
A Systematic Review of Extracellular Matrix-Related Alterations in Parkinson’s Disease

Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Transcriptomics Studies
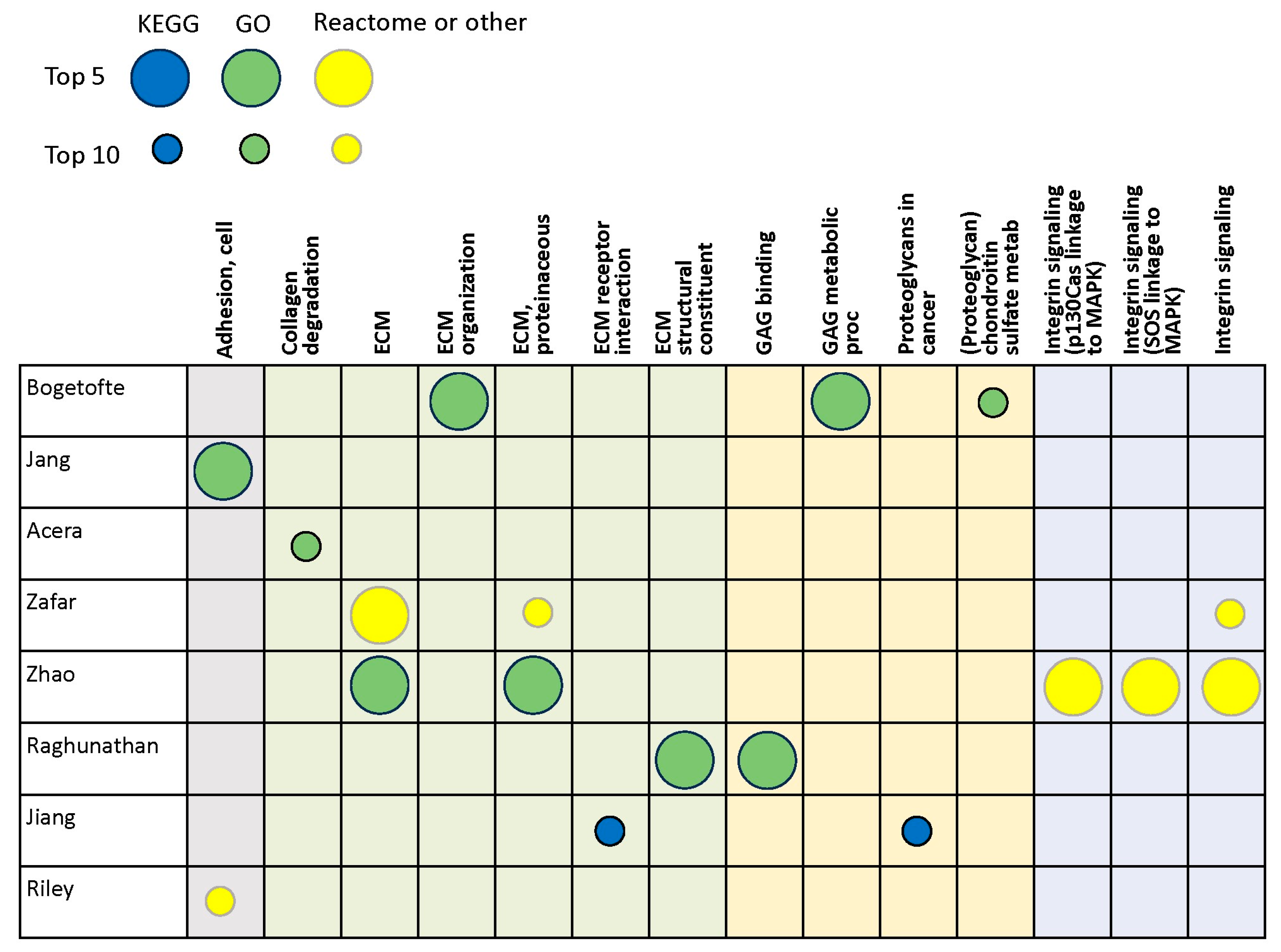
3.2. Proteomics Studies
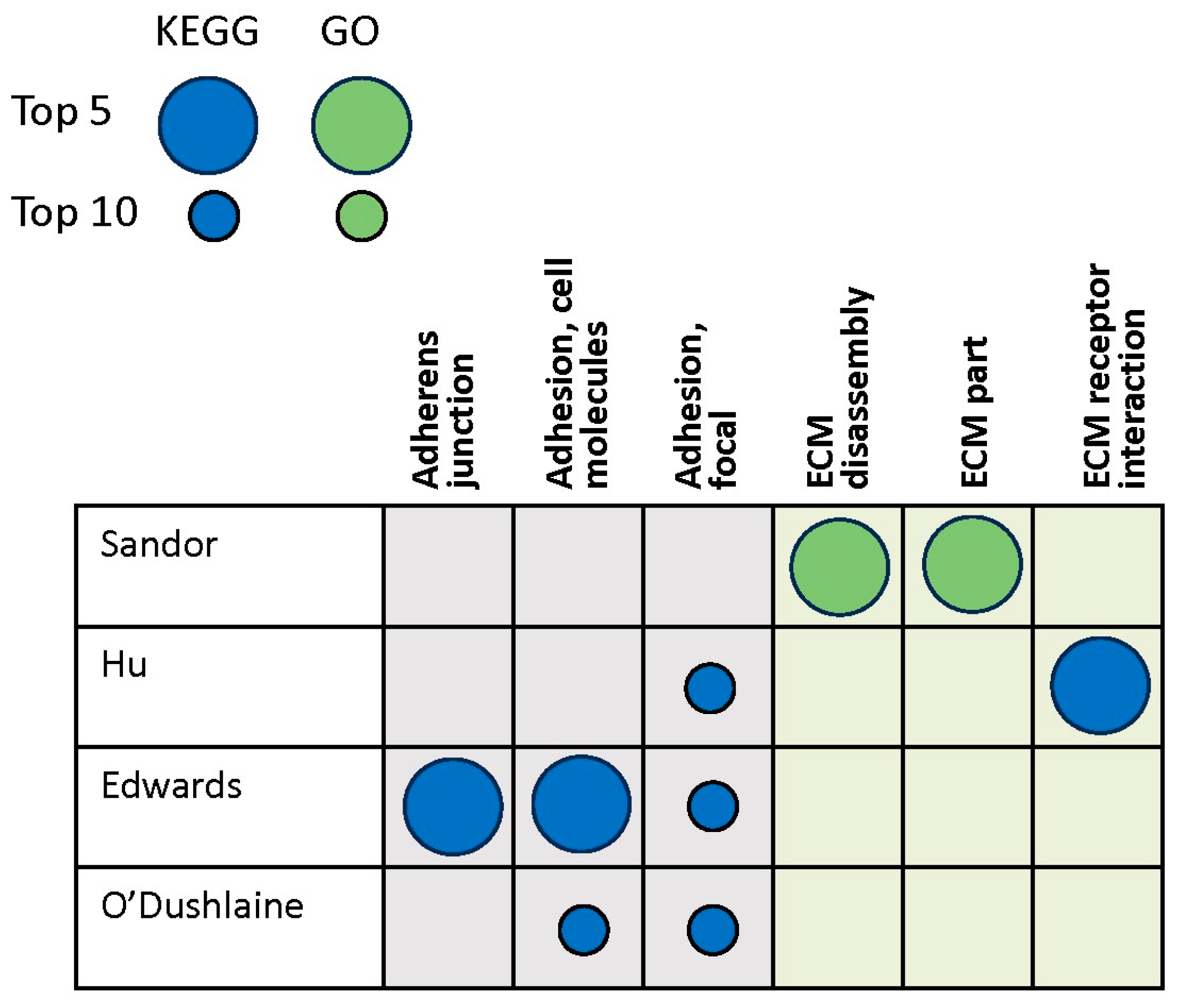
3.3. Genomics Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hynes, R.O.; Naba, A. Overview of the matrisome—An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, N.K.; Theocharis, A.D.; Piperigkou, Z.; Manou, D.; Passi, A.; Skandalis, S.S.; Vynios, D.H.; Orian-Rousseau, V.; Ricard-Blum, S.; Schmelzer, C.E.H.; et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021, 288, 6850–6912. [Google Scholar] [CrossRef] [PubMed]
- Meredith, J.E., Jr.; Fazeli, B.; Schwartz, M.A. The extracellular matrix as a cell survival factor. Mol. Biol. Cell 1993, 4, 953–961. [Google Scholar] [CrossRef]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Yayon, A.; Klagsbrun, M.; Esko, J.D.; Leder, P.; Ornitz, D.M. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991, 64, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.W.; Fisher, C.E.; Perona-Wright, G.; Davies, J.A. Signalling by glial cell line-derived neurotrophic factor (GDNF) requires heparan sulphate glycosaminoglycan. J. Cell Sci. 2002, 115, 4495–4503. [Google Scholar] [CrossRef] [PubMed]
- Bespalov, M.M.; Sidorova, Y.A.; Tumova, S.; Ahonen-Bishopp, A.; Magalhães, A.C.; Kulesskiy, E.; Paveliev, M.; Rivera, C.; Rauvala, H.; Saarma, M. Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J. Cell Biol. 2011, 192, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Sorg, B.A.; Berretta, S.; Blacktop, J.M.; Fawcett, J.W.; Kitagawa, H.; Kwok, J.C.; Miquel, M. Casting a Wide Net: Role of Perineuronal Nets in Neural Plasticity. J. Neurosci. 2016, 36, 11459–11468. [Google Scholar] [CrossRef] [PubMed]
- Chaunsali, L.; Tewari, B.P.; Sontheimer, H. Perineuronal Net Dynamics in the Pathophysiology of Epilepsy. Epilepsy Curr. 2021, 21, 273–281. [Google Scholar] [CrossRef]
- Fawcett, J.W.; Fyhn, M.; Jendelova, P.; Kwok, J.C.F.; Ruzicka, J.; Sorg, B.A. The extracellular matrix and perineuronal nets in memory. Mol. Psychiatry 2022, 27, 3192–3203. [Google Scholar] [CrossRef]
- Fawcett, J.W.; Oohashi, T.; Pizzorusso, T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat. Rev. Neurosci. 2019, 20, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Pizzorusso, T.; Medini, P.; Berardi, N.; Chierzi, S.; Fawcett, J.W.; Maffei, L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science 2002, 298, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Cacquevel, M.; Saksida, L.M.; Bussey, T.J.; Schneider, B.L.; Aebischer, P.; Melani, R.; Pizzorusso, T.; Fawcett, J.W.; Spillantini, M.G. Perineuronal net digestion with chondroitinase restores memory in mice with tau pathology. Exp. Neurol. 2015, 265, 48–58. [Google Scholar] [CrossRef]
- Bruckner, G.; Morawski, M.; Arendt, T. Aggrecan-based extracellular matrix is an integral part of the human basal ganglia circuit. Neuroscience 2008, 151, 489–504. [Google Scholar] [CrossRef]
- Bekku, Y.; Oohashi, T. Neurocan contributes to the molecular heterogeneity of the perinodal ECM. Arch. Histol. Cytol. 2010, 73, 95–102. [Google Scholar] [CrossRef]
- Bonneh-Barkay, D.; Wiley, C.A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009, 19, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Santiago, R.; Esteve-Codina, A.; Fernández, M.; Valldeoriola, F.; Sanchez-Gómez, A.; Muñoz, E.; Compta, Y.; Tolosa, E.; Ezquerra, M.; Martí, M.J. Transcriptome analysis in LRRK2 and idiopathic Parkinson’s disease at different glucose levels. NPJ Parkinsons Dis. 2021, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Raghunathan, R.; Hogan, J.D.; Labadorf, A.; Myers, R.H.; Zaia, J. A glycomics and proteomics study of aging and Parkinson’s disease in human brain. Sci. Rep. 2020, 10, 12804. [Google Scholar] [CrossRef]
- Jung, S.Y.; Choi, J.M.; Rousseaux, M.W.C.; Malovannaya, A.; Kim, J.J.; Kutzera, J.; Wang, Y.; Huang, Y.; Zhu, W.; Maity, S.; et al. An Anatomically Resolved Mouse Brain Proteome Reveals Parkinson Disease-relevant Pathways. Mol. Cell Proteom. 2017, 16, 581–593. [Google Scholar] [CrossRef]
- Villadiego, J.; García-Swinburn, R.; García-González, D.; Lebrón-Galán, R.; Murcia-Belmonte, V.; García-Roldán, E.; Suárez-Luna, N.; Nombela, C.; Marchena, M.; de Castro, F.; et al. Extracellular matrix protein anosmin-1 overexpression alters dopaminergic phenotype in the CNS and the PNS with no pathogenic consequences in a MPTP model of Parkinson’s disease. Brain Struct. Funct. 2023, 228, 907–920. [Google Scholar] [CrossRef]
- Khan, A.H.; Lee, L.K.; Smith, D.J. Single-cell analysis of gene expression in the substantia nigra pars compacta of a pesticide-induced mouse model of Parkinson’s disease. Transl. Neurosci. 2022, 13, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Kim, Y.S.; Bok, E.; Yune, T.Y.; Maeng, S.; Jin, B.K. MMP-3 contributes to nigrostriatal dopaminergic neuronal loss, BBB damage, and neuroinflammation in an MPTP mouse model of Parkinson’s disease. Mediators Inflamm. 2013, 2013, 370526. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Joh, T.H. Matrix metalloproteinases, new insights into the understanding of neurodegenerative disorders. Biomol. Ther. 2012, 20, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Annese, V.; Herrero, M.-T.; Di Pentima, M.; Gomez, A.; Lombardi, L.; Ros, C.M.; De Pablos, V.; Fernandez-Villalba, E.; De Stefano, M.E. Metalloproteinase-9 contributes to inflammatory glia activation and nigro-striatal pathway degeneration in both mouse and monkey models of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. Brain Struct. Funct. 2015, 220, 703–727. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, G.; Sehgal, A.; Bhardwaj, S.; Singh, S.; Buhas, C.; Judea-Pusta, C.; Uivarosan, D.; Munteanu, M.A.; Bungau, S. Multifaceted Role of Matrix Metalloproteinases in Neurodegenerative Diseases: Pathophysiological and Therapeutic Perspectives. Int. J. Mol. Sci. 2021, 22, 1413. [Google Scholar] [CrossRef] [PubMed]
- Rosh, I.; Tripathi, U.; Hussein, Y.; Rike, W.A.; Djamus, J.; Shklyar, B.; Manole, A.; Houlden, H.; Winkler, J.; Gage, F.H.; et al. Synaptic dysfunction and extracellular matrix dysregulation in dopaminergic neurons from sporadic and E326K-GBA1 Parkinson’s disease patients. NPJ Parkinsons Dis. 2024, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Akrioti, E.; Karamitros, T.; Gkaravelas, P.; Kouroupi, G.; Matsas, R.; Taoufik, E. Early Signs of Molecular Defects in iPSC-Derived Neural Stems Cells from Patients with Familial Parkinson’s Disease. Biomolecules 2022, 12, 876. [Google Scholar] [CrossRef] [PubMed]
- Stern, S.; Lau, S.; Manole, A.; Rosh, I.; Percia, M.M.; Ben Ezer, R.; Shokhirev, M.N.; Qiu, F.; Schafer, S.; Mansour, A.A.; et al. Reduced synaptic activity and dysregulated extracellular matrix pathways in midbrain neurons from Parkinson’s disease patients. NPJ Parkinsons Dis. 2022, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, S.M.J.; Swart, P.; Womersely, J.S.; Ovenden, E.S.; van den Heuvel, L.L.; McGregor, N.W.; Meier, S.; Bardien, S.; Abrahams, S.; Tromp, G.; et al. RNA-seq analysis of gene expression profiles in posttraumatic stress disorder, Parkinson’s disease and schizophrenia identifies roles for common and distinct biological pathways. Discov. Ment. Health 2022, 2, 6. [Google Scholar] [CrossRef]
- Cho, E.; Park, J.; Kim, K.; Kim, M.G.; Cho, S.R. Reelin Alleviates Mesenchymal Stem Cell Senescence and Reduces Pathological alpha-Synuclein Expression in an In Vitro Model of Parkinson’s Disease. Genes 2021, 12, 1066. [Google Scholar] [CrossRef]
- Yang, M.; Wu, X.Q.; Ding, C.B.; Zhang, G.F.; Li, M.; Lv, L.N.; Li, Y.H.; Sun, D.W.; Zhao, J.J. Weighted gene co-expression network analysis identifies specific modules and hub genes related to Parkinson’s disease. Neuroreport 2021, 32, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Wessely, F.; Connor-Robson, N.; Rinaldi, F.; Vowles, J.; Browne, C.; Evetts, S.G.; Hu, M.T.; Cowley, S.A.; Webber, C.; et al. RNA sequencing reveals MMP2 and TGFB1 downregulation in LRRK2 G2019S Parkinson’s iPSC-derived astrocytes. Neurobiol. Dis. 2019, 129, 56–66. [Google Scholar] [CrossRef] [PubMed]
- González-Casacuberta, I.; Moren, C.; Juarez-Flores, D.L.; Esteve-Codina, A.; Sierra, C.; Catalan-Garcia, M.; Guitart-Mampel, M.; Tobias, E.; Milisenda, J.C.; Pont-Sunyer, C.; et al. Transcriptional alterations in skin fibroblasts from Parkinson’s disease patients with parkin mutations. Neurobiol. Aging 2018, 65, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Liu, X.; Chen, J. Microarray Analysis of the Molecular Mechanism Involved in Parkinson’s Disease. Parkinsons Dis. 2018, 2018, 1590465. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.; Prieto, C.; Sierra, M.; Sanchez-Juan, P.; Gonzalez-Aramburu, I.; Sanchez-Quintana, C.; Berciano, J.; Combarros, O.; Sainz, J. Comparative blood transcriptome analysis in idiopathic and LRRK2 G2019S-associated Parkinson’s disease. Neurobiol. Aging 2016, 38, 214.e1–214.e5. [Google Scholar] [CrossRef] [PubMed]
- Riley, B.E.; Gardai, S.J.; Emig-Agius, D.; Bessarabova, M.; Ivliev, A.E.; Schule, B.; Alexander, J.; Wallace, W.; Halliday, G.M.; Langston, J.W.; et al. Systems-based analyses of brain regions functionally impacted in Parkinson’s disease reveals underlying causal mechanisms. PLoS ONE 2014, 9, e102909. [Google Scholar] [CrossRef] [PubMed]
- Durrenberger, P.F.; Grunblatt, E.; Fernando, F.S.; Monoranu, C.M.; Evans, J.; Riederer, P.; Reynolds, R.; Dexter, D.T. Inflammatory Pathways in Parkinson’s Disease; A BNE Microarray Study. Parkinsons Dis. 2012, 2012, 214714. [Google Scholar] [CrossRef]
- Zhang, B.; Xia, C.; Lin, Q.; Huang, J. Identification of key pathways and transcription factors related to Parkinson disease in genome wide. Mol. Biol. Rep. 2012, 39, 10881–10887. [Google Scholar] [CrossRef] [PubMed]
- Edwards, Y.J.; Beecham, G.W.; Scott, W.K.; Khuri, S.; Bademci, G.; Tekin, D.; Martin, E.R.; Jiang, Z.; Mash, D.C.; Ffrench-Mullen, J.; et al. Identifying consensus disease pathways in Parkinson’s disease using an integrative systems biology approach. PLoS ONE 2011, 6, e16917. [Google Scholar] [CrossRef]
- Grunblatt, E.; Mandel, S.; Jacob-Hirsch, J.; Zeligson, S.; Amariglo, N.; Rechavi, G.; Li, J.; Ravid, R.; Roggendorf, W.; Riederer, P.; et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J. Neural Transm. 2004, 111, 1543–1573. [Google Scholar] [CrossRef]
- Mandel, S.; Grunblatt, E.; Riederer, P.; Amariglio, N.; Jacob-Hirsch, J.; Rechavi, G.; Youdim, M.B. Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann. N. Y. Acad. Sci. 2005, 1053, 356–375. [Google Scholar] [PubMed]
- Bogetofte, H.; Ryan, B.J.; Jensen, P.; Schmidt, S.I.; Vergoossen, D.L.E.; Barnkob, M.B.; Kiani, L.N.; Chughtai, U.; Heon-Roberts, R.; Caiazza, M.C.; et al. Post-translational proteomics platform identifies neurite outgrowth impairments in Parkinson’s disease GBA-N370S dopamine neurons. Cell Rep. 2023, 42, 112180. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Pletnikova, O.; Troncoso, J.C.; Pantelyat, A.Y.; Dawson, T.M.; Rosenthal, L.S.; Na, C.H. Mass Spectrometry-Based Proteomics Analysis of Human Substantia Nigra From Parkinson’s Disease Patients Identifies Multiple Pathways Potentially Involved in the Disease. Mol. Cell Proteom. 2023, 22, 100452. [Google Scholar]
- Acera, A.; Gomez-Esteban, J.C.; Murueta-Goyena, A.; Galdos, M.; Azkargorta, M.; Elortza, F.; Ruzafa, N.; Ibarrondo, O.; Pereiro, X.; Vecino, E. Potential Tear Biomarkers for the Diagnosis of Parkinson’s Disease-A Pilot Study. Proteomes 2022, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Zafar, S.; Noor, A.; Younas, N.; Shafiq, M.; Schmitz, M.; Wurster, I.; Brockmann, K.; Gasser, T.; Zerr, I. SWATH Mass Spectrometry-Based CSF Proteome Profile of GBA-Linked Parkinson’s Disease Patients. Int. J. Mol. Sci. 2022, 23, 14166. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, Y.; Zhang, J.; Yang, G. Plasma proteome profiling using tandem mass tag labeling technology reveals potential biomarkers for Parkinson’s disease: A preliminary study. Proteom. Clin. Appl. 2022, 16, e2100010. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Rong, C.; Ke, R.; Meng, S.; Yan, X.; Ke, H.; Wu, S. Differential proteomic analysis of serum exosomes reveals alterations in progression of Parkinson disease. Medicine 2019, 98, e17478. [Google Scholar] [CrossRef] [PubMed]
- Sandor, C.; Honti, F.; Haerty, W.; Szewczyk-Krolikowski, K.; Tomlinson, P.; Evetts, S.; Millin, S.; Keane, T.; McCarthy, S.A.; Durbin, R.; et al. Whole-exome sequencing of 228 patients with sporadic Parkinson’s disease. Sci. Rep. 2017, 7, 41188. [Google Scholar] [CrossRef]
- Hu, Y.; Deng, L.; Zhang, J.; Fang, X.; Mei, P.; Cao, X.; Lin, J.; Wei, Y.; Zhang, X.; Xu, R. A Pooling Genome-Wide Association Study Combining a Pathway Analysis for Typical Sporadic Parkinson’s Disease in the Han Population of Chinese Mainland. Mol. Neurobiol. 2016, 53, 4302–4318. [Google Scholar] [CrossRef]
- O’Dushlaine, C.; Kenny, E.; Heron, E.A.; Segurado, R.; Gill, M.; Morris, D.W.; Corvin, A. The SNP ratio test: Pathway analysis of genome-wide association datasets. Bioinformatics 2009, 25, 2762–2763. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef]
- Jellinger, K.A. Synuclein deposition and non-motor symptoms in Parkinson disease. J. Neurol. Sci. 2011, 310, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Bang, D.; Lim, S.; Lee, S.; Kim, S. Biomedical knowledge graph learning for drug repurposing by extending guilt-by-association to multiple layers. Nat. Commun. 2023, 14, 3570. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.N.; Chew, E.G.Y.; Chung, S.J.; Peng, R.; Blauwendraat, C.; Nalls, M.A.; Mok, K.Y.; Satake, W.; Toda, T.; Chao, Y.; et al. Identification of Risk Loci for Parkinson Disease in Asians and Comparison of Risk Between Asians and Europeans: A Genome-Wide Association Study. JAMA Neurol. 2020, 77, 746–754. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.S.; Sawalha, A.H. Glycoprotein nonmetastatic melanoma protein B: A key mediator and an emerging therapeutic target in autoimmune diseases. FASEB J. 2020, 34, 8810–8823. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, U.; Rosh, I.; Ben Ezer, R.; Nayak, R.; Chowdhary, A.; Djamus, J.; Manole, A.; Haulden, H.; Gage, F.H.; Stern, S. Upregulated extracellular matrix-related genes and impaired synaptic activity in dopaminergic and hippocampal neurons derived from Parkinson’s disease patients with PINK1 and PARK2 mutations. bioRxiv 2022. [Google Scholar] [CrossRef]
- Rosh, I.; Tripathi, U.; Hussein, Y.; Rike, W.A.; Manole, A.; Cordeiro, D.; Houlden, H.; Winkler, J.; Gage, F.; Stern, S. Synaptic dysfunction and dysregulation of extracellular matrix-related genes in dopaminergic neurons derived from Parkinson’s disease sporadic patients and with GBA1 mutations. bioRxiv 2023. [Google Scholar] [CrossRef]
- Rike, W.A.; Stern, S. Proteins and Transcriptional Dysregulation of the Brain Extracellular Matrix in Parkinson’s Disease: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 7435. [Google Scholar] [CrossRef]
- Teves, J.M.Y.; Bhargava, V.; Kirwan, K.R.; Corenblum, M.J.; Justiniano, R.; Wondrak, G.T.; Anandhan, A.; Flores, A.J.; Schipper, D.A.; Khalpey, Z.; et al. Parkinson’s Disease Skin Fibroblasts Display Signature Alterations in Growth, Redox Homeostasis, Mitochondrial Function, and Autophagy. Front. Neurosci. 2017, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Browser. Extracelullar Matrix Organization. Available online: https://www.informatics.jax.org/vocab/gene_ontology/GO:0030198# (accessed on 15 October 2023).
- Liu, C.Z.; Guo, D.S.; Ma, J.J.; Dong, L.R.; Chang, Q.Q.; Yang, H.Q.; Liang, K.K.; Li, X.H.; Yang, D.W.; Fan, Y.Y.; et al. Correlation of matrix metalloproteinase 3 and matrix metalloproteinase 9 levels with non-motor symptoms in patients with Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 889257. [Google Scholar] [CrossRef] [PubMed]
- Downs, M.; Sethi, M.K.; Raghunathan, R.; Layne, M.D.; Zaia, J. Matrisome changes in Parkinson’s disease. Anal. Bioanal. Chem. 2022, 414, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.; Aroso, M.; Rocha, S.; Ferreira, R.; Vitorino, R.; Gomez-Lazaro, M. Bioinformatic analysis of the human brain extracellular matrix proteome in neurodegenerative disorders. Eur. J. Neurosci. 2021, 53, 4016–4033. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Singh, M.K.; Garg, R.K.; Pant, K.K.; Khattri, S. Evaluation of peripheral matrix metalloproteinase-1 in Parkinson’s disease: A case-control study. Int. J. Neurosci. 2014, 124, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; De Jong, G.I.; de Vos, R.A.; Jansen Steur, E.N.; Luiten, P.G. Pathological features of cerebral cortical capillaries are doubled in Alzheimer’s disease and Parkinson’s disease. Acta Neuropathol. 2000, 100, 395–402. [Google Scholar] [CrossRef]
- Downs, M.; Zaia, J.; Sethi, M.K. Mass spectrometry methods for analysis of extracellular matrix components in neurological diseases. Mass. Spectrom. Rev. 2023, 42, 1848–1875. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.; Pandini, C.; Messa, L.; Launi, R.; Barzaghini, B.; Zangaglia, R.; Raimondi, M.T.; Gagliardi, S.; Cereda, C.; Zuccotti, G.V.; et al. alpha-Synuclein antisense transcript SNCA-AS1 regulates synapses- and aging-related genes suggesting its implication in Parkinson’s disease. Aging Cell 2021, 20, e13504. [Google Scholar] [CrossRef]
- Huang, Y.; Wen, D.; Yuan, Y.; Chen, W. Gene Set Enrichment Analysis and Genetic Experiment Reveal Changes in Cell Signaling Pathways Induced by alpha-Synuclein Overexpression. Biomedicines 2023, 11, 263. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. alpha-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Riederer, P.; Berg, D.; Casadei, N.; Cheng, F.; Classen, J.; Dresel, C.; Jost, W.; Krüger, R.; Müller, T.; Reichmann, H.; et al. alpha-Synuclein in Parkinson’s disease: Causal or bystander? J. Neural Transm. 2019, 126, 815–840. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Chalapathi, A.V.; Balagurunathan, K. Investigating the Roles of Heparan Sulfate Structures in Alpha-Synuclein Aggregation in Cell Culture Models. Methods Mol. Biol. 2022, 2303, 807–820. [Google Scholar] [PubMed]
- Mehra, S.; Ghoshn, D.; Kumar, R.; Mondal, M.; Gadhe, L.G.; Das, S.; Anoop, A.; Jha, N.N.; Jacob, R.S.; Chatterjee, D.; et al. Glycosaminoglycans have variable effects on alpha-synuclein aggregation and differentially affect the activities of the resulting amyloid fibrils. J. Biol. Chem. 2018, 293, 12975–12991. [Google Scholar] [CrossRef] [PubMed]
- Maiza, A.; Chantepie, S.; Vera, C.; Fifre, A.; Huynh, M.B.; Stettler, O.; Ouidja, M.O.; Papy-Garcia, D. The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. 2018, 592, 3806–3818. [Google Scholar] [CrossRef] [PubMed]
- Ihse, E.; Yamakado, H.; van Wijk, X.M.; Lawrence, R.; Esko, J.D.; Masliah, E. Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci. Rep. 2017, 7, 9008. [Google Scholar] [CrossRef] [PubMed]
- Adulla, A.; Patel, U.; Ashok, A.; Katiyar, P.; Kaulakis, M.; Kritikos, A.E.; Pillai, S.; Lee, H.; Lindner, E.; Rhee, D.J.; et al. Alpha-Synuclein modulates fibronectin expression in the trabecular meshwork independent of TGFbeta2. Exp. Eye Res. 2023, 226, 109351. [Google Scholar] [CrossRef] [PubMed]
- Paiva, I.; Jain, G.; Lazaro, D.F.; Jercic, K.G.; Hentrich, T.; Kerimoglu, C.; Pinho, R.; Szegő, M.; Burkhardt, S.; Capece, V.; et al. Alpha-synuclein deregulates the expression of COL4A2 and impairs ER-Golgi function. Neurobiol. Dis. 2018, 119, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Cohlberg, J.A.; Li, J.; Uversky, V.N.; Fink, A.L. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 2002, 41, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Gao, J.; Hou, L.; Wang, L.; Zhang, F.; Sun, F.; Zhang, T.; Xu, P.; Shi, Z.; Hu, F.; et al. Neuroprotective effect of chondroitin sulfate on SH-SY5Y cells overexpressing wild-type or A53T mutant alpha-synuclein. Mol. Med. Rep. 2017, 16, 8721–8728. [Google Scholar] [CrossRef]
- Tsuboi, K.; Grzesiak, J.J.; Bouvet, M.; Hashimoto, M.; Masliah, E.; Shults, C.W. Alpha-synuclein overexpression in oligodendrocytic cells results in impaired adhesion to fibronectin and cell death. Mol. Cell Neurosci. 2005, 29, 259–268. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef]
- Yamada, K.; Iwatsubo, T. Extracellular alpha-synuclein levels are regulated by neuronal activity. Mol. Neurodegener. 2018, 13, 9. [Google Scholar] [CrossRef]
- Ferreira, S.A.; Romero-Ramos, M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front. Cell Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef]
- Surguchev, A.A.; Surguchov, A. Integrins-A missing link in synuclein’s pathogenic mechanism. J. Neurosci. Res. 2019, 97, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates alpha-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- DeWitt, D.A.; Richey, P.L.; Praprotnik, D.; Silver, J.; Perry, G. Chondroitin sulfate proteoglycans are a common component of neuronal inclusions and astrocytic reaction in neurodegenerative diseases. Brain Res. 1994, 656, 205–209. [Google Scholar] [CrossRef]
- Lehri-Boufala, S.; Ouidja, M.O.; Barbier-Chassefiere, V.; Henault, E.; Raisman-Vozari, R.; Garrigue-Antar, L.; Papy-Garcia, D.; Morin, C. New roles of glycosaminoglycans in alpha-synuclein aggregation in a cellular model of Parkinson disease. PLoS ONE 2015, 10, e0116641. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Sun, Y.; Lv, S.; Xia, W.; Zhao, K.; Xu, Q.; Zhao, Q.; He, L.; Le, W.; Wang, Y.; et al. Heparin induces alpha-synuclein to form new fibril polymorphs with attenuated neuropathology. Nat. Commun. 2022, 13, 4226. [Google Scholar] [CrossRef]
- Nikolaus, S.; Antke, C.; Muller, H.W. In vivo imaging of synaptic function in the central nervous system: I. Movement disorders and dementia. Behav. Brain Res. 2009, 204, 1–31. [Google Scholar] [CrossRef]
- Sousa, V.L.; Bellani, S.; Giannandrea, M.; Yousuf, M.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. {alpha}-synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol. Biol. Cell 2009, 20, 3725–3739. [Google Scholar] [CrossRef] [PubMed]
- Dankovich, T.M.; Rizzoli, S.O. The Synaptic Extracellular Matrix: Long-Lived, Stable, and Still Remarkably Dynamic. Front. Synaptic Neurosci. 2022, 14, 854956. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, N.; Klingenberg, S.; Schweiger, S.; Gerber, S. Common Factors in Neurodegeneration: A Meta-Study Revealing Shared Patterns on a Multi-Omics Scale. Cells 2020, 9, 2642. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nie, Y.; Yu, J. An Effective Method to Identify Shared Pathways and Common Factors among Neurodegenerative Diseases. PLoS ONE 2015, 10, e0143045. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sakaguchi, M.; Sabit, H.; Tamai, S.; Ichinose, T.; Tanaka, S.; Kinoshita, M.; Uchida, Y.; Ohtsuki, S.; Nakada, M. COL1A2 inhibition suppresses glioblastoma cell proliferation and invasion. J. Neurosurg. 2023, 138, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.; Keon, M.; Liu, B.; Su, Z.; Saksena, N.K. Panoramic Visualization of Circulating MicroRNAs Across Neurodegenerative Diseases in Humans. Mol. Neurobiol. 2019, 56, 7380–7407. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C. Biochemistry of Parkinson’s disease with special reference to the dopaminergic systems. Mol. Neurobiol. 1994, 9, 135–142. [Google Scholar] [CrossRef]
- Aguila, J.; Cheng, S.; Kee, N.; Cao, M.; Wang, M.; Deng, Q.; Hedlund, E. Spatial RNA Sequencing Identifies Robust Markers of Vulnerable and Resistant Human Midbrain Dopamine Neurons and Their Expression in Parkinson’s Disease. Front. Mol. Neurosci. 2021, 14, 699562. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef]
- Thalhammer, A.; Cingolani, L.A. Cell adhesion and homeostatic synaptic plasticity. Neuropharmacology 2014, 78, 23–30. [Google Scholar] [CrossRef]
- Dityatev, A.; Seidenbecher, C.I.; Schachner, M. Compartmentalization from the outside: The extracellular matrix and functional microdomains in the brain. Trends Neurosci. 2010, 33, 503–512. [Google Scholar] [CrossRef]
- Soles, A.; Selimovic, A.; Sbrocco, K.; Ghannoum, F.; Hamel, K.; Moncada, E.L.; Gilliat, S.; Cvetanovic, M. Extracellular Matrix Regulation in Physiology and in Brain Disease. Int. J. Mol. Sci. 2023, 24, 7049. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Plowey, E.D.; Chu, C.T. Synaptic dysfunction in genetic models of Parkinson’s disease: A role for autophagy? Neurobiol. Dis. 2011, 43, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Ng, X.Y.; Cao, M. Dysfunction of synaptic endocytic trafficking in Parkinson’s disease. Neural Regen. Res. 2024, 19, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Dityatev, A.; Schachner, M.; Sonderegger, P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 2010, 11, 735–746. [Google Scholar] [CrossRef]
- Mehdi, S.J.; Rosas-Hernandez, H.; Cuevas, E.; Lantz, S.M.; Barger, S.W.; Sarkar, S.; Paule, M.G.; Ali, S.F.; Imam, S.Z. Protein Kinases and Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 1585. [Google Scholar] [CrossRef] [PubMed]
- Kingwell, K. LRRK2-targeted Parkinson disease drug advances into phase III. Nat. Rev. Drug Discov. 2023, 22, 3–5. [Google Scholar] [CrossRef]
- Chapman, M.A. Interactions between cell adhesion and the synaptic vesicle cycle in Parkinson’s disease. Med. Hypotheses 2014, 83, 203–207. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, L.; Liu, H.; Yang, J.; Yu, F.; Cui, C.; Huang, D. A Diagnostic Model for Parkinson’s Disease Based on Anoikis-Related Genes. Mol. Neurobiol. 2024, 61, 3641–3656. [Google Scholar] [CrossRef]
- Pantaleo, E.; Monaco, A.; Amoroso, N.; Lombardi, A.; Bellantuono, L.; Urso, D.; Giudice, C.L.; Picardi, E.; Tafuri, B.; Nigro, S.; et al. A Machine Learning Approach to Parkinson’s Disease Blood Transcriptomics. Genes 2022, 13, 727. [Google Scholar] [CrossRef]
- Uehara, Y.; Ueno, S.I.; Amano-Takeshige, H.; Suzuki, S.; Imamichi, Y.; Fujimaki, M.; Ota, N.; Murase, T.; Inoue, T.; Saiki, S.; et al. Non-invasive diagnostic tool for Parkinson’s disease by sebum RNA profile with machine learning. Sci. Rep. 2021, 11, 18550. [Google Scholar] [CrossRef]
- Odumpatta, R.; Arumugam, M. Integrative Analysis of Gene Expression and Regulatory Network Interaction Data Reveals the Protein Kinase C Family of Serine/Threonine Receptors as a Significant Druggable Target for Parkinson’s Disease. J. Mol. Neurosci. 2021, 71, 466–480. [Google Scholar] [CrossRef]
- Hu, L.; Dong, M.X.; Huang, Y.L.; Lu, C.Q.; Qian, Q.; Zhang, C.C.; Xu, X.M.; Liu, Y.; Chen, G.H.; Wei, Y.D. Integrated Metabolomics and Proteomics Analysis Reveals Plasma Lipid Metabolic Disturbance in Patients With Parkinson’s Disease. Front. Mol. Neurosci. 2020, 13, 80. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhang, S.; Li, J.; Xiao, J.; Li, X.; Yang, J.; Lu, D.; Wang, Y. Comprehensive analysis of core genes and key pathways in Parkinson’s disease. Am. J. Transl. Res. 2020, 12, 5630–5639. [Google Scholar]
- Moni, M.A.; Rana, H.K.; Islam, M.B.; Ahmed, M.B.; Xu, H.; Hasan, M.A.M.; Lei, Y.; Quinn, J.M. A computational approach to identify blood cell-expressed Parkinson’s disease biomarkers that are coordinately expressed in brain tissue. Comput. Biol. Med. 2019, 113, 103385. [Google Scholar] [CrossRef]
- Dong, N.; Zhang, X.; Liu, Q. Identification of therapeutic targets for Parkinson’s disease via bioinformatics analysis. Mol. Med. Rep. 2017, 15, 731–735. [Google Scholar] [CrossRef]
- Rezaei-Tavirani, M.; Zamanian-Azodi, M.; Rajabi, S.; Masoudi-Nejad, A.; Rostami-Nejad, M.; Rahmatirad, S. Protein Clustering and Interactome Analysis in Parkinson and Alzheimer’s Diseases. Arch. Iran. Med. 2016, 19, 101–109. [Google Scholar]
- Dumitriu, A.; Golji, J.; Labadorf, A.T.; Gao, B.; Beach, T.G.; Myers, R.H.; Longo, K.A.; Latourelle, J.C. Integrative analyses of proteomics and RNA transcriptomics implicate mitochondrial processes, protein folding pathways and GWAS loci in Parkinson disease. BMC Med. Genom. 2016, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, R.; Banica, M.; Roncaglia, P.; Robotti, E.; Finaurini, S.; Vlachouli, C.; Antonutti, L.; Iorio, F.; Carissimo, A.; Cattaruzza, T.; et al. Blood transcriptomics of drug-naive sporadic Parkinson’s disease patients. BMC Genom. 2015, 16, 876. [Google Scholar] [CrossRef] [PubMed]
- Hao, B.; Chen, X.; Dai, D.; Zou, C.; Wu, X.; Chen, J. Bioinformatic analysis of microRNA expression in Parkinson’s disease. Mol. Med. Rep. 2015, 11, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Licker, V.; Cote, M.; Lobrinus, J.A.; Rodrigo, N.; Kovari, E.; Hochstrasser, D.F.; Turck, N.; Sanchez, J.C.; Burkhard, P.R. Proteomic profiling of the substantia nigra demonstrates CNDP2 overexpression in Parkinson’s disease. J. Proteom. 2012, 75, 4656–4667. [Google Scholar] [CrossRef] [PubMed]
- Botta-Orfila, T.; Sanchez-Pla, A.; Fernandez, M.; Carmona, F.; Ezquerra, M.; Tolosa, E. Brain transcriptomic profiling in idiopathic and LRRK2-associated Parkinson’s disease. Brain Res. 2012, 1466, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Jin, J.; Wang, Y.; Beyer, R.P.; Kitsou, E.; Albin, R.L.; Gearing, M.; Pan, C.; Zhang, J. Mortalin: A protein associated with progression of Parkinson disease? J. Neuropathol. Exp. Neurol. 2008, 67, 117–124. [Google Scholar] [CrossRef]
- Ferraro, F.; Fevga, C.; Bonifati, V.; Mandemakers, W.; Mahfouz, A.; Reinders, M. Correcting Differential Gene Expression Analysis for Cyto-Architectural Alterations in Substantia Nigra of Parkinson’s Disease Patients Reveals Known and Potential Novel Disease-Associated Genes and Pathways. Cells 2022, 11, 198. [Google Scholar] [CrossRef] [PubMed]
- Kurvits, L.; Lattekivi, F.; Reimann, E.; Kadastik-Eerme, L.; Kasterpalu, K.M.; Koks, S.; Taba, P.; Planken, A. Transcriptomic profiles in Parkinson’s disease. Exp. Biol. Med. 2021, 246, 584–595. [Google Scholar] [CrossRef]
- Licker, V.; Turck, N.; Kovari, E.; Burkhardt, K.; Cote, M.; Surini-Demiri, M.; Lobrinus, J.A.; Sanchez, J.C.; Burkhard, P.R. Proteomic analysis of human substantia nigra identifies novel candidates involved in Parkinson’s disease pathogenesis. Proteomics 2014, 14, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Cerdan, A.; Andreu, Z.; Hidalgo, M.R.; Grillo-Risco, R.; Catala-Senent, J.F.; Soler-Saez, I.; Neva-Alejo, A.; Gordillo, F.; de la Iglesia-Vayá, M.; García-García, F. Unveiling sex-based differences in Parkinson’s disease: A comprehensive meta-analysis of transcriptomic studies. Biol. Sex. Differ. 2022, 13, 68. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Vialle, R.A.; Navarro, E.; Udine, E.; de Paiva Lopes, K.; Humphrey, J.; Allan, A.; Parks, M.; Henderson, B.; Astudillo, K.; et al. Transcriptome deregulation of peripheral monocytes and whole blood in GBA-related Parkinson’s disease. Mol. Neurodegener. 2022, 17, 52. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors, Year | Tissue | Method |
|---|---|---|
| Transcriptomics Studies | ||
| Rosh et al., 2024 [26] | iPSC-derived DA neurons | RNA sequencing |
| Akrioti et al., 2022 [27] | iPSC-derived neural progenitor cells | RNA sequencing |
| Stern et al., 2022 [28] | iPSC-derived DA neurons | RNA sequencing |
| Hemmings et al., 2022 [29] | Blood | RNA sequencing |
| Cho et al., 2021 [30] | MSCs from adipose tissue | RNA sequencing |
| Fernandez-Santiago et al., 2021 [17] | Dermal fibroblasts | RNA sequencing |
| Yang et al., 2021 [31] | Blood | RNA sequencing |
| Booth et al., 2019 [32] | iPSC-derived, midbrain-patterned astrocytes generated from skin | RNA sequencing |
| Gonzalez-Cascuberta et al., 2018 [33] | Dermal fibroblasts | RNA sequencing |
| Tan et al., 2018 [34] | Blood | Microarray |
| Infante et al., 2016 [35] | Whole blood | RNA sequencing |
| Riley et al., 2014 [36] | Substantia nigra, striatum, cortex | Microarray |
| Durrenberger et al., 2012 [37] | Substantia nigra | Microarray |
| Zhang et al., 2012 [38] | Substantia nigra | Microarray |
| Edwards et al., 2011 [39] | Dorsal motor nucleus of vagus, locus coeruleus, substantia nigra, putamen, insula | Microarray |
| Grunblatt et al., 2004 [40]; Mandel et al., 2005 [41] | Substantia nigra, pars compacta | Microarray |
| Proteomics studies | ||
| Bogetofte et al., 2023 [42] | iPSC-derived DA neurons | LC/MS |
| Jang et al., 2023 [43] | Substantia nigra | LC/MS |
| Acera et al., 2022 [44] | Tears | LC/MS |
| Zafar et al., 2022 [45] | Cerebrospinal fluid | LC/MS |
| Zhao et al., 2022 [46] | Plasma | LC/MS |
| Raghunathan et al., 2020 [18] | Prefrontal cortex | LC/MS |
| Jiang et al., 2019 [47] | Serum exosomes | LC/MS |
| Riley et al., 2014 [36] | Striatum, cortex | LC/MS |
| Genomics studies | ||
| Sandor et al., 2017 [48] | Not reported | Whole exome sequencing |
| Hu et al., 2016 [49] | White blood cells | GWAS |
| Edwards et al., 2011 [39] | Not reported | GWAS |
| O’Dushlaine et al., 2009 [50] | Not reported | GWAS/SNP ratio test |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chapman, M.A.; Sorg, B.A. A Systematic Review of Extracellular Matrix-Related Alterations in Parkinson’s Disease. Brain Sci. 2024, 14, 522. https://doi.org/10.3390/brainsci14060522
Chapman MA, Sorg BA. A Systematic Review of Extracellular Matrix-Related Alterations in Parkinson’s Disease. Brain Sciences. 2024; 14(6):522. https://doi.org/10.3390/brainsci14060522
Chicago/Turabian StyleChapman, Mary Ann, and Barbara A. Sorg. 2024. "A Systematic Review of Extracellular Matrix-Related Alterations in Parkinson’s Disease" Brain Sciences 14, no. 6: 522. https://doi.org/10.3390/brainsci14060522