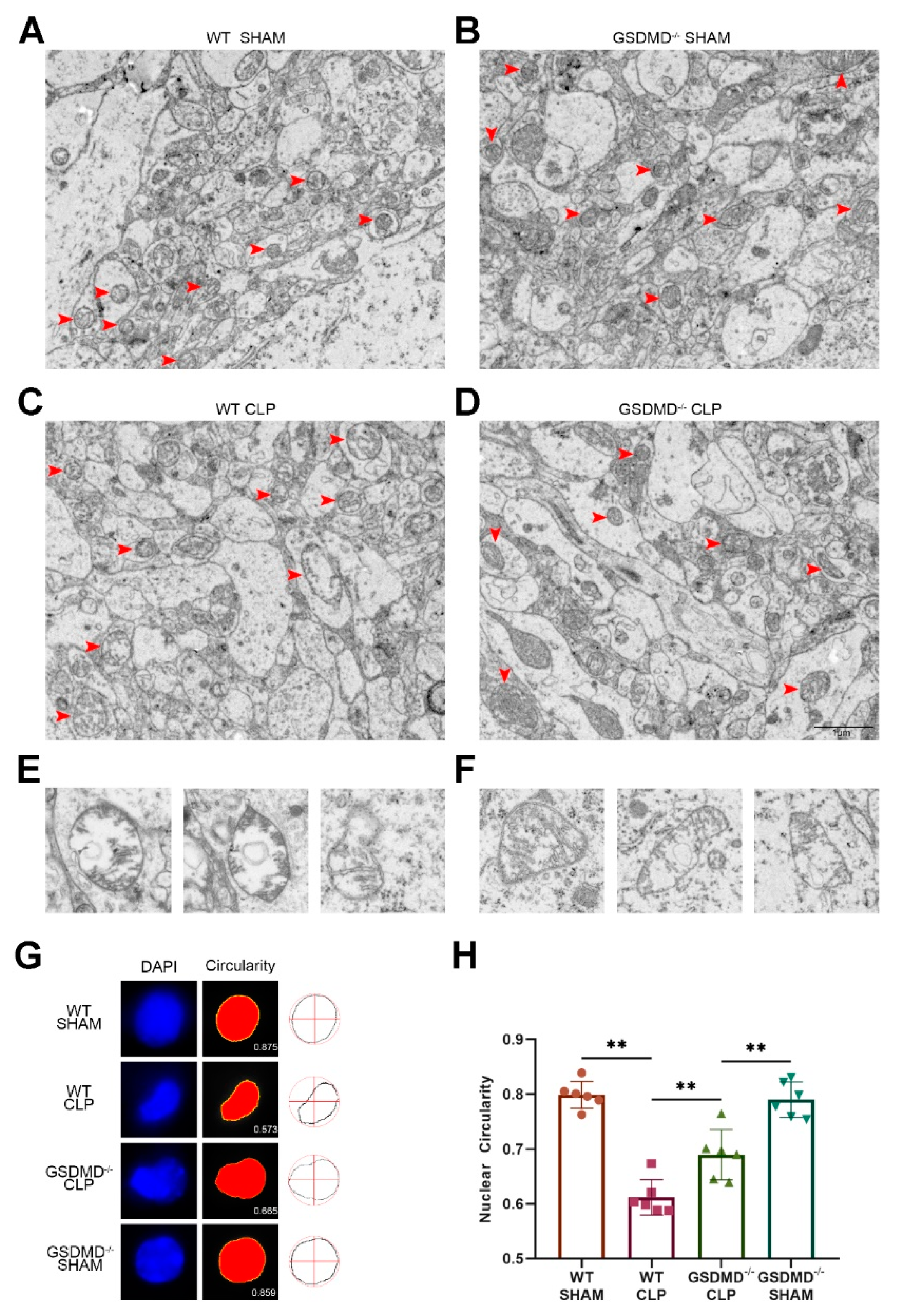
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fu, Q.; Wu, J.; Zhou, X.Y.; Ji, M.H.; Mao, Q.H.; Li, Q.; Zong, M.M.; Zhou, Z.Q.; Yang, J.J. NLRP3/Caspase-1 Pathway-Induced Pyroptosis Mediated Cognitive Deficits in a Mouse Model of Sepsis-Associated Encephalopathy. Inflammation 2019, 42, 306–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, G.; Zuo, J.; Fang, Q.; Yuan, M.; Xia, Y.; Jin, Q.; Liu, Y.; Wang, Y.; Zhang, Z.; Liu, W.; et al. Erbin protects against sepsis-associated encephalopathy by attenuating microglia pyroptosis via IRE1α/Xbp1s-Ca(2+) axis. J. Neuroinflamm. 2022, 19, 237. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Wang, Q.; Yang, X. Roles of the pyroptosis signaling pathway in a sepsis-associated encephalopathy cell model. J. Int. Med. Res. 2020, 48, 300060520949767. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.E.; Liu, L.; Wang, Y.C.; Wang, C.T.; Zheng, Q.; Liu, Q.X.; Li, Z.F.; Bai, X.J.; Liu, X.H. Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain Behav. Immun. 2019, 80, 859–870. [Google Scholar] [CrossRef]
- Zhong, X.; Chen, Z.; Wang, Y.; Mao, M.; Deng, Y.; Shi, M.; Xu, Y.; Chen, L.; Cao, W. JQ1 attenuates neuroinflammation by inhibiting the inflammasome-dependent canonical pyroptosis pathway in SAE. Brain Res. Bull. 2022, 189, 174–183. [Google Scholar] [CrossRef]
- Tauber, S.C.; Djukic, M.; Gossner, J.; Eiffert, H.; Brück, W.; Nau, R. Sepsis-associated encephalopathy and septic encephalitis: An update. Expert Rev. Anti-Infect. Ther. 2020, 19, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, J.R. Delirium in the acute care setting: Characteristics, diagnosis and treatment. Crit. Care Clin. 2008, 24, 657–722, vii. [Google Scholar] [CrossRef]
- Pan, S.; Lv, Z.; Wang, R.; Shu, H.; Yuan, S.; Yu, Y. Sepsis-Induced Brain Dysfunction: Pathogenesis, Diagnosis, and Treatment. Oxid. Med. Cell Longev. 2022, 2022, 1328729. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Karmakar, M.; Minns, M. N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat. Commun. 2020, 11, 2212. [Google Scholar] [CrossRef]
- Booty, L.M.; Bryant, C.E. Gasdermin D and Beyond—Gasdermin-mediated Pyroptosis in Bacterial Infections. J. Mol. Biol. 2022, 434, 167409. [Google Scholar] [CrossRef] [PubMed]
- Brokatzky, D.; Mostowy, S. Pyroptosis in host defence against bacterial infection. Dis. Model Mech. 2022, 15, dmm049414. [Google Scholar] [CrossRef] [PubMed]
- Chai, Q.; Yu, S. A bacterial phospholipid phosphatase inhibits host pyroptosis by hijacking ubiquitin. Science 2022, 378, eabq0132. [Google Scholar] [CrossRef]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Xu, M.; Xie, J.; Liu, T.; Xu, X.; Gao, W.; Li, Z.; Bai, X.; Liu, X. Inhibition of Ferroptosis Attenuates Glutamate Excitotoxicity and Nuclear Autophagy in a CLP Septic Mouse Model. Shock 2022, 57, 694–702. [Google Scholar] [CrossRef]
- Luo, M.; Zhao, X.; Song, Y.; Cheng, H.; Zhou, R. Nuclear autophagy: An evolutionarily conserved mechanism of nuclear degradation in the cytoplasm. Autophagy 2016, 12, 1973–1983. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, A.; Yue, Z. Autophagy and its normal and pathogenic states in the brain. Annu. Rev. Neurosci. 2014, 37, 55–78. [Google Scholar] [CrossRef] [Green Version]
- Pfister, A.S. Emerging Role of the Nucleolar Stress Response in Autophagy. Front. Cell. Neurosci. 2019, 13, 156. [Google Scholar] [CrossRef] [Green Version]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [Green Version]
- Iershov, A.; Nemazanyy, I.; Alkhoury, C.; Girard, M. The class 3 PI3K coordinates autophagy and mitochondrial lipid catabolism by controlling nuclear receptor PPARα. Nat. Commun. 2019, 10, 1566. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Lee, J.M. Transcriptional Regulation of Hepatic Autophagy by Nuclear Receptors. Cells 2022, 11, 620. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Nakatogawa, H. Degradation of nuclear components via different autophagy pathways. Trends Cell Biol. 2022, 32, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Montanaro, L. The Ribosome Biogenesis-Cancer Connection. Cells 2019, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Zhou, Y.; Casanova, E.; Chai, M.; Kiss, E.; Gröne, H.J.; Schütz, G.; Grummt, I. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol. Cell 2005, 19, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, M.; Rempala, G.; Nelson, P.T.; Zheng, J.J.; Hetman, M. Epigenetic silencing of nucleolar rRNA genes in Alzheimer’s disease. PLoS ONE 2011, 6, e22585. [Google Scholar] [CrossRef]
- Baron, O.; Boudi, A.; Dias, C.; Schilling, M.; Nölle, A.; Vizcay-Barrena, G.; Rattray, I.; Jungbluth, H.; Scheper, W.; Fleck, R.A.; et al. Stall in Canonical Autophagy-Lysosome Pathways Prompts Nucleophagy-Based Nuclear Breakdown in Neurodegeneration. Curr. Biol. CB 2017, 27, 3626–3642.e3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.K.; Gordon, J.; Glauser, G.M.; Myers, V.D.; Feldman, A.M.; Cheung, J.Y.; Khalili, K. Lamin B is a target for selective nuclear PQC by BAG3: Implication for nuclear envelopathies. Cell Death Dis. 2019, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Mareninova, O.A.; Jia , W.; Gretler, S.R.; Holthaus, C.L.; Thomas, D.D.H.; Pimienta, M.; Dillon, D.L.; Gukovskaya, A.S.; Gukovsky, I.; Groblewski, G.E. Transgenic expression of GFP-LC3 perturbs autophagy in exocrine pancreas and acute pancreatitis responses in mice. Autophagy 2020, 16, 2084–2097. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. (Clifton N.J.) 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciuffa, R.; Lamark, T.; Tarafder, A.K.; Guesdon, A.; Rybina, S.; Hagen, W.J.; Johansen, T.; Sachse, C. The selective autophagy receptor p62 forms a flexible filamentous helical scaffold. Cell Rep. 2015, 11, 748–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Li, J.; Zhang, G.; Da, Q.; Chen, L.; Yu, S.; Zhou, Q.; Weng, Z.; Xin, Z.; Shi, L.; et al. HMGB1-Induced p62 Overexpression Promotes Snail-Mediated Epithelial-Mesenchymal Transition in Glioblastoma Cells via the Degradation of GSK-3β. Theranostics 2019, 9, 1909–1922. [Google Scholar] [CrossRef]
- Deng, S.; Ai, Y.; Gong, H.; Feng, Q.; Li, X.; Chen, C.; Liu, Z.; Wang, Y.; Peng, Q.; Zhang, L. Mitochondrial dynamics and protective effects of a mitochondrial division inhibitor, Mdivi-1, in lipopolysaccharide-induced brain damage. Biochem. Biophys. Res. Commun. 2018, 496, 865–871. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Z.; Zhang, Y.; Fang, S.; Zeng, Q. Mitochondrial biogenesis of astrocytes is increased under experimental septic conditions. Chin. Med. J. 2014, 127, 1837–1842. [Google Scholar]
- Zhang, L.; Jiang, Y.; Deng, S.; Mo, Y.; Huang, Y.; Li, W.; Ge, C.; Ren, X.; Zhang, H.; Zhang, X.; et al. S100B/RAGE/Ceramide signaling pathway is involved in sepsis-associated encephalopathy. Life Sci. 2021, 277, 119490. [Google Scholar] [CrossRef]
- Zhao, L.; Song, Y.; Zhang, Y.; Liu, H.; Shen, Y.; Fan, Y.; Li, Y.; Xie, K. HIF-1α/BNIP3L induced cognitive deficits in a mouse model of sepsis-associated encephalopathy. Front. Immunol. 2022, 13, 1095427. [Google Scholar] [CrossRef]
- Huang, S.X.; Partridge, M.A.; Ghandhi, S.A.; Davidson, M.M.; Amundson, S.A.; Hei, T.K. Mitochondria-derived reactive intermediate species mediate asbestos-induced genotoxicity and oxidative stress-responsive signaling pathways. Environ. Health Perspect. 2012, 120, 840–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Sun, M. The Improvement of Sepsis-Associated Encephalopathy by P2X7R Inhibitor through Inhibiting the Omi/HtrA2 Apoptotic Signaling Pathway. Behav. Neurol. 2022, 2022, 3777351. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Zhao, Y.; Yu, F. Puerarin ameliorates acute lung injury by modulating NLRP3 inflammasome-induced pyroptosis. Cell Death Discov. 2022, 8, 368. [Google Scholar] [CrossRef]
- Tanaka, M.; Szabó, Á.; Spekker, E.; Polyák, H.; Tóth, F.; Vécsei, L. Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan-Kynurenine Metabolic System. Cells 2022, 11, 2607. [Google Scholar] [CrossRef]
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734. [Google Scholar] [CrossRef]
- Battaglia, S.; Cardellicchio, P.; Di Fazio, C.; Nazzi, C.; Fracasso, A.; Borgomaneri, S. Stopping in (e)motion: Reactive action inhibition when facing valence-independent emotional stimuli. Front. Behav. Neurosci. 2022, 16, 998714. [Google Scholar] [CrossRef]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef] [Green Version]
- Rittirsch, D.; Huber-Lang, M.S.; Flierl, M.A.; Ward, P.A. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 2009, 4, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, I.; Diniz, L.P.; Damico, I.V.; Araujo, A.P.B.; Neves, L.D.S.; Vargas, G.; Leite, R.E.P.; Suemoto, C.K.; Nitrini, R.; Jacob-Filho, W.; et al. Loss of lamin-B1 and defective nuclear morphology are hallmarks of astrocyte senescence in vitro and in the aging human hippocampus. Aging Cell 2022, 21, e13521. [Google Scholar] [CrossRef] [PubMed]
- Sturman, O.; Germain, P.L.; Bohacek, J. Exploratory rearing: A context- and stress-sensitive behavior recorded in the open-field test. Stress (Amst. Neth.) 2018, 21, 443–452. [Google Scholar] [CrossRef]
- Caputi, A.; Liu, X.; Fuchs, E.C.; Liu, Y.C.; Monyer, H. Medial entorhinal cortex commissural input regulates the activity of spatially and object-tuned cells contributing to episodic memory. Neuron 2022, 110, 3389–3405.e3387. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yao, J.; Liu, Y.; Huang, L. Targeting the gasdermin D as a strategy for ischemic stroke therapy. Biochem. Pharmacol. 2021, 188, 114585. [Google Scholar] [CrossRef]
- de Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oami, T.; Watanabe, E.; Hatano, M.; Teratake, Y.; Fujimura, L.; Sakamoto, A.; Ito, C.; Toshimori, K.; Swanson, P.E.; Oda, S. Blocking Liver Autophagy Accelerates Apoptosis and Mitochondrial Injury in Hepatocytes and Reduces Time to Mortality in a Murine Sepsis Model. Shock 2018, 50, 427–434. [Google Scholar] [CrossRef]
- Karagiannidis, I.; Kataki, A.; Glustianou, G.; Memos, N.; Papalois, A.; Alexakis, N.; Zografos, G.C.; Konstadoulakis, M.M. Extended cytoprotective effect of autophagy in the late stages of sepsis and fluctuations in signal transduction pathways in a rat experimental model of kidney injury. Shock 2016, 45, 139–147. [Google Scholar] [CrossRef]
- Pu, Q.; Gan, C.; Li, R.; Li, Y.; Tan, S.; Li, X.; Wei, Y.; Lan, L.; Deng, X.; Liang, H.; et al. Atg7 Deficiency Intensifies Inflammasome Activation and Pyroptosis in Pseudomonas Sepsis. J. Immunol. (Baltim. Md. 1950) 2017, 198, 3205–3213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finethy, R.; Dockterman, J.; Kutsch, M.; Orench-Rivera, N.; Wallace, G.D.; Piro, A.S.; Luoma, S.; Haldar, A.K.; Hwang, S.; Martinez, J.; et al. Dynamin-related Irgm proteins modulate LPS-induced caspase-11 activation and septic shock. EMBO Rep. 2020, 21, e50830. [Google Scholar] [CrossRef] [PubMed]
- Eren, E.; Planès, R.; Bagayoko, S.; Bordignon, P.J.; Chaoui, K.; Hessel, A.; Santoni, K.; Pinilla, M.; Lagrange, B.; Burlet-Schiltz, O.; et al. Irgm2 and Gate-16 cooperatively dampen Gram-negative bacteria-induced caspase-11 response. EMBO Rep. 2020, 21, e50829. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, W.; Xie, Z.; Bai, X.; Li, Z.; Liu, X. The Absence of Gasdermin D Reduces Nuclear Autophagy in a Cecal Ligation and Puncture-Induced Sepsis-Associated Encephalopathy Mouse Model. Brain Sci. 2023, 13, 478. https://doi.org/10.3390/brainsci13030478
Su W, Xie Z, Bai X, Li Z, Liu X. The Absence of Gasdermin D Reduces Nuclear Autophagy in a Cecal Ligation and Puncture-Induced Sepsis-Associated Encephalopathy Mouse Model. Brain Sciences. 2023; 13(3):478. https://doi.org/10.3390/brainsci13030478
Chicago/Turabian StyleSu, Wei, Zhenxing Xie, Xiangjun Bai, Zhanfei Li, and Xinghua Liu. 2023. "The Absence of Gasdermin D Reduces Nuclear Autophagy in a Cecal Ligation and Puncture-Induced Sepsis-Associated Encephalopathy Mouse Model" Brain Sciences 13, no. 3: 478. https://doi.org/10.3390/brainsci13030478