Indoleamine 2,3-Dioxygenase as a Therapeutic Target for Alzheimer’s Disease and Geriatric Depression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
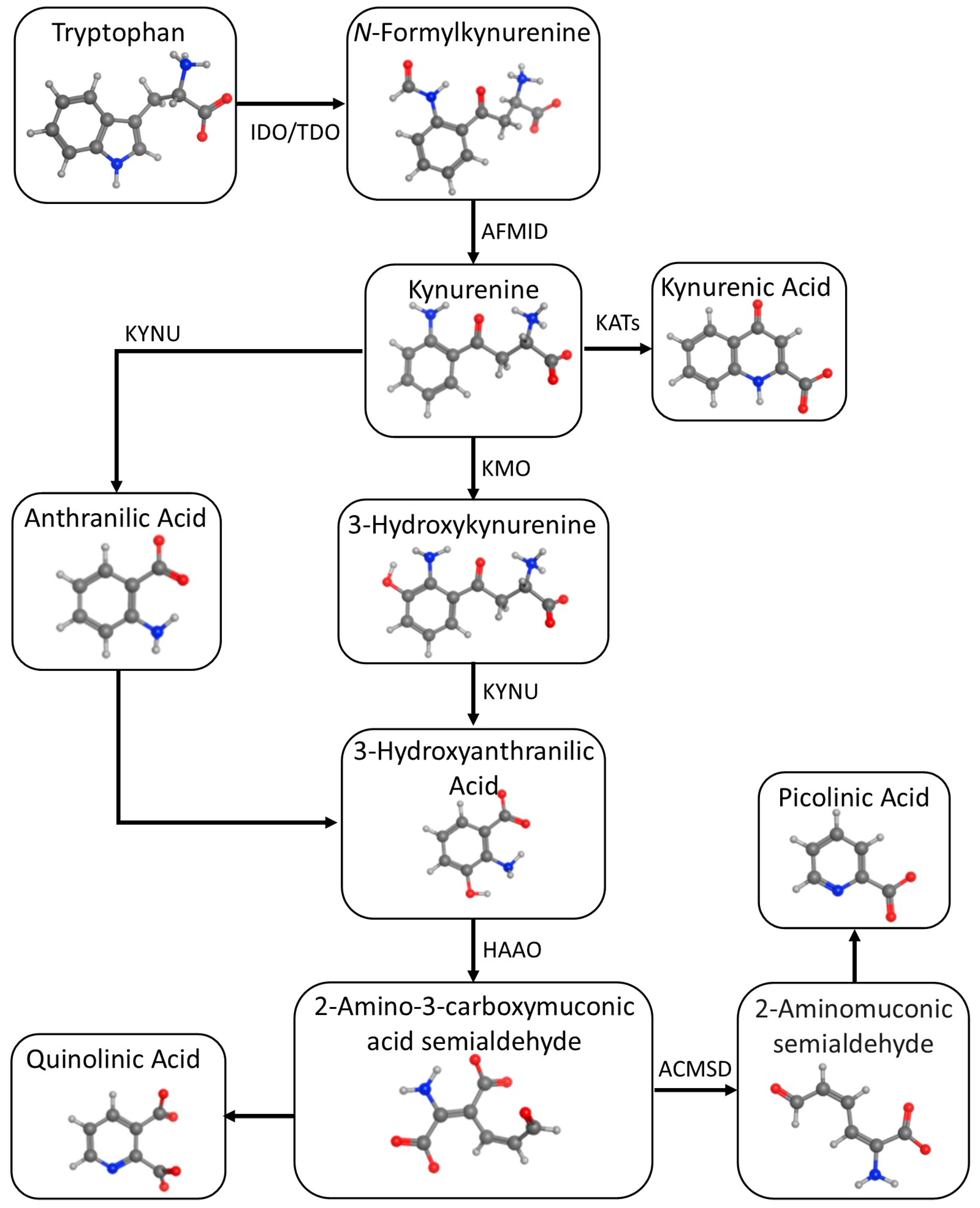
2. Tryptophan Metabolism
2.1. Serotonergic Pathway
2.2. Kynurenic Pathway
3. Function and Structure of IDO
4. Drugability of IDO
5. IDO and Depression
Geriatric Depression and Inflammation
6. IDO and Dementia
6.1. Depression-Dementia Spectrum Disorder
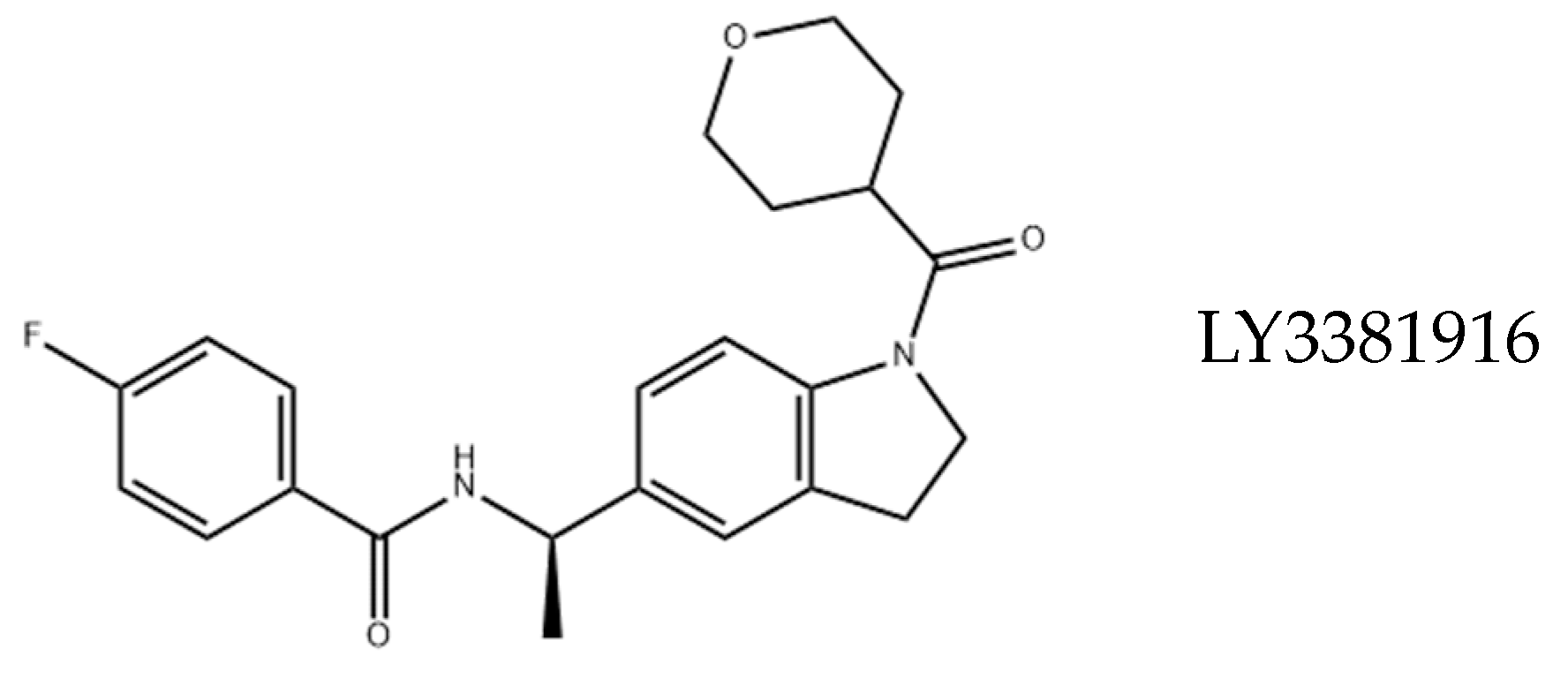
6.2. Future of IDO-1 Inhibitors for Treating Dementia and Depression
7. Other Neurological Disorders
7.1. Parkinson’s Disease
7.2. Multiple Sclerosis (MS)
7.3. Schizophrenia
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Comai, S.; Bertazzo, A.; Brughera, M.; Crotti, S. Chapter Five—Tryptophan in health and disease. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 165–218. [Google Scholar]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2022, 1, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.P.; Pan, Z.Z.; Luo, D.Y. TDO as a therapeutic target in brain diseases. Metab. Brain Dis. 2016, 31, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, P.; Grohmann, U. IDO and regulatory T cells: A role for reverse signalling and non-canonical NF-kappaB activation. Nat. Rev. Immunol. 2007, 7, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Espey, M.G.; Namboodiri, M.A. Antibodies to quinolinic acid and the determination of its cellular distribution within the rat immune system. Cell Tissue Res. 1994, 278, 461–469. [Google Scholar] [CrossRef]
- Yamazaki, F.; Kuroiwa, T.; Takikawa, O.; Kido, R. Human indolylamine 2,3-dioxygenase. Its tissue distribution, and characterization of the placental enzyme. Biochem. J. 1985, 230, 635–638. [Google Scholar] [CrossRef]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-brain barrier transport of kynurenines: Implications for brain synthesis and metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef]
- el-Defrawy, S.R.; Boegman, R.J.; Jhamandas, K.; Beninger, R.J. The neurotoxic actions of quinolinic acid in the central nervous system. Can. J. Phys. Pharmacol. 1986, 64, 369–375. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, J.; Zhang, Z.; Guo, Y.; Wu, Y.; Wang, R.; Wang, L.; Mao, S.; Yao, X. Overexpression of Indoleamine 2,3-Dioxygenase 1 Promotes Epithelial-Mesenchymal Transition by Activation of the IL-6/STAT3/PD-L1 Pathway in Bladder Cancer. Transl. Oncol. 2019, 12, 485–492. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease hippocampus. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; DuHadaway, J.B.; Montgomery, J.D.; Peng, W.D.; Murray, P.J.; Prendergast, G.C.; Caton, A.J.; Muller, A.J.; Mandik-Nayak, L. Differential Roles of IDO1 and IDO2 in T and B Cell Inflammatory Immune Responses. Front. Immunol. 2020, 11, 1861. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Nicolazzo, J.A.; Wen, L.; Chung, R.; Stankovic, R.; Bao, S.S.; Lim, C.K.; Brew, B.J.; Cullen, K.M.; Guillemin, G.J. Expression of tryptophan 2,3-dioxygenase and production of kynurenine pathway metabolites in triple transgenic mice and human Alzheimer’s disease brain. PLoS ONE 2013, 8, e59749. [Google Scholar] [CrossRef]
- Myint, A.M.; Kim, Y.K.; Verkerk, R.; Scharpé, S.; Steinbusch, H.; Leonard, B. Kynurenine pathway in major depression: Evidence of impaired neuroprotection. J. Affect. Disord. 2007, 98, 143–151. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.C.; Lawson, M.A.; André, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 2009, 14, 511–522. [Google Scholar] [CrossRef]
- Savitz, J.; Drevets, W.C.; Wurfel, B.E.; Ford, B.N.; Bellgowan, P.S.; Victor, T.A.; Bodurka, J.; Teague, T.K.; Dantzer, R. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav. Immun. 2015, 46, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Bay-Richter, C.; Linderholm, K.R.; Lim, C.K.; Samuelsson, M.; Träskman-Bendz, L.; Guillemin, G.J.; Erhardt, S.; Brundin, L. A role for inflammatory metabolites as modulators of the glutamate N-methyl-D-aspartate receptor in depression and suicidality. Brain Behav. Immun. 2015, 43, 110–117. [Google Scholar] [CrossRef]
- O’Connor, J.C.; André, C.; Wang, Y.; Lawson, M.A.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J. Neuroci. 2009, 29, 4200–4209. [Google Scholar] [CrossRef]
- Lotrich, F.E. Major depression during interferon-alpha treatment: Vulnerability and prevention. Dialogues Clin. Neurosci. 2009, 11, 417–425. [Google Scholar] [CrossRef]
- Höglund, E.; Øverli, Ø.; Winberg, S. Tryptophan Metabolic Pathways and Brain Serotonergic Activity: A Comparative Review. Front. Endocrinol. 2019, 10, 158. [Google Scholar] [CrossRef]
- Stasi, C.; Sadalla, S.; Milani, S. The Relationship Between the Serotonin Metabolism, Gut-Microbiota and the Gut-Brain Axis. Curr. Drug. Metab. 2019, 20, 646–655. [Google Scholar] [CrossRef]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.J.; Yuasa, H.J.; Austin, C.J.; Weiser, S.; Hunt, N.H. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int. J. Biochem. Cell Biol. 2009, 41, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Alvarado, D.M.; Iticovici, M.; Kau, N.S.; Park, H.; Parikh, P.J.; Thotala, D.; Ciorba, M.A. Interferon-Induced IDO1 Mediates Radiation Resistance and Is a Therapeutic Target in Colorectal Cancer. Cancer Immunol Res. 2020, 8, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; Wainwright, D.A. IDO1 in cancer: A Gemini of immune checkpoints. Cell. Mol. Immunol. 2018, 15, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, M.T.; Rossini, S.; Suvieri, C.; Coletti, A.; Orabona, C.; Macchiarulo, A.; Volpi, C.; Grohmann, U. Indoleamine 2,3-dioxygenase 1 (IDO1): An up-to-date overview of an eclectic immunoregulatory enzyme. FEBS J. 2022, 289, 6099–6118. [Google Scholar] [CrossRef]
- Salminen, A. Role of indoleamine 2,3-dioxygenase 1 (IDO1) and kynurenine pathway in the regulation of the aging process. Ageing Res. Rev. 2022, 75, 101573. [Google Scholar] [CrossRef]
- Sari, S.; Tomek, P.; Leung, E.; Reynisson, J. Discovery and Characterisation of Dual Inhibitors of Tryptophan 2,3-Dioxygenase (TDO2) and Indoleamine 2,3-Dioxygenase 1 (IDO1) Using Virtual Screening. Molecules 2019, 24, 4346. [Google Scholar] [CrossRef]
- Sugimoto, H.; Oda, S.; Otsuki, T.; Hino, T.; Yoshida, T.; Shiro, Y. Crystal structure of human indoleamine 2,3-dioxygenase: Catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Nat. Acad. Sci. USA 2006, 103, 2611–2616. [Google Scholar] [CrossRef]
- Lewis-Ballester, A.; Pham, K.N.; Batabyal, D.; Karkashon, S.; Bonanno, J.B.; Poulos, T.L.; Yeh, S.R. Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat. Commun. 2017, 8, 1693. [Google Scholar] [CrossRef]
- Serafini, M.; Torre, E.; Aprile, S.; Grosso, E.D.; Gesù, A.; Griglio, A.; Colombo, G.; Travelli, C.; Paiella, S.; Adamo, A.; et al. Discovery of Highly Potent Benzimidazole Derivatives as Indoleamine 2,3-Dioxygenase-1 (IDO1) Inhibitors: From Structure-Based Virtual Screening to in Vivo Pharmacodynamic Activity. J. Med. Chem. 2020, 63, 3047–3065. [Google Scholar] [CrossRef]
- Röhrig, U.F.; Michielin, O.; Zoete, V. Structure and Plasticity of Indoleamine 2,3-Dioxygenase 1 (IDO1). J. Med. Chem. 2021, 64, 17690–17705. [Google Scholar] [CrossRef] [PubMed]
- Basran, J.; Efimov, I.; Chauhan, N.; Thackray, S.J.; Krupa, J.L.; Eaton, G.; Griffith, G.A.; Mowat, C.G.; Handa, S.; Raven, E.L. The mechanism of formation of N-formylkynurenine by heme dioxygenases. J. Am. Chem. Soc. 2011, 133, 16251–16257. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.; Ambler, B.R.; Wherritt, D.; Griffith, W.P.; Maldonado, A.C.; Altman, R.A.; Liu, A. Stepwise O-Atom Transfer in Heme-Based Tryptophan Dioxygenase: Role of Substrate Ammonium in Epoxide Ring Opening. J. Am. Chem. Soc. 2018, 140, 4372–4379. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2020, 9, 1777625. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef]
- Kjeldsen, J.W.; Lorentzen, C.L.; Martinenaite, E.; Ellebaek, E.; Donia, M.; Holmstroem, R.B.; Klausen, T.W.; Madsen, C.O.; Ahmed, S.M.; Weis-Banke, S.E.; et al. A phase 1/2 trial of an immune-modulatory vaccine against IDO/PD-L1 in combination with nivolumab in metastatic melanoma. Nat. Med. 2021, 27, 2212–2223. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Zakharia, Y.; McWilliams, R.R.; Rixe, O.; Drabick, J.; Shaheen, M.F.; Grossmann, K.F.; Kolhe, R.; Pacholczyk, R.; Sadek, R.; Tennant, L.L.; et al. Phase II trial of the IDO pathway inhibitor indoximod plus pembrolizumab for the treatment of patients with advanced melanoma. J. Immunother. Cancer 2021, 9, e002057. [Google Scholar] [CrossRef]
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.J.; Hellmann, M.D.; Cervantes, A.; Ochoa de Olza, M.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood-Brain Barrier (BBB) Score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Stafford, P.M.; Stover, K.R.; Mohan, D.C.; Gupta, M.; Keske, E.C.; Schiavini, P.; Villar, L.; Wu, F.; Kreft, A.; et al. A Series of 2-((1-Phenyl-1H-imidazol-5-yl)methyl)-1H-indoles as Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. ChemMedChem 2021, 16, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.M.; Tanabe, A.; Iwahori, Y. A systematic review of the effect of L-tryptophan supplementation on mood and emotional functioning. J. Diet. Suppl. 2021, 18, 316–333. [Google Scholar] [CrossRef] [PubMed]
- Edinoff, A.N.; Akuly, H.A.; Hanna, T.A.; Ochoa, C.O.; Patti, S.J.; Ghaffar, Y.A.; Kaye, A.D.; Viswanath, O.; Urits, I.; Boyer, A.G.; et al. Selective Serotonin Reuptake Inhibitors and Adverse Effects: A Narrative Review. Neurol. Int. 2021, 13, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Ogyu, K.; Kubo, K.; Noda, Y.; Iwata, Y.; Tsugawa, S.; Omura, Y.; Wada, M.; Tarumi, R.; Plitman, E.; Moriguchi, S.; et al. Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2018, 90, 16–25. [Google Scholar] [CrossRef]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef]
- Smith, R.A.; Norris, F.; Palmer, D.; Bernhardt, L.; Wills, R.J. Distribution of alpha interferon in serum and cerebrospinal fluid after systemic administration. Clin. Pharmacol. Ther. 1985, 37, 85–88. [Google Scholar] [CrossRef]
- Greig, N.H.; Fredericks, W.R.; Holloway, H.W.; Soncrant, T.T.; Rapoport, S.I. Delivery of human interferon-alpha to brain by transient osmotic blood-brain barrier modification in the rat. J. Pharmacol. Exp. Ther. 1988, 245, 581–586. [Google Scholar]
- Gál, E.M.; Sherman, A.D. L-kynurenine: Its synthesis and possible regulatory function in brain. Neurochem. Res. 1980, 5, 223–239. [Google Scholar] [CrossRef]
- Connor, T.J.; Starr, N.; O’Sullivan, J.B.; Harkin, A. Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: A role for IFN-gamma? Neurosci. Lett. 2008, 441, 29–34. [Google Scholar] [CrossRef]
- Morimoto, S.S.; Alexopoulos, G.S. Immunity, aging, and geriatric depression. Psychiatr. Clin. N. Am. 2011, 34, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Sanada, F.; Taniyama, Y.; Muratsu, J.; Otsu, R.; Shimizu, H.; Rakugi, H.; Morishita, R. Source of Chronic Inflammation in Aging. Front. Cardiovasc. Med. 2018, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, G.S.; Morimoto, S.S. The inflammation hypothesis in geriatric depression. Int. J. Geriatr. Psychiatry 2011, 26, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Baharikhoob, P.; Kolla, N.J. Microglial Dysregulation and Suicidality: A Stress-Diathesis Perspective. Front. Psychiatry 2020, 11, 781. [Google Scholar] [CrossRef] [PubMed]
- Fenn, A.M.; Gensel, J.C.; Huang, Y.; Popovich, P.G.; Lifshitz, J.; Godbout, J.P. Immune activation promotes depression 1 month after diffuse brain injury: A role for primed microglia. Biol. Psychiatry 2014, 76, 575–584. [Google Scholar] [CrossRef]
- Corona, A.W.; Norden, D.M.; Skendelas, J.P.; Huang, Y.; O’Connor, J.C.; Lawson, M.; Dantzer, R.; Kelley, K.W.; Godbout, J.P. Indoleamine 2,3-dioxygenase inhibition attenuates lipopolysaccharide induced persistent microglial activation and depressive-like complications in fractalkine receptor (CX(3)CR1)-deficient mice. Brain Behav. Immun. 2013, 31, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Hugo, J.; Ganguli, M. Dementia and cognitive impairment: Epidemiology, diagnosis, and treatment. Clin. Geriatr. Med. 2014, 30, 421–442. [Google Scholar] [CrossRef]
- Oh, M.; Weaver, D.F. Alzheimer’s disease as a fundamental disease of information processing systems: An information theory perspective. Front. Neurosci. 2023, 17, 1106623. [Google Scholar] [CrossRef]
- Katsel, P.; Haroutunian, V. Is Alzheimer disease a failure of mobilizing immune defense? Lessons from cognitively fit oldest-old. Dialog. Clin. Neurosci. 2019, 21, 7–19. [Google Scholar] [CrossRef]
- Wang, C.; Yu, J.T.; Miao, D.; Wu, Z.C.; Tan, M.S.; Tan, L. Targeting the mTOR signaling network for Alzheimer’s disease therapy. Mol. Neurobiol. 2014, 49, 120–135. [Google Scholar] [CrossRef]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef]
- Fertan, E.; Stover, K.R.J.; Brant, M.G.; Stafford, P.M.; Kelly, B.; Diez-Cecilia, E.; Wong, A.A.; Weaver, D.F.; Brown, R.E. Effects of the Novel IDO Inhibitor DWG-1036 on the Behavior of Male and Female 3xTg-AD Mice. Front. Pharmacol. 2019, 10, 1044. [Google Scholar] [CrossRef] [PubMed]
- Meier-Stephenson, F.S.; Meier-Stephenson, V.C.; Carter, M.D.; Meek, A.R.; Wang, Y.; Pan, L.; Chen, Q.; Jacobo, S.; Wu, F.; Lu, E.; et al. Alzheimer’s disease as an autoimmune disorder of innate immunity endogenously modulated by tryptophan metabolites. Alzheimers Dement. 2022, 8, e12283. [Google Scholar] [CrossRef] [PubMed]
- Byers, A.L.; Yaffe, K. Depression and risk of developing dementia. Nat. Rev. Neurol. 2011, 7, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Hakim, A. Perspectives on the complex links between depression and dementia. Front. Aging Neurosci. 2022, 14, 821866. [Google Scholar] [CrossRef]
- Bennett, S.; Thomas, A.J. Depression and dementia: Cause, consequence or coincidence? Maturitas 2014, 79, 184–190. [Google Scholar] [CrossRef]
- Gutzmann, H.; Qazi, A. Depression associated with dementia. Z. Gerontol. Geriatr. 2015, 48, 305–311. [Google Scholar] [CrossRef]
- Kepp, K.P.; Robakis, N.K.; Høilund-Carlsen, P.F.; Sensi, S.L.; Vissel, B. The amyloid cascade hypothesis: An updated critical review. Brain 2023, 15, 59. [Google Scholar] [CrossRef]
- Maoz, H. Failure of first SSRI for depression--what is the next step? Isr. J. Psychiatry Relat. Sci. 2007, 44, 327–329. [Google Scholar]
- Li, J.; O, W.; Li, W.; Jiang, Z.G.; Ghanbari, H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guillemin, G.J. Kynurenine pathway metabolites in humans: Disease and healthy States. Int. J. Tryptophan Res. 2009, 2, IJTR-S2097. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, R.K.; Bansal, Y.; Singh, R.; Saroj, P.; Bhandari, R.; Kumar, B.; Kuhad, A. IDO-1 inhibition protects against neuroinflammation, oxidative stress and mitochondrial dysfunction in 6-OHDA induced murine model of Parkinson’s disease. Neurotoxicology 2021, 84, 184–197. [Google Scholar] [CrossRef]
- Bjelobaba, I.; Savic, D.; Lavrnja, I. Multiple Sclerosis and Neuroinflammation: The Overview of Current and Prospective Therapies. Curr. Pharm. Des. 2017, 23, 693–730. [Google Scholar] [CrossRef]
- Naegele, M.; Martin, R. The good and the bad of neuroinflammation in multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 59–87. [Google Scholar] [CrossRef]
- Bar-Or, A.; Li, R. Cellular immunology of relapsing multiple sclerosis: Interactions, checks, and balances. Lancet Neurol. 2021, 20, 470–483. [Google Scholar] [CrossRef]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, H.; Yang, G.; Yang, Y.; Li, W.; Song, M.; Shao, M.; Su, X.; Lv, L. Associations between expression of indoleamine 2, 3-dioxygenase enzyme and inflammatory cytokines in patients with first-episode drug-naive Schizophrenia. Transl. Psychiatry 2021, 11, 595. [Google Scholar] [CrossRef]
- Li, G.; Dai, J.; Liu, H.; Lin, Y.; Liu, Q.; Zheng, K.; Li, S.; Chen, S.; Ye, Y. Association study between genetic variants and the risk of schizophrenia in the Chinese population based on GWAS-implicated 6p21.3-23.1 human genome region: A case-control study. BMC Psychiatry 2021, 21, 483. [Google Scholar] [CrossRef]
- De Picker, L.; Fransen, E.; Coppens, V.; Timmers, M.; de Boer, P.; Oberacher, H.; Fuchs, D.; Verkerk, R.; Sabbe, B.; Morrens, M. Immune and Neuroendocrine Trait and State Markers in Psychotic Illness: Decreased Kynurenines Marking Psychotic Exacerbations. Front. Immunol. 2020, 10, 2971. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.K.; Linderholm, K.R.; Engberg, G.; Paulson, L.; Blennow, K.; Lindström, L.H.; Nordin, C.; Karanti, A.; Persson, P.; Erhardt, S. Elevated levels of kynurenic acid in the cerebrospinal fluid of male patients with schizophrenia. Schizophr. Res. 2005, 80, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Araújo, T.; Maia Chaves Filho, A.J.; Monte, A.S.; de Góis Queiroz, A.; Cordeiro, R.C.; de Jesus Souza Machado, M.; de Freitas Lima, R.; Freitas de Lucena, D.; Maes, M.; Macêdo, D. Reversal of schizophrenia-like symptoms and immune alterations in mice by immunomodulatory drugs. J. Psychiatr. Res. 2017, 84, 49–58. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savonije, K.; Meek, A.; Weaver, D.F. Indoleamine 2,3-Dioxygenase as a Therapeutic Target for Alzheimer’s Disease and Geriatric Depression. Brain Sci. 2023, 13, 852. https://doi.org/10.3390/brainsci13060852
Savonije K, Meek A, Weaver DF. Indoleamine 2,3-Dioxygenase as a Therapeutic Target for Alzheimer’s Disease and Geriatric Depression. Brain Sciences. 2023; 13(6):852. https://doi.org/10.3390/brainsci13060852
Chicago/Turabian StyleSavonije, Karl, Autumn Meek, and Donald F. Weaver. 2023. "Indoleamine 2,3-Dioxygenase as a Therapeutic Target for Alzheimer’s Disease and Geriatric Depression" Brain Sciences 13, no. 6: 852. https://doi.org/10.3390/brainsci13060852
APA StyleSavonije, K., Meek, A., & Weaver, D. F. (2023). Indoleamine 2,3-Dioxygenase as a Therapeutic Target for Alzheimer’s Disease and Geriatric Depression. Brain Sciences, 13(6), 852. https://doi.org/10.3390/brainsci13060852