
Role of Glial Cells in Neuronal Function, Mood Disorders, and Drug Addiction

 ,
,  , ,
, , 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Major Depressive Disorder (MDD)
2.1. Neurotrophic Factors and MDD
2.2. Neuroinflammation and MDD
2.3. Gut Microbiota and MDD
3. Drug Addiction
3.1. Neurotrophic Factors and Drug Addiction
3.2. Neuroinflammation and Drug Addiction
3.3. Gut Microbiota and Drug Addiction
4. Glial Cells
4.1. Microglia
4.1.1. Microglia–Depression
4.1.2. Microglia–Drug Addiction
4.2. Astroglia (Astrocytes)
4.2.1. Astrocytes–Depression
4.2.2. Astrocytes–Drug Addiction
4.3. Oligodendrocytes
4.3.1. Oligodendrocytes–Depression
4.3.2. Oligodendrocytes–Drug Addiction
4.4. Synantocytes (NG2 Cells)
4.4.1. NG2 Cells–Depression
4.4.2. NG2 Cells–Drug Addiction
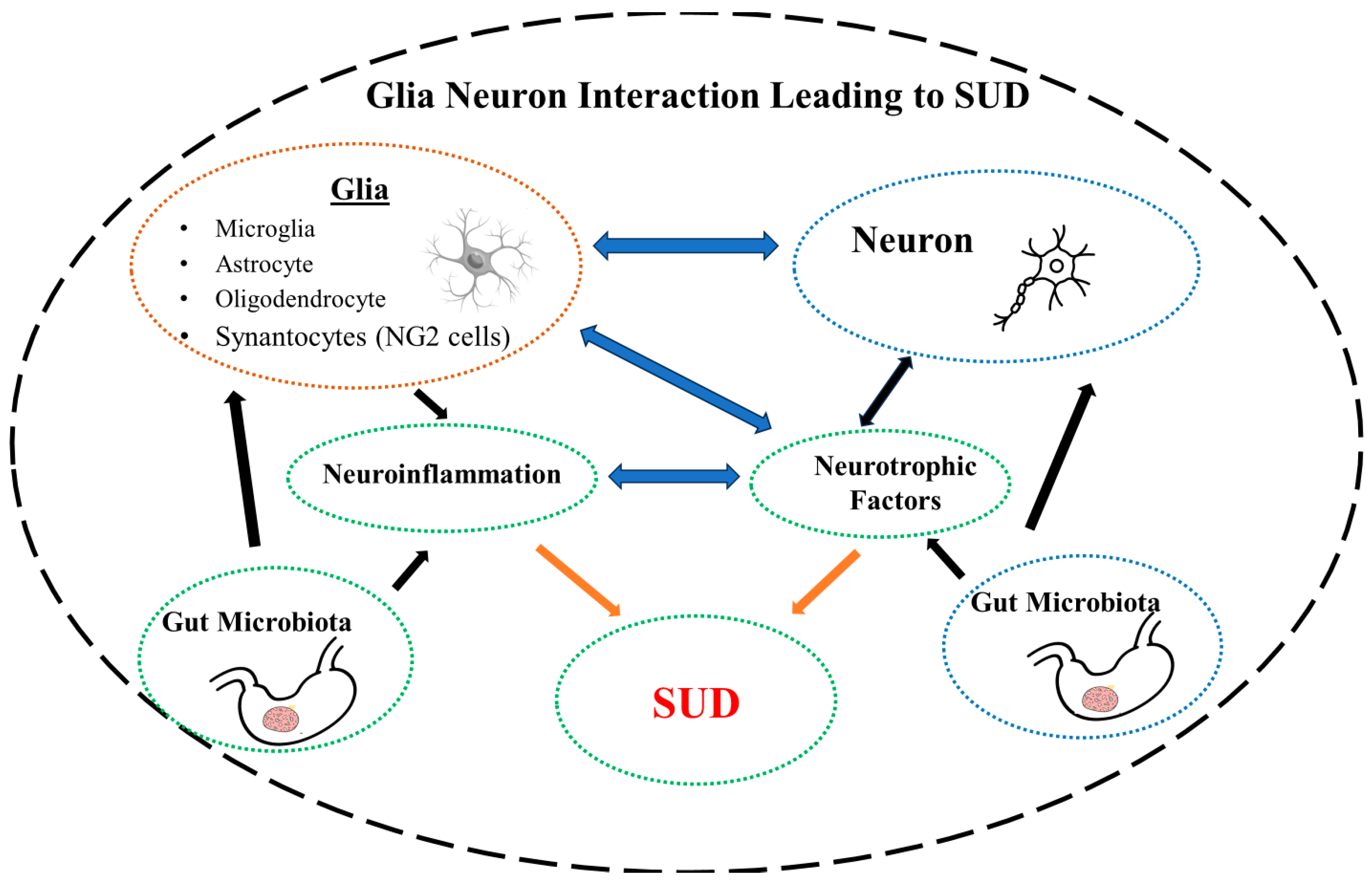
5. Glia–Neuron Interaction–SUD
Therapeutic Interventions
6. Microbiota–Neurotrophic Factors–Neuroinflammation
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Greenberg, P.E.; Fournier, A.A.; Sisitsky, T.; Simes, M.; Berman, R.; Koenigsberg, S.H.; Kessler, R.C. The Economic Burden of Adults with Major Depressive Disorder in the United States (2010 and 2018). Pharmacoeconomics 2021, 39, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Formánek, T.; Krupchanka, D.; Mladá, K.; Winkler, P.; Jones, P.B. Mortality and life-years lost following subsequent physical comorbidity in people with pre-existing substance use disorders: A national registry-based retrospective cohort study of hospitalised individuals in Czechia. Lancet Psychiatry 2022, 9, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.; Li, M.; Xu, L.; Mikosz, C.A.; Luo, F. Assessment of Annual Cost of Substance Use Disorder in US Hospitals. JAMA Netw. Open 2021, 4, e210242. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, K.; Manza, P.; Chapman, M.; Nawal, N.; Biesecker, E.; McPherson, K.; Dennis, E.; Johnson, A.; Volkow, N.D.; Joseph, P.V. Inflammatory Markers in Substance Use and Mood Disorders: A Neuroimaging Perspective. Front. Psychiatry 2022, 13, 863734. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Exosomes What Do We Love So Much About Them? Circ. Res. 2016, 119, 1280–1282. [Google Scholar] [CrossRef] [PubMed]
- Leza, L.; Haro, B.; López-Goñi, J.J.; Fernández-Montalvo, J. Substance use disorder and lifetime suicidal behaviour: A scoping review. Psychiatry Res. 2024, 334, 115830. [Google Scholar] [CrossRef] [PubMed]
- Schildkraut, J.J. The catecholamine hypothesis of affective disorders: A review of supporting evidence. Am. J. Psychiatry 1965, 122, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jeong, J.; Kwak, Y.; Park, S.K. Depression research: Where are we now? Mol. Brain 2010, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.S.; Bell, C.E.; Pollard, D.A. Revisiting the monoamine hypothesis of depression: A new perspective. Perspect. Medicin. Chem. 2014, 6, PMC-S11375. [Google Scholar] [CrossRef]
- Hillhouse, T.M.; Porter, J.H. A brief history of the development of antidepressant drugs: From monoamines to glutamate. Exp. Clin. Psychopharmacol. 2015, 23, 1–21. [Google Scholar] [CrossRef]
- Matveychuk, D.; Thomas, R.K.; Swainson, J.; Khullar, A.; MacKay, M.A.; Baker, G.B.; Dursun, S.M. Ketamine as an antidepressant: Overview of its mechanisms of action and potential predictive biomarkers. Ther. Adv. Psychopharmacol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Kaye, A.P.; Jefferson, S.; Girgenti, M.J.; Wilkinson, S.T.; Sanacora, G.; Esterlis, I. Ketamine and the neurobiology of depression: Toward next-generation rapid-acting antidepressant treatments. Proc. Natl. Acad. Sci. USA 2023, 120, e2305772120. [Google Scholar] [CrossRef] [PubMed]
- Elmeseiny, O.S.A.; Müller, H.K. A molecular perspective on mGluR5 regulation in the antidepressant effect of ketamine. Pharmacol. Res. 2024, 200, 107081. [Google Scholar] [CrossRef] [PubMed]
- Craft, J.M.; Watterson, D.M.; Van Eldik, L.J. Neuroinflammation: A potential therapeutic target. Expert Opin. Ther. Targets 2005, 9, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Salim, S.; Chugh, G.; Asghar, M. Inflammation in anxiety. Adv. Protein Chem. Struct. Biol. 2012, 88, 1–25. [Google Scholar] [CrossRef]
- Matrisciano, F.; Pinna, G. The Strategy of Targeting Peroxisome Proliferator-Activated Receptor (PPAR) in the Treatment of Neuropsychiatric Disorders. Adv. Exp. Med. Biol. 2023, 1411, 513–535. [Google Scholar] [CrossRef]
- Friedrich, M.J. Depression Is the Leading Cause of Disability Around the World. JAMA 2017, 317, 1517. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Yamagata, H.; Seki, T.; Watanabe, Y. Epigenetic mechanisms of major depression: Targeting neuronal plasticity. Psychiatry Clin. Neurosci. 2018, 72, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Burgunder, J.M. Mechanisms underlying phenotypic variation in neurogenetic disorders. Nat. Rev. Neurol. 2023, 19, 363–370. [Google Scholar] [CrossRef]
- Lee, J.; Duan, W.; Mattson, M.P. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002, 82, 1367–1375. [Google Scholar] [CrossRef]
- Hagg, T. From neurotransmitters to neurotrophic factors to neurogenesis. Neuroscientist 2009, 15, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Manji, H.K.; Drevets, W.C.; Charney, D.S. The cellular neurobiology of depression. Nat. Med. 2001, 7, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Lee, S.Y.; Hu, M.C.; Chen, S.L.; Chang, Y.H.; Chu, C.H.; Lin, S.H.; Li, C.L.; Wang, L.J.; Chen, P.S.; et al. More inflammation but less brain-derived neurotrophic factor in antisocial personality disorder. Psychoneuroendocrinology 2017, 85, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Poo, M.M. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2001, 2, 24–32. [Google Scholar] [CrossRef]
- Dremencov, E.; Jezova, D.; Barak, S.; Gaburjakova, J.; Gaburjakova, M.; Kutna, V.; Ovsepian, S.V. Trophic factors as potential therapies for treatment of major mental disorders. Neurosci. Lett. 2021, 764, 136194. [Google Scholar] [CrossRef] [PubMed]
- Berton, O.; McClung, C.A.; DiLeone, R.J.; Krishnan, V.; Renthal, W.; Russo, S.J.; Graham, D.; Tsankova, N.M.; Bolanos, C.A.; Rios, M.; et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 2006, 311, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the pathophysiology and treatment of depression: Activity-dependent effects distinguish rapid-acting antidepressants. Eur. J. Neurosci. 2021, 53, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Amidfar, M.; Réus, G.Z.; de Moura, A.B.; Quevedo, J.; Kim, Y.K. The Role of Neurotrophic Factors in Pathophysiology of Major Depressive Disorder. Adv. Exp. Med. Biol. 2021, 1305, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Kajihara, R. Involvement of brain-derived neurotrophic factor signaling in the pathogenesis of stress-related brain diseases. Front. Mol. Neurosci. 2023, 16, 1247422. [Google Scholar] [CrossRef]
- Zelada, M.I.; Garrido, V.; Liberona, A.; Jones, N.; Zúñiga, K.; Silva, H.; Nieto, R.R. Brain-Derived Neurotrophic Factor (BDNF) as a Predictor of Treatment Response in Major Depressive Disorder (MDD): A Systematic Review. Int. J. Mol. Sci. 2023, 24, 14810. [Google Scholar] [CrossRef]
- Fang, S.; Wu, Z.; Guo, Y.; Zhu, W.; Wan, C.; Yuan, N.; Chen, J.; Hao, W.; Mo, X.; Guo, X.; et al. Roles of microglia in adult hippocampal neurogenesis in depression and their therapeutics. Front. Immunol. 2023, 14, 1193053. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zhang, Y.; Tu, M.; Ye, Y.; Li, M.; Ran, R.; Zou, Z. Brain-derived neurotrophic factor levels across psychiatric disorders: A systemic review and network meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2024, 131, 110954. [Google Scholar] [CrossRef] [PubMed]
- Porter, G.A.; O’Connor, J.C. Brain-derived neurotrophic factor and inflammation in depression: Pathogenic partners in crime? World J. Psychiatry 2022, 12, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H. Mammalian neural stem cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H. Neurogenesis in the adult brain. J. Neurosci. 2002, 22, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Gomar, I.; Geribaldi-Doldán, N.; Santos-Rosendo, C.; Sanguino-Caneva, C.; Carrillo-Chapman, C.; Fiorillo-Moreno, O.; Villareal Camacho, J.L.; Quiroz, E.N.; Verástegui, C. Exploring the Intricacies of Neurogenic Niches: Unraveling the Anatomy and Neural Microenvironments. Biomolecules 2024, 14, 335. [Google Scholar] [CrossRef] [PubMed]
- Burgess, N.; Maguire, E.A.; O’Keefe, J. The human hippocampus and spatial and episodic memory. Neuron 2002, 35, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Wen, Z.; Song, H.; Christian, K.M.; Ming, G.L. Adult Neurogenesis and Psychiatric Disorders. Cold Spring Harb. Perspect. Biol. 2016, 8, a019026. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef]
- Guillin, O.; Diaz, J.; Carroll, P.; Griffon, N.; Schwartz, J.C.; Sokoloff, P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 2001, 411, 86–89. [Google Scholar] [CrossRef]
- Staley, J.K.; Mash, D.C. Adaptive increase in D3 dopamine receptors in the brain reward circuits of human cocaine fatalities. J. Neurosci. 1996, 16, 6100–6106. [Google Scholar] [CrossRef] [PubMed]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, P.; Saarma, M. Cerebral dopamine neurotrophic factor protects and repairs dopamine neurons by novel mechanism. Mol. Psychiatry 2021, 27, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, P.; Zhang, M.; Yao, Z. The roles of neuron-NG2 glia synapses in promoting oligodendrocyte development and remyelination. Cell Tissue Res. 2020, 381, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Küppers, E.; Beyer, C. Dopamine regulates brain-derived neurotrophic factor (BDNF) expression in cultured embryonic mouse striatal cells. Neuroreport 2001, 12, 1175–1179. [Google Scholar] [CrossRef]
- Williams, S.N.; Undieh, A.S. Dopamine D1-like receptor activation induces brain-derived neurotrophic factor protein expression. Neuroreport 2009, 20, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Colucci-D’amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Andreska, T.; Lüningschrör, P.; Wolf, D.; McFleder, R.L.; Ayon-Olivas, M.; Rattka, M.; Drechsler, C.; Perschin, V.; Blum, R.; Aufmkolk, S.; et al. DRD1 signaling modulates TrkB turnover and BDNF sensitivity in direct pathway striatal medium spiny neurons. Cell Rep. 2023, 42, 112575. [Google Scholar] [CrossRef]
- Martinowich, K.; Lu, B. Interaction between BDNF and Serotonin: Role in Mood Disorders. Neuropsychopharmacol. 2007, 33, 73–83. [Google Scholar] [CrossRef]
- Maes, M. The cytokine hypothesis of depression: Inflammation, oxidative & nitrosative stress (IO&NS) and leaky gut as new targets for adjunctive treatments in depression. Neuro Endocrinol. Lett. 2008, 29, 287–291. [Google Scholar]
- Hurley, L.L.; Tizabi, Y. Neuroinflammation, Neurodegeneration, and Depression. Neurotox. Res. 2012, 23, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Audet, M.C.; McQuaid, R.J.; Merali, Z.; Anisman, H. Cytokine variations and mood disorders: Influence of social stressors and social support. Front. Neurosci. 2014, 8, 119363. [Google Scholar] [CrossRef]
- Dhabhar, F.S.; Burke, H.M.; Epel, E.S.; Mellon, S.H.; Rosser, R.; Reus, V.I.; Wolkowitz, O.M. Low serum IL-10 concentrations and loss of regulatory association between IL-6 and IL-10 in adults with major depression. J. Psychiatr. Res. 2009, 43, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Ferrari, C.; Uher, R.; Bocchio-Chiavetto, L.; Riva, M.A.; Pariante, C.M. Absolute Measurements of Macrophage Migration Inhibitory Factor and Interleukin-1-β mRNA Levels Accurately Predict Treatment Response in Depressed Patients. Int. J. Neuropsychopharmacol. 2016, 19, pyw045. [Google Scholar] [CrossRef]
- Hashioka, S.; Inoue, K.; Hayashida, M.; Wake, R.; Oh-Nishi, A.; Miyaoka, T. Implications of systemic inflammation and periodontitis for major depression. Front. Neurosci. 2018, 12, 380928. [Google Scholar] [CrossRef]
- Kouba, B.R.; de Araujo Borba, L.; Borges de Souza, P.; Gil-Mohapel, J.; Rodrigues, A.L.S. Role of Inflammatory Mechanisms in Major Depressive Disorder: From Etiology to Potential Pharmacological Targets. Cells 2024, 13, 423. [Google Scholar] [CrossRef]
- Orsolini, L.; Pompili, S.; Valenta, S.T.; Salvi, V.; Volpe, U. C-Reactive Protein as a Biomarker for Major Depressive Disorder? Int. J. Mol. Sci. 2022, 23, 1616. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Jin, J.; Tang, J. Inflammatory pathophysiological mechanisms implicated in postpartum depression. Front. Pharmacol. 2022, 13, 955672. [Google Scholar] [CrossRef]
- Zhu, H.; Guan, A.; Liu, J.; Peng, L.; Zhang, Z.; Wang, S. Noteworthy perspectives on microglia in neuropsychiatric disorders. J. Neuroinflamm. 2023, 20, 223. [Google Scholar] [CrossRef]
- Gal, Z.; Torok, D.; Gonda, X.; Eszlari, N.; Anderson, I.M.; Deakin, B.; Juhasz, G.; Bagdy, G.; Petschner, P. Inflammation and Blood-Brain Barrier in Depression: Interaction of CLDN5 and IL6 Gene Variants in Stress-Induced Depression. Int. J. Neuropsychopharmacol. 2023, 26, 189–197. [Google Scholar] [CrossRef]
- Dadkhah, M.; Baziar, M.; Rezaei, N. The regulatory role of BDNF in neuroimmune axis function and neuroinflammation induced by chronic stress: A new therapeutic strategies for neurodegenerative disorders. Cytokine 2024, 174, 156477. [Google Scholar] [CrossRef]
- Jing, D.; Hou, X.; Guo, X.; Zhao, X.; Zhang, K.; Zhang, J.; Kan, C.; Han, F.; Liu, J.; Sun, X. Astrocytes in Post-Stroke Depression: Roles in Inflammation, Neurotransmission, and Neurotrophin Signaling. Cell. Mol. Neurobiol. 2023, 43, 3301–3313. [Google Scholar] [CrossRef]
- Feng, X.; Ma, X.; Li, J.; Zhou, Q.; Liu, Y.; Song, J.; Liu, J.; Situ, Q.; Wang, L.; Zhang, J.; et al. Inflammatory Pathogenesis of Post-stroke Depression. Aging Dis. 2024. [Google Scholar] [CrossRef]
- Akinfiresoye, L.; Tizabi, Y. Antidepressant effects of AMPA and ketamine combination: Role of hippocampal BDNF, synapsin, and mTOR. Psychopharmacology 2013, 230, 291–298. [Google Scholar] [CrossRef]
- Kim, J.; He, M.J.; Widmann, A.K.; Lee, F.S. The role of neurotrophic factors in novel, rapid psychiatric treatments. Neuropsychopharmacology 2023, 49, 227–245. [Google Scholar] [CrossRef]
- Quintanilla, B.; Zarate, C.A.; Pillai, A. Ketamine’s mechanism of action with an emphasis on neuroimmune regulation: Can the complement system complement ketamine’s antidepressant effects? Mol. Psychiatry 2024. [Google Scholar] [CrossRef]
- Lewandowska-Pietruszka, Z.; Figlerowicz, M.; Mazur-Melewska, K. The History of the Intestinal Microbiota and the Gut-Brain Axis. Pathogens 2022, 11, 1540. [Google Scholar] [CrossRef]
- Shoemaker, W.R.; Chen, D.; Garud, N.R. Comparative Population Genetics in the Human Gut Microbiome. Genome Biol. Evol. 2022, 14, evab116. [Google Scholar] [CrossRef]
- Chatterjee, G.; Negi, S.; Basu, S.; Faintuch, J.; O’Donovan, A.; Shukla, P. Microbiome systems biology advancements for natural well-being. Sci. Total Environ. 2022, 838, 155915. [Google Scholar] [CrossRef]
- VanEvery, H.; Franzosa, E.A.; Nguyen, L.H.; Huttenhower, C. Microbiome epidemiology and association studies in human health. Nat. Rev. Genet. 2022, 24, 109–124. [Google Scholar] [CrossRef]
- Radjabzadeh, D.; Bosch, J.A.; Uitterlinden, A.G.; Zwinderman, A.H.; Ikram, M.A.; van Meurs, J.B.J.; Luik, A.I.; Nieuwdorp, M.; Lok, A.; van Duijn, C.M.; et al. Gut microbiome-wide association study of depressive symptoms. Nat. Commun. 2022, 13, 7128. [Google Scholar] [CrossRef]
- Tizabi, Y.; Bennani, S.; El Kouhen, N.; Getachew, B.; Aschner, M. Interaction of Heavy Metal Lead with Gut Microbiota: Implications for Autism Spectrum Disorder. Biomolecules 2023, 13, 1549. [Google Scholar] [CrossRef]
- Niemela, L.; Lamoury, G.; Carroll, S.; Morgia, M.; Yeung, A.; Oh, B. Exploring gender differences in the relationship between gut microbiome and depression—A scoping review. Front. Psychiatry 2024, 15, 1361145. [Google Scholar] [CrossRef]
- Xie, Z.; Huang, J.; Sun, G.; He, S.; Luo, Z.; Zhang, L.; Li, L.; Yao, M.; Du, C.; Yu, W.; et al. Integrated multi-omics analysis reveals gut microbiota dysbiosis and systemic disturbance in major depressive disorder. Psychiatry Res. 2024, 334, 115804. [Google Scholar] [CrossRef]
- Zhang, J.C.; Yao, W.; Dong, C.; Yang, C.; Ren, Q.; Ma, M.; Hashimoto, K. Blockade of interleukin-6 receptor in the periphery promotes rapid and sustained antidepressant actions: A possible role of gut-microbiota-brain axis. Transl. Psychiatry 2017, 7, e1138. [Google Scholar] [CrossRef]
- Bahmani, M.; Mehrtabar, S.; Jafarizadeh, A.; Zoghi, S.; Heravi, F.S.; Abbasi, A.; Sanaie, S.; Rahnemayan, S.; Leylabadlo, H.E. The Gut Microbiota and Major Depressive Disorder: Current Understanding and Novel Therapeutic Strategies. Curr. Pharm. Biotechnol. 2024, 25. [Google Scholar] [CrossRef]
- Kolobaric, A.; Andreescu, C.; Jašarević, E.; Hong, C.H.; Roh, H.W.; Cheong, J.Y.; Kim, Y.K.; Shin, T.S.; Kang, C.S.; Kwon, C.O.; et al. Gut microbiome predicts cognitive function and depressive symptoms in late life. Mol. Psychiatry 2023, 19, e076910. [Google Scholar] [CrossRef]
- United Nations Office on Drugs and Crime. Executive Summary, World Drug Report; United Nations Office on Drugs and Crime: Vienna, Austria, 2023; Volume 2012. [Google Scholar]
- García-Cabrerizo, R.; Cryan, J.F. A gut (microbiome) feeling about addiction: Interactions with stress and social systems. Neurobiol. Stress 2024, 30, 100629. [Google Scholar] [CrossRef]
- Goldstein, R.Z.; Volkow, N.D. Dysfunction of the prefrontal cortex in addiction: Neuroimaging findings and clinical implications. Nat. Rev. Neurosci. 2011, 12, 652–669. [Google Scholar] [CrossRef]
- Russo, S.J.; Mazei-Robison, M.S.; Ables, J.L.; Nestler, E.J. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology 2009, 56 (Suppl. S1), 73–82. [Google Scholar] [CrossRef]
- Koskela, M.; Bäck, S.; Võikar, V.; Richie, C.T.; Domanskyi, A.; Harvey, B.K.; Airavaara, M. Update of neurotrophic factors in neurobiology of addiction and future directions. Neurobiol. Dis. 2017, 97, 189–200. [Google Scholar] [CrossRef]
- Chen, W.; Meng, S.; Han, Y.; Shi, J. Astrocytes: The neglected stars in the central nervous system and drug addiction. Med. Rev. 2022, 2, 417–426. [Google Scholar] [CrossRef]
- Mann, L.G.; Claassen, D.O. Mesial temporal dopamine: From biology to behaviour. Eur. J. Neurosci. 2024, 59, 1141–1152. [Google Scholar] [CrossRef]
- Salamone, J.D.; Correa, M. The Neurobiology of Activational Aspects of Motivation: Exertion of Effort, Effort-Based Decision Making, and the Role of Dopamine. Annu. Rev. Psychol. 2024, 75, 1–32. [Google Scholar] [CrossRef]
- Nestler, E.J.; Lüscher, C. The Molecular Basis of Drug Addiction: Linking Epigenetic to Synaptic and Circuit Mechanisms. Neuron 2019, 102, 48–59. [Google Scholar] [CrossRef]
- Volkow, N.D. Drugs, Brains, and Behavior: The Science of Addiction: Preface. Available online: https://nida.nih.gov/research-topics/addiction-science/drugs-brain-behavior-science-of-addiction (accessed on 27 April 2024).
- Getachew, B.; Hauser, S.R.; Bennani, S.; El Kouhen, N.; Sari, Y.; Tizabi, Y. Adolescent alcohol drinking interaction with the gut microbiome: Implications for adult alcohol use disorder. Adv. Drug Alcohol Res. 2024, 4, 11881. [Google Scholar] [CrossRef]
- Hatoum, A.S.; Colbert, S.M.C.; Johnson, E.C.; Huggett, S.B.; Deak, J.D.; Pathak, G.A.; Jennings, M.V.; Paul, S.E.; Karcher, N.R.; Hansen, I.; et al. Multivariate genome-wide association meta-analysis of over 1 million subjects identifies loci underlying multiple substance use disorders. Nat. Ment. Health 2023, 1, 210–223. [Google Scholar] [CrossRef]
- Koob, G.F. Anhedonia, Hyperkatifeia, and Negative Reinforcement in Substance Use Disorders. Curr. Top. Behav. Neurosci. 2022, 58, 147–165. [Google Scholar] [CrossRef]
- Tijani, A.O.; Garg, J.; Frempong, D.; Verana, G.; Kaur, J.; Joga, R.; Sabanis, C.D.; Kumar, S.; Kumar, N.; Puri, A. Sustained drug delivery strategies for treatment of common substance use disorders: Promises and challenges. J. Control. Release 2022, 348, 970–1003. [Google Scholar] [CrossRef]
- Barker, J.M.; Taylor, J.R.; De Vries, T.J.; Peters, J. Brain-derived neurotrophic factor and addiction: Pathological versus therapeutic effects on drug seeking. Brain Res. 2015, 1628, 68–81. [Google Scholar] [CrossRef]
- Ornell, F.; Hansen, F.; Schuch, F.B.; Pezzini Rebelatto, F.; Tavares, A.L.; Scherer, J.N.; Valerio, A.G.; Pechansky, F.; Paim Kessler, F.H.; von Diemen, L. Brain-derived neurotrophic factor in substance use disorders: A systematic review and meta-analysis. Drug Alcohol Depend. 2018, 193, 91–103. [Google Scholar] [CrossRef]
- Peregud, D.I.; Baronets, V.Y.; Terebilina, N.N.; Gulyaeva, N.V. Role of BDNF in Neuroplasticity Associated with Alcohol Dependence. Biochemistry 2023, 88, 404–416. [Google Scholar] [CrossRef]
- Rodrigues, L.C.M.; Gobira, P.H.; De Oliveira, A.C.; Pelição, R.; Teixeira, A.L.; Moreira, F.A.; Campos, A.C. Neuroinflammation as a possible link between cannabinoids and addiction. Acta Neuropsychiatr. 2014, 26, 334–346. [Google Scholar] [CrossRef]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2015, 16, 22–34. [Google Scholar] [CrossRef]
- Cuitavi, J.; Torres-Pérez, J.V.; Lorente, J.D.; Campos-Jurado, Y.; Andrés-Herrera, P.; Polache, A.; Agustín-Pavón, C.; Hipólito, L. Crosstalk between Mu-Opioid receptors and neuroinflammation: Consequences for drug addiction and pain. Neurosci. Biobehav. Rev. 2023, 145, 105011. [Google Scholar] [CrossRef]
- Friedman, H.; Newton, C.; Klein, T.W. Microbial Infections, Immunomodulation, and Drugs of Abuse. Clin. Microbiol. Rev. 2003, 16, 209. [Google Scholar] [CrossRef]
- Sarkar, D.; Jung, M.K.; Wang, H.J. Alcohol and the Immune System. Alcohol Res. 2015, 37, 153. [Google Scholar]
- Kohno, M.; Link, J.; Dennis, L.E.; McCready, H.; Huckans, M.; Hoffman, W.F.; Loftis, J.M. Neuroinflammation in addiction: A review of neuroimaging studies and potential immunotherapies. Pharmacol. Biochem. Behav. 2019, 179, 34–42. [Google Scholar] [CrossRef]
- Salavrakos, M.; Leclercq, S.; De Timary, P.; Dom, G. Microbiome and substances of abuse. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 105, 110113. [Google Scholar] [CrossRef]
- Chivero, E.T.; Sil, S.; Kumar, M.; Buch, S. Substance use, microbiome and psychiatric disorders. Pharmacol. Biochem. Behav. 2022, 219, 173432. [Google Scholar] [CrossRef]
- Gervasi, T.; Mandalari, G. The Interplay Between Gut Microbiota and Central Nervous System. Curr. Pharm. Des. 2023, 29, 3274–3281. [Google Scholar] [CrossRef]
- Ransom, B.R.; Kettenmann, H. Studying Human Glial Cells: Where Are We Today? Available online: https://pubmed.ncbi.nlm.nih.gov/32057156/ (accessed on 27 April 2024).
- Ndubaku, U.; de Bellard, M.E. Glial cells: Old cells with new twists. Acta Histochem. 2008, 110, 182–195. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. The Human Brain in Numbers: A Linearly Scaled-up Primate Brain. Front. Hum. Neurosci. 2009, 3, 857. [Google Scholar] [CrossRef]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef]
- Shi, J.; Huang, S. Comparative Insight into Microglia/Macrophages-Associated Pathways in Glioblastoma and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 25, 16. [Google Scholar] [CrossRef]
- Souza, D.G.; Almeida, R.F.; Souza, D.O.; Zimmer, E.R. The astrocyte biochemistry. Semin. Cell Dev. Biol. 2019, 95, 142–150. [Google Scholar] [CrossRef]
- Bonvento, G.; Bolaños, J.P. Astrocyte-neuron metabolic cooperation shapes brain activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef]
- Ebling, F.J.P.; Lewis, J.E. Tanycytes and hypothalamic control of energy metabolism. Glia 2018, 66, 1176–1184. [Google Scholar] [CrossRef]
- Chamberlain, K.A.; Huang, N.; Xie, Y.; LiCausi, F.; Li, S.; Li, Y.; Sheng, Z.H. Oligodendrocytes enhance axonal energy metabolism by deacetylation of mitochondrial proteins through transcellular delivery of SIRT2. Neuron 2021, 109, 3456–3472.e8. [Google Scholar] [CrossRef]
- Sanchez-Petidier, M.; Guerri, C.; Moreno-Manzano, V. Toll-like receptors 2 and 4 differentially regulate the self-renewal and differentiation of spinal cord neural precursor cells. Stem Cell Res. Ther. 2022, 13, 117. [Google Scholar] [CrossRef]
- Wies Mancini, V.S.B.; Mattera, V.S.; Pasquini, J.M.; Pasquini, L.A.; Correale, J.D. Microglia-derived extracellular vesicles in homeostasis and demyelination/remyelination processes. J. Neurochem. 2024, 168, 3–25. [Google Scholar] [CrossRef]
- Manu, D.R.; Slevin, M.; Barcutean, L.; Forro, T.; Boghitoiu, T.; Balasa, R. Astrocyte Involvement in Blood–Brain Barrier Function: A Critical Update Highlighting Novel, Complex, Neurovascular Interactions. Int. J. Mol. Sci. 2023, 24, 17146. [Google Scholar] [CrossRef]
- Fernandes, V.M.; Auld, V.; Klämbt, C. Glia as Functional Barriers and Signaling Intermediaries. Cold Spring Harb. Perspect. Biol. 2024, 16, a041423. [Google Scholar] [CrossRef]
- Savtchouk, I.; Volterra, A. Gliotransmission: Beyond Black-and-White. J. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef]
- Lalo, U.; Koh, W.; Lee, C.J.; Pankratov, Y. The tripartite glutamatergic synapse. Neuropharmacology 2021, 199, 108758. [Google Scholar] [CrossRef]
- Rasia-Filho, A.A.; Calcagnotto, M.E.; von Bohlen und Halbach, O. Glial Cell Modulation of Dendritic Spine Structure and Synaptic Function. Adv. Neurobiol. 2023, 34, 255–310. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Novikov, N.I.; Brazhnik, E.S.; Kitchigina, V.F. Pathological Correlates of Cognitive Decline in Parkinson’s Disease: From Molecules to Neural Networks. Biochemistry 2023, 88, 1890–1904. [Google Scholar] [CrossRef]
- Reed, M.M.; Blazer-Yost, B. Channels and Transporters in Astrocyte Volume Regulation in Health and Disease. Cell. Physiol. Biochem. 2022, 56, 12–30. [Google Scholar] [CrossRef]
- Clayton, R.W.; Lovell-Badge, R.; Galichet, C. The Properties and Functions of Glial Cell Types of the Hypothalamic Median Eminence. Front. Endocrinol. 2022, 13, 953995. [Google Scholar] [CrossRef]
- Kofler, J.; Wiley, C.A. Microglia: Key innate immune cells of the brain. Toxicol. Pathol. 2011, 39, 103–114. [Google Scholar] [CrossRef]
- Chen, X.; Holtzman, D.M. Emerging roles of innate and adaptive immunity in Alzheimer’s disease. Immunity 2022, 55, 2236–2254. [Google Scholar] [CrossRef]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2014, 40, 2570–2582. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef]
- Rahman, S.; Alzarea, S. Glial mechanisms underlying major depressive disorder: Potential therapeutic opportunities. Prog. Mol. Biol. Transl. Sci. 2019, 167, 159–178. [Google Scholar] [CrossRef]
- Scuderi, C.; Verkhratsky, A.; Parpura, V.; Li, B. Neuroglia in Psychiatric Disorders. Adv. Neurobiol. 2021, 26, 3–19. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Marino, K.M.; Ulland, T.K. Modulation of Glial Function in Health, Aging, and Neurodegenerative Disease. Front. Cell. Neurosci. 2021, 15, 718324. [Google Scholar] [CrossRef]
- Zhao, G. Shared and disease-specific glial gene expression changes in neurodegenerative diseases. Nat. Aging 2023, 3, 246–247. [Google Scholar] [CrossRef]
- Soares, É.N.; Carla, A.; Costa, S.; De, G.; Ferrolho, J.; Ureshino, R.P.; Getachew, B.; Lima Costa, S.; Diogenes Amaral Da Silva, V.; Tizabi, Y. Nicotinic Acetylcholine Receptors in Glial Cells as Molecular Target for Parkinson’s Disease. Cells 2024, 13, 474. [Google Scholar] [CrossRef]
- Magni, G.; Riboldi, B.; Ceruti, S. Human Glial Cells as Innovative Targets for the Therapy of Central Nervous System Pathologies. Cells 2024, 13, 606. [Google Scholar] [CrossRef]
- Sanadgol, N.; Miraki Feriz, A.; Lisboa, S.F.; Joca, S.R.L. Putative role of glial cells in treatment resistance depression: An updated critical literation review and evaluation of single-nuclei transcriptomics data. Life Sci. 2023, 331, 122025. [Google Scholar] [CrossRef]
- Saba, W. Glial dysfunction in substance use disorders. New insights from PET and MR imaging. Addict. Neurosci. 2023, 9, 100135. [Google Scholar] [CrossRef]
- Bernier, L.P.; Bohlen, C.J.; York, E.M.; Choi, H.B.; Kamyabi, A.; Dissing-Olesen, L.; Hefendehl, J.K.; Collins, H.Y.; Stevens, B.; Barres, B.A.; et al. Nanoscale Surveillance of the Brain by Microglia via cAMP-Regulated Filopodia. Cell Rep. 2019, 27, 2895–2908.e4. [Google Scholar] [CrossRef]
- Nebeling, F.C.; Poll, S.; Justus, L.C.; Steffen, J.; Keppler, K.; Mittag, M.; Fuhrmann, M. Microglial motility is modulated by neuronal activity and correlates with dendritic spine plasticity in the hippocampus of awake mice. eLife 2023, 12, e83176. [Google Scholar] [CrossRef]
- Pathak, D.; Sriram, K. Molecular Mechanisms Underlying Neuroinflammation Elicited by Occupational Injuries and Toxicants. Int. J. Mol. Sci. 2023, 24, 2272. [Google Scholar] [CrossRef]
- Darwish, S.F.; Elbadry, A.M.M.; Elbokhomy, A.S.; Salama, G.A.; Salama, R.M. The dual face of microglia (M1/M2) as a potential target in the protective effect of nutraceuticals against neurodegenerative diseases. Front. Aging 2023, 4, 1231706. [Google Scholar] [CrossRef]
- Wu, Q.; Zhao, M.; Li, D.; He, X.; Zang, W. Cholinergic drugs reduce metabolic inflammation and diabetic myocardial injury by regulating the gut bacterial component lipopolysaccharide-induced ERK/Egr-1 pathway. FASEB J. 2023, 37, e22917. [Google Scholar] [CrossRef]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 2022, 24, 23–39. [Google Scholar] [CrossRef]
- Saitgareeva, A.R.; Bulygin, K.V.; Gareev, I.F.; Beylerli, O.A.; Akhmadeeva, L.R. The role of microglia in the development of neurodegeneration. Neurol. Sci. 2020, 41, 3609–3615. [Google Scholar] [CrossRef]
- Costa, T.; Fernandez-Villalba, E.; Izura, V.; Lucas-Ochoa, A.; Menezes-Filho, N.; Santana, R.; de Oliveira, M.; Araújo, F.; Estrada, C.; Silva, V.; et al. Combined 1-Deoxynojirimycin and Ibuprofen Treatment Decreases Microglial Activation, Phagocytosis and Dopaminergic Degeneration in MPTP-Treated Mice. J. Neuroimmune Pharmacol. 2021, 16, 390–402. [Google Scholar] [CrossRef]
- De Marchi, F.; Munitic, I.; Vidatic, L.; Papić, E.; Rački, V.; Nimac, J.; Jurak, I.; Novotni, G.; Rogelj, B.; Vuletic, V.; et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines 2023, 11, 2793. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Tremblay, M.È.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The Role of Microglia in the Healthy Brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef]
- Nimmerjahn, A. Two-Photon Imaging of Microglia in the Mouse Cortex In Vivo. Cold Spring Harb. Protoc. 2012, 2012, pdb.prot069294. [Google Scholar] [CrossRef]
- Leyh, J.; Paeschke, S.; Mages, B.; Michalski, D.; Nowicki, M.; Bechmann, I.; Winter, K. Classification of Microglial Morphological Phenotypes Using Machine Learning. Front. Cell. Neurosci. 2021, 15, 701673. [Google Scholar] [CrossRef]
- Ziebell, J.M.; Taylor, S.E.; Cao, T.; Harrison, J.L.; Lifshitz, J. Rod microglia: Elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J. Neuroinflamm. 2012, 9, 247. [Google Scholar] [CrossRef]
- Taylor, S.E.; Morganti-Kossmann, C.; Lifshitz, J.; Ziebell, J.M. Rod Microglia: A Morphological Definition. PLoS ONE 2014, 9, e97096. [Google Scholar] [CrossRef]
- Fatoba, O.; Itokazu, T.; Yamashita, T. Microglia as therapeutic target in central nervous system disorders. J. Pharmacol. Sci. 2020, 144, 102–118. [Google Scholar] [CrossRef]
- Pathak, D.; Sriram, K. Neuron-astrocyte omnidirectional signaling in neurological health and disease. Front. Mol. Neurosci. 2023, 16, 1169320. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.X.; Wu, R. Toll-Like Receptor 4: A Novel Target to Tackle Drug Addiction? Handb. Exp. Pharmacol. 2022, 276, 275–290. [Google Scholar] [CrossRef]
- Heidari, A.; Yazdanpanah, N.; Rezaei, N. The role of Toll-like receptors and neuroinflammation in Parkinson’s disease. J. Neuroinflamm. 2022, 19, 135. [Google Scholar] [CrossRef]
- Yirmiya, R.; Rimmerman, N.; Reshef, R. Depression as a microglial disease. Trends Neurosci. 2015, 38, 637–658. [Google Scholar] [CrossRef]
- Wang, F.; Wu, H.; Hu, A.; Dong, L.; Lin, X.; Li, M.; Wang, Y.; Li, W.; Chang, L.; Chang, Y.; et al. Ultrasound combined with glial cell line-derived neurotrophic factor-loaded microbubbles for the targeted treatment of drug addiction. Front. Bioeng. Biotechnol. 2022, 10, 961728. [Google Scholar] [CrossRef]
- Brites, D.; Fernandes, A. Neuroinflammation and Depression: Microglia Activation, Extracellular Microvesicles and microRNA Dysregulation. Front. Cell. Neurosci. 2015, 9, 476. [Google Scholar] [CrossRef]
- Scheepstra, K.W.F.; Mizee, M.R.; van Scheppingen, J.; Adelia, A.; Wever, D.D.; Mason, M.R.J.; Dubbelaar, M.L.; Hsiao, C.C.; Eggen, B.J.L.; Hamann, J.; et al. Microglia Transcriptional Profiling in Major Depressive Disorder Shows Inhibition of Cortical Gray Matter Microglia. Biol. Psychiatry 2023, 94, 619–629. [Google Scholar] [CrossRef]
- Böttcher, C.; Fernández-Zapata, C.; Snijders, G.J.L.; Schlickeiser, S.; Sneeboer, M.A.M.; Kunkel, D.; De Witte, L.D.; Priller, J. Single-cell mass cytometry of microglia in major depressive disorder reveals a non-inflammatory phenotype with increased homeostatic marker expression. Transl. Psychiatry 2020, 10, 310. [Google Scholar] [CrossRef]
- Li, H.; Watkins, L.R.; Wang, X. Microglia in neuroimmunopharmacology and drug addiction. Mol. Psychiatry 2024. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Prinz, M. Microglia in steady state. J. Clin. Investig. 2017, 127, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Engel, P. Mechanisms underlying gut microbiota-host interactions in insects. J. Exp. Biol. 2021, 224, jeb207696. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.L.; Chen, J.G.; Wang, F. Microglia: A Central Player in Depression. Curr. Med. Sci. 2020, 40, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Bisagno, V. Glial-neuronal ensembles: Partners in drug addiction- associated synaptic plasticity. Front. Pharmacol. 2014, 5, 111326. [Google Scholar] [CrossRef] [PubMed]
- Lacagnina, M.J.; Rivera, P.D.; Bilbo, S.D. Glial and Neuroimmune Mechanisms as Critical Modulators of Drug Use and Abuse. Neuropsychopharmacology 2016, 42, 156–177. [Google Scholar] [CrossRef]
- Li, L.; Acioglu, C.; Heary, R.F.; Elkabes, S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain. Behav. Immun. 2021, 91, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Reverte, I.; Marchetti, C.; Pezza, S.; Zenoni, S.F.; Scaringi, G.; Ferrucci, L.; D’Ottavio, G.; Pignataro, A.; Andolina, D.; Raspa, M.; et al. Microglia-mediated calcium-permeable AMPAR accumulation in the nucleus accumbens drives hyperlocomotion during cocaine withdrawal. Brain. Behav. Immun. 2024, 115, 535–542. [Google Scholar] [CrossRef]
- Stoklund Dittlau, K.; Freude, K. Astrocytes: The Stars in Neurodegeneration? Biomolecules 2024, 14, 289. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Kotliarova, A.; Sidorova, Y.A. Glial Cell Line-Derived Neurotrophic Factor Family Ligands, Players at the Interface of Neuroinflammation and Neuroprotection: Focus Onto the Glia. Front. Cell. Neurosci. 2021, 15, 679034. [Google Scholar] [CrossRef] [PubMed]
- Zinchuk, M.S.; Guekht, A.B.; Druzhkova, T.A.; Gulyaeva, N.V.; Shpak, A.A. Glial cell line-derived neurotrophic factor (GDNF) in blood serum and lacrimal fluid of patients with a current depressive episode. J. Affect. Disord. 2022, 318, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Jurga, A.M.; Paleczna, M.; Kadluczka, J.; Kuter, K.Z. Beyond the GFAP-Astrocyte Protein Markers in the Brain. Biomolecules 2021, 11, 1361. [Google Scholar] [CrossRef]
- Heir, R.; Abbasi, Z.; Komal, P.; Altimimi, H.F.; Franquin, M.; Moschou, D.; Chambon, J.; Stellwagen, D. Astrocytes Are the Source of TNF Mediating Homeostatic Synaptic Plasticity. J. Neurosci. 2024, 44, e2278222024. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Guerrero, S.; Guardo-Maya, S.; Medina-Rincón, G.J.; Orrego-González, E.E.; Cabezas-Pérez, R.; González-Reyes, R.E. Taurine and Astrocytes: A Homeostatic and Neuroprotective Relationship. Front. Mol. Neurosci. 2022, 15, 937789. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front. Neurosci. 2022, 16, 824888. [Google Scholar] [CrossRef]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Rajkowska, G.; Stockmeier, C. Astrocyte pathology in major depressive disorder: Insights from human postmortem brain tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef]
- Czéh, B.; Di Benedetto, B. Antidepressants act directly on astrocytes: Evidences and functional consequences. Eur. Neuropsychopharmacol. 2013, 23, 171–185. [Google Scholar] [CrossRef]
- Zhou, X.; Xiao, Q.; Xie, L.; Yang, F.; Wang, L.; Tu, J. Astrocyte, a Promising Target for Mood Disorder Interventions. Front. Mol. Neurosci. 2019, 12, 136. [Google Scholar] [CrossRef]
- Tsai, S.F.; Hsu, P.L.; Chen, Y.W.; Hossain, M.S.; Chen, P.C.; Tzeng, S.F.; Chen, P.S.; Kuo, Y.M. High-fat diet induces depression-like phenotype via astrocyte-mediated hyperactivation of ventral hippocampal glutamatergic afferents to the nucleus accumbens. Mol. Psychiatry 2022, 27, 4372–4384. [Google Scholar] [CrossRef]
- O’Leary, L.A.; Mechawar, N. Implication of cerebral astrocytes in major depression: A review of fine neuroanatomical evidence in humans. Glia 2021, 69, 2077–2099. [Google Scholar] [CrossRef] [PubMed]
- Marshak, D. S100 beta as a neurotrophic factor. Prog. Brain Res. 1990, 86, 169–181. [Google Scholar]
- Du, J.; Yi, M.; Zhou, F.; He, W.; Yang, A.; Qiu, M.; Huang, H. S100B is selectively expressed by gray matter protoplasmic astrocytes and myelinating oligodendrocytes in the developing CNS. Mol. Brain 2021, 14, 154. [Google Scholar] [CrossRef]
- Charvériat, M.; Guiard, B.P. Serotonergic neurons in the treatment of mood disorders: The dialogue with astrocytes. Prog. Brain Res. 2021, 259, 197–228. [Google Scholar] [CrossRef]
- Quesseveur, G.; David, D.J.; Gaillard, M.C.; Pla, P.; Wu, M.V.; Nguyen, H.T.; Nicolas, V.; Auregan, G.; David, I.; Dranovsky, A.; et al. BDNF overexpression in mouse hippocampal astrocytes promotes local neurogenesis and elicits anxiolytic-like activities. Transl. Psychiatry 2013, 3, e253. [Google Scholar] [CrossRef]
- Malik, A.R.; Szydlowska, K.; Nizinska, K.; Asaro, A.; van Vliet, E.A.; Popp, O.; Dittmar, G.; Fritsche-Guenther, R.; Kirwan, J.A.; Nykjaer, A.; et al. SorCS2 Controls Functional Expression of Amino Acid Transporter EAAT3 and Protects Neurons from Oxidative Stress and Epilepsy-Induced Pathology. Cell Rep. 2019, 26, 2792–2804.e6. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Guan, X.; Liu, Z.; Pan, L.; Wang, Y.; Jia, X.; Yang, J.; Hou, T. SorCS2 is involved in promoting periodontitis-induced depression-like behaviour in mice. Oral Dis 2024. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.F.; Verkhratsky, A.; Tang, Y.; Illes, P. Astrocytes and major depression: The purinergic avenue. Neuropharmacology 2022, 220, 109252. [Google Scholar] [CrossRef]
- 194 González-Arias, C.; Sánchez-Ruiz, A.; Esparza, J.; Sánchez-Puelles, C.; Arancibia, L.; Ramírez-Franco, J.; Gobbo, D.; Kirchhoff, F.; Perea, G. Dysfunctional serotonergic neuron-astrocyte signaling in depressive-like states. Mol. Psychiatry 2023, 28, 3856–3873. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, M.M.; Korshunov, K.S.; Grant, R.A.; Martin, M.E.; Valencia, H.A.; Budinger, G.R.S.; Radulovic, J.; Prakriya, M. Astrocyte reactivity and inflammation-induced depression-like behaviors are regulated by Orai1 calcium channels. Nat. Commun. 2023, 14, 5500. [Google Scholar] [CrossRef]
- Yao, J.; Chen, C.; Guo, Y.; Yang, Y.; Liu, X.; Chu, S.; Ai, Q.; Zhang, Z.; Lin, M.; Yang, S.; et al. A Review of Research on the Association between Neuron–Astrocyte Signaling Processes and Depressive Symptoms. Int. J. Mol. Sci. 2023, 24, 6985. [Google Scholar] [CrossRef] [PubMed]
- Vestring, S.; Dorner, A.; Scholliers, J.; Ehrenberger, K.; Kiss, A.; Arenz, L.; Theiss, A.; Rossner, P.; Frase, S.; Du Vinage, C.; et al. D-Cycloserine enhances the bidirectional range of NMDAR-dependent hippocampal synaptic plasticity. Transl. Psychiatry 2024, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, S.S.; Buskila, Y. Astrocytic modulation of neuronal signalling. Front. Netw. Physiol. 2023, 3, 1205544. [Google Scholar] [CrossRef]
- Wang, J.; Holt, L.M.; Huang, H.H.; Sesack, S.R.; Nestler, E.J.; Dong, Y. Astrocytes in cocaine addiction and beyond. Mol. Psychiatry 2022, 27, 652–668. [Google Scholar] [CrossRef]
- Scofield, M.D.; Kalivas, P.W. Astrocytic dysfunction and addiction: Consequences of impaired glutamate homeostasis. Neuroscientist 2014, 20, 610–622. [Google Scholar] [CrossRef]
- Kruyer, A.; Kalivas, P.W. Astrocytes as cellular mediators of cue reactivity in addiction. Curr. Opin. Pharmacol. 2021, 56, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Holt, L.M.; Nestler, E.J. Astrocytic transcriptional and epigenetic mechanisms of drug addiction. J. Neural Transm. 2023, 131, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Michalski, J.P.; Kothary, R. Oligodendrocytes in a nutshell. Front. Cell. Neurosci. 2015, 9, 158759. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, J. Neuronal activity and remyelination: New insights into the molecular mechanisms and therapeutic advancements. Front. Cell Dev. Biol. 2023, 11, 1221890. [Google Scholar] [CrossRef]
- Bsibsi, M.; Nomden, A.; van Noort, J.M.; Baron, W. Toll-like receptors 2 and 3 agonists differentially affect oligodendrocyte survival, differentiation, and myelin membrane formation. J. Neurosci. Res. 2012, 90, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Toll-like receptors in the pathogenesis of neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Drevets, W.C.; Price, J.L. Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol. Psychiatry 2004, 55, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Miguel-Hidalgo, J. Gliogenesis and glial pathology in depression. CNS Neurol. Disord. Drug Targets 2007, 6, 219–233. [Google Scholar] [CrossRef]
- Zhou, B.; Zhu, Z.; Ransom, B.R.; Tong, X. Oligodendrocyte lineage cells and depression. Mol. Psychiatry 2020, 26, 103–117. [Google Scholar] [CrossRef]
- Szebeni, A.; Szebeni, K.; DiPeri, T.P.; Johnson, L.A.; Stockmeier, C.A.; Crawford, J.D.; Chandley, M.J.; Hernandez, L.J.; Burgess, K.C.; Brown, R.W.; et al. Elevated DNA Oxidation and DNA Repair Enzyme Expression in Brain White Matter in Major Depressive Disorder. Int. J. Neuropsychopharmacol. 2017, 20, 363–373. [Google Scholar] [CrossRef]
- Kokkosis, A.G.; Madeira, M.M.; Mullahy, M.R.; Tsirka, S.E. Chronic stress disrupts the homeostasis and progeny progression of oligodendroglial lineage cells, associating immune oligodendrocytes with prefrontal cortex hypomyelination. Mol. Psychiatry 2022, 27, 2833–2848. [Google Scholar] [CrossRef] [PubMed]
- Chandley, M.J.; Szebeni, A.; Szebeni, K.; Wang-Heaton, H.; Garst, J.; Stockmeier, C.A.; Lewis, N.H.; Ordway, G.A. Markers of elevated oxidative stress in oligodendrocytes captured from the brainstem and occipital cortex in major depressive disorder and suicide. Prog. Neuropsychopharmacol. Biol. Psychiatry 2022, 117, 110559. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yuan, J.; Dong, Y.; Jiang, S.; Zhang, M.; Zhao, X.; Ramos-Rodríguez, J.; Bermudez, B.; Liu, Y.; Yuan, J.; et al. Interaction between Oligodendrocytes and Interneurons in Brain Development and Related Neuropsychiatric Disorders. Int. J. Mol. Sci. 2024, 25, 3620. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fang, Y.; Cui, L.; Wang, Z.; Luo, Y.; Gao, C.; Ge, W.; Huang, T.; Wen, J.; Zhou, T. Butyrate emerges as a crucial effector of Zhi-Zi-Chi decoctions to ameliorate depression via multiple pathways of brain-gut axis. Biomed. Pharmacother. 2022, 149, 112861. [Google Scholar] [CrossRef] [PubMed]
- Reissner, K.J.; Pletnikov, M.V. Contributions of nonneuronal brain cells in substance use disorders. Neuropsychopharmacology 2020, 45, 224–225. [Google Scholar] [CrossRef] [PubMed]
- Velasco, B.; Mohamed, E.; Sato-Bigbee, C. Endogenous and exogenous opioid effects on oligodendrocyte biology and developmental brain myelination. Neurotoxicol. Teratol. 2021, 86, 107002. [Google Scholar] [CrossRef] [PubMed]
- Marguet, F.; Brosolo, M.; Friocourt, G.; Sauvestre, F.; Marcorelles, P.; Lesueur, C.; Marret, S.; Gonzalez, B.J.; Laquerrière, A. Oligodendrocyte lineage is severely affected in human alcohol-exposed foetuses. Acta Neuropathol. Commun. 2022, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.A.; Nishiyama, A. NG2 cells (polydendrocytes): Listeners to the neural network with diverse properties. Glia 2014, 62, 1195–1210. [Google Scholar] [CrossRef] [PubMed]
- Kirdajova, D.; Anderova, M. NG2 cells and their neurogenic potential. Curr. Opin. Pharmacol. 2020, 50, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.P.; Zhao, J.; Li, S. Roles of NG2 glial cells in diseases of the central nervous system. Neurosci. Bull. 2011, 27, 413–421. [Google Scholar] [CrossRef]
- Dimou, L.; Gallo, V. NG2-glia and their functions in the central nervous system. Glia 2015, 63, 1429–1451. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; Errede, M.; Girolamo, F.; Morando, S.; Ivaldi, F.; Panini, N.; Bendotti, C.; Perris, R.; Furlan, R.; Virgintino, D.; et al. NG2, a common denominator for neuroinflammation, blood–brain barrier alteration, and oligodendrocyte precursor response in EAE, plays a role in dendritic cell activation. Acta Neuropathol. 2016, 132, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Z.; Wang, Q.Q.; Yang, Q.Q.; Gu, H.Y.; Yin, Y.Q.; Li, Y.D.; Hou, J.C.; Chen, R.; Sun, Q.Q.; Sun, Y.F.; et al. NG2 glia regulate brain innate immunity via TGF-β2/TGFBR2 axis. BMC Med. 2019, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- 225 Hu, X.; Geng, P.; Zhao, X.; Wang, Q.; Liu, C.; Guo, C.; Dong, W.; Jin, X. The NG2-glia is a potential target to maintain the integrity of neurovascular unit after acute ischemic stroke. Neurobiol. Dis. 2023, 180, 106076. [Google Scholar] [CrossRef] [PubMed]
- Vélez-Fort, M.; Audinat, E.; Angulo, M.C. Functional α7-containing nicotinic receptors of NG2-expressing cells in the hippocampus. Glia 2009, 57, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, A.; Tascio, D.; Jabs, R.; Boehlen, A.; Domingos, C.; Skubal, M.; Huang, W.; Kirchhoff, F.; Henneberger, C.; Bilkei-Gorzo, A.; et al. Dysfunction of NG2 glial cells affects neuronal plasticity and behavior. Glia 2023, 71, 1481–1501. [Google Scholar] [CrossRef] [PubMed]
- Poggi, G.; Wennström, M.; Müller, M.B.; Treccani, G. NG2-glia: Rising stars in stress-related mental disorders? Mol. Psychiatry 2022, 28, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.A.; Wallis, G.J.; Wilcox, K.S. Reactivity and increased proliferation of NG2 cells following central nervous system infection with Theiler’s murine encephalomyelitis virus. J. Neuroinflamm. 2020, 17, 369. [Google Scholar] [CrossRef] [PubMed]
- Janeckova, L.; Knotek, T.; Kriska, J.; Hermanova, Z.; Kirdajova, D.; Kubovciak, J.; Berkova, L.; Tureckova, J.; Camacho Garcia, S.; Galuskova, K.; et al. Astrocyte-like subpopulation of NG2 glia in the adult mouse cortex exhibits characteristics of neural progenitor cells. Glia 2024, 72, 245–273. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, T.; Zhao, C.; Li, G. The Regulation of Exosome Generation and Function in Physiological and Pathological Processes. Int. J. Mol. Sci. 2023, 25, 255. [Google Scholar] [CrossRef]
- Sun, T.; Li, M.; Liu, Q.; Yu, A.; Cheng, K.; Ma, J.; Murphy, S.; McNutt, P.M.; Zhang, Y. Insights into optimizing exosome therapies for acute skin wound healing and other tissue repair. Front. Med. 2024, 18, 258–284. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Li, X.; Wang, Z.; Li, J.; Shahzad, K.; Zheng, J. Clinical applications of stem cell-derived exosomes. Signal Transduct. Target. Ther. 2024, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Somkuwar, S.S.; Staples, M.C.; Galinato, M.H.; Fannon, M.J.; Mandyam, C.D. Role of NG2 expressing cells in addiction: A new approach for an old problem. Front. Pharmacol. 2014, 5, 279. [Google Scholar] [CrossRef]
- Darbinian, N.; Darbinyan, A.; Merabova, N.; Bajwa, A.; Tatevosian, G.; Martirosyan, D.; Zhao, H.; Selzer, M.E.; Goetzl, L. Ethanol-mediated alterations in oligodendrocyte differentiation in the developing brain. Neurobiol. Dis. 2021, 148, 105181. [Google Scholar] [CrossRef] [PubMed]
- Moreira, F.P.; Medeiros, J.R.C.; Lhullier, A.C.; de Mattos Souza, L.D.; Jansen, K.; Portela, L.V.; Lara, D.R.; da Silva, R.A.; Wiener, C.D.; Oses, J.P. Cocaine abuse and effects in the serum levels of cytokines IL-6 and IL-10. Drug Alcohol Depend. 2016, 158, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Linker, K.E.; Cross, S.J.; Leslie, F.M. Glial mechanisms underlying substance use disorders. Eur. J. Neurosci. 2019, 50, 2574. [Google Scholar] [CrossRef] [PubMed]
- Agharahimi, M.; Badisa, R.B.; Mazzio, E.; Soliman, K.F.; Goodman, C.B. Cocaine potentiates an inflammatory response in C6 astroglia-like cells. Biomed. Rep. 2021, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.; Magalhães, C.; Andrade, E.B.; Magalhães, A.; Summavielle, T. The impact of psychostimulants on central and peripheral neuro-immune regulation: A scoping review of cytokine profiles and their implications for addiction. Front. Cell. Neurosci. 2023, 17, 1109611. [Google Scholar] [CrossRef]
- Niciu, M.J.; Henter, I.D.; Sanacora, G.; Zarate, C.A. Glial abnormalities in substance use disorders and depression: Does shared glutamatergic dysfunction contribute to comorbidity? World J. Biol. Psychiatry 2014, 15, 2–16. [Google Scholar] [CrossRef]
- Kruyer, A.; Scofield, M.D. Astrocytes in Addictive Disorders. Adv. Neurobiol. 2021, 26, 231–254. [Google Scholar] [CrossRef] [PubMed]
- Rebec, G.V.; Sun, W. Neuronal substrates of relapse to cocaine-seeking behavior: Role of prefrontal cortex. J. Exp. Anal. Behav. 2005, 84, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Kalivas, P.W.; LaLumiere, R.T.; Knackstedt, L.; Shen, H. Glutamate transmission in addiction. Neuropharmacology 2009, 56 (Suppl. S1), 169–173. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D. Potential of Glial Cell Modulators in the Management of Substance Use Disorders. CNS Drugs 2020, 34, 697–722. [Google Scholar] [CrossRef] [PubMed]
- Miguel-Hidalgo, J.J. The role of glial cells in drug abuse. Curr. Drug Abuse Rev. 2009, 2, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.; Kalivas, P.W. Glutamate Transport: A New Bench to Bedside Mechanism for Treating Drug Abuse. Int. J. Neuropsychopharmacol. 2017, 20, 797–812. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Patel, S.; Regan, M.R.; Haenggeli, C.; Huang, Y.H.; Bergles, D.E.; Jin, L.; Hoberg, M.D.; Vidensky, S.; Chung, D.S.; et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005, 433, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Das, S.C.; Yamamoto, B.K.; Hristov, A.M.; Sari, Y. Ceftriaxone attenuates ethanol drinking and restores extracellular glutamate concentration through normalization of GLT-1 in nucleus accumbens of male alcohol-preferring rats. Neuropharmacology 2015, 97, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Sari, Y.; Smith, K.D.; Ali, P.K.; Rebec, G.V. Upregulation of GLT1 attenuates cue-induced reinstatement of cocaine-seeking behavior in rats. J. Neurosci. 2009, 29, 9239–9243. [Google Scholar] [CrossRef]
- Sari, Y. Potential therapeutic role of glutamate transporter 1 for the treatment of alcohol dependence. OA Alcohol 2013, 1, 6. [Google Scholar] [CrossRef]
- Travaglianti, S.; Alotaibi, A.; Wong, W.; Abou-Gharbia, M.; Childers, W.; Sari, Y. Effects of novel GLT-1 modulator, MC-100093, on neuroinflammatory and neurotrophic biomarkers in mesocorticolimbic brain regions of male alcohol preferring rats exposed chronically to ethanol. Brain Res. Bull. 2024, 211, 110935. [Google Scholar] [CrossRef] [PubMed]
- McGinty, J.F.; Whitfield, T.W.; Berglind, W.J. Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 2010, 1314, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Berglind, W.J.; Whitfield, T.W.; Lalumiere, R.T.; Kalivas, P.W.; Mcginty, J.F. A single intra-PFC infusion of BDNF prevents cocaineinduced alterations in extracellular glutamate within the nucleus accumbens. J. Neurosci. 2009, 29, 3715–3719. [Google Scholar] [CrossRef]
- Nohesara, S.; Abdolmaleky, H.M.; Zhou, J.R.; Thiagalingam, S. Microbiota-Induced Epigenetic Alterations in Depressive Disorders Are Targets for Nutritional and Probiotic Therapies. Genes 2023, 14, 2217. [Google Scholar] [CrossRef] [PubMed]
- Soret, R.; Chevalier, J.; De Coppet, P.; Poupeau, G.; Derkinderen, P.; Segain, J.P.; Neunlist, M. Short-chain fatty acids regulate the enteric neurons and control gastrointestinal motility in rats. Gastroenterology 2010, 138, 1772–1782. [Google Scholar] [CrossRef]
- Candido, E.P.M.; Reeves, R.; Davie, J.R. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 1978, 14, 105–113. [Google Scholar] [CrossRef]
- Sealy, L.; Chalkley, R. The effect of sodium butyrate on histone modification. Cell 1978, 14, 115–121. [Google Scholar] [CrossRef]
- Schroeder, F.A.; Lin, C.L.; Crusio, W.E.; Akbarian, S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol. Psychiatry 2007, 62, 55–64. [Google Scholar] [CrossRef]
- Valvassori, S.; Varela, R.; Arent, C.; Dal-Pont, G.; Bobsin, T.; Budni, J.; Reus, G.; Quevedo, J. Sodium butyrate functions as an antidepressant and improves cognition with enhanced neurotrophic expression in models of maternal deprivation and chronic mild stress. Curr. Neurovasc. Res. 2014, 11, 359–366. [Google Scholar] [CrossRef]
- Valvassori, S.S.; Varela, R.B.; Resende, W.R.; Della, T.P.; Borba, L.A.; Behenck, J.P.; Réus, G.Z.; Quevedo, J. Antidepressant Effect of Sodium Butyrate is Accompanied by Brain Epigenetic Modulation in Rats Subjected to Early or Late Life Stress. Curr. Neurovasc. Res. 2023, 20, 586–598. [Google Scholar] [CrossRef]
- Palasz, E.; Wilkaniec, A.; Stanaszek, L.; Andrzejewska, A.; Adamczyk, A. Glia-Neurotrophic Factor Relationships: Possible Role in Pathobiology of Neuroinflammation-Related Brain Disorders. Int. J. Mol. Sci. 2023, 24, 6321. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Rossetti, A.C.; Racagni, G.; Gass, P.; Riva, M.A.; Molteni, R. Brain-derived neurotrophic factor: A bridge between inflammation and neuroplasticity. Front. Cell. Neurosci. 2014, 8, 430. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Zhang, J. Neuroinflammation, memory, and depression: New approaches to hippocampal neurogenesis. J. Neuroinflamm. 2023, 20, 283. [Google Scholar] [CrossRef] [PubMed]
- Luqman, A.; He, M.; Hassan, A.; Ullah, M.; Zhang, L.; Rashid Khan, M.; Din, A.U.; Ullah, K.; Wang, W.; Wang, G. Mood and microbes: A comprehensive review of intestinal microbiota’s impact on depression. Front. Psychiatry 2024, 15, 1295766. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Song, Z.; Lai, S.; Tang, F.; Dou, L.; Yang, F. Depression-associated gut microbes, metabolites and clinical trials. Front. Microbiol. 2024, 15, 1292004. [Google Scholar] [CrossRef]
- Garg, K.; Mohajeri, M.H. Potential effects of the most prescribed drugs on the microbiota-gut-brain-axis: A review. Brain Res. Bull. 2024, 207, 110883. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tizabi, Y.; Getachew, B.; Hauser, S.R.; Tsytsarev, V.; Manhães, A.C.; da Silva, V.D.A. Role of Glial Cells in Neuronal Function, Mood Disorders, and Drug Addiction. Brain Sci. 2024, 14, 558. https://doi.org/10.3390/brainsci14060558
Tizabi Y, Getachew B, Hauser SR, Tsytsarev V, Manhães AC, da Silva VDA. Role of Glial Cells in Neuronal Function, Mood Disorders, and Drug Addiction. Brain Sciences. 2024; 14(6):558. https://doi.org/10.3390/brainsci14060558
Chicago/Turabian StyleTizabi, Yousef, Bruk Getachew, Sheketha R. Hauser, Vassiliy Tsytsarev, Alex C. Manhães, and Victor Diogenes Amaral da Silva. 2024. "Role of Glial Cells in Neuronal Function, Mood Disorders, and Drug Addiction" Brain Sciences 14, no. 6: 558. https://doi.org/10.3390/brainsci14060558
APA StyleTizabi, Y., Getachew, B., Hauser, S. R., Tsytsarev, V., Manhães, A. C., & da Silva, V. D. A. (2024). Role of Glial Cells in Neuronal Function, Mood Disorders, and Drug Addiction. Brain Sciences, 14(6), 558. https://doi.org/10.3390/brainsci14060558