
Research Progress on the Pathogenesis, Diagnosis, and Drug Therapy of Alzheimer’s Disease

Abstract
:1. Introduction
2. Pathogenesis of Alzheimer’s Disease
2.1. Amyloid Cascade Hypothesis
2.2. Tau Protein Hyperphosphorylation Hypothesis
2.3. Inflammatory Hypothesis
2.4. Other Hypotheses
2.4.1. Abnormal Mitochondrial Autophagy
2.4.2. Cholinergic Theory
2.4.3. Insulin Resistance
2.4.4. Abnormal Gut Microflora
2.4.5. Presenilin Hypothesis
2.4.6. Calcium Hypothesis
2.4.7. Oxidative Stress
3. Diagnosis of Alzheimer’s Disease
3.1. Cerebrospinal Fluid Molecular Diagnosis
3.2. PET Neuroimaging Diagnosis
3.3. MRI Neuroimaging Diagnosis
3.4. Blood Tests
3.4.1. Plasma Testing
3.4.2. Blood–Brain Barrier Testing
3.4.3. Serum Testing
3.5. Artificial Intelligence Diagnosis
4. Therapy of Alzheimer’s Disease
4.1. Acetylcholinesterase Inhibitors
4.2. Glutamate Receptor Antagonists
4.3. Aβ Monoclonal Antibody
4.4. The Drug Targeting the Brain-Gut Axis
4.5. Other Drugs under Study
4.6. Nonpharmacological Therapy
5. Summary and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of Early Alzheimer’s Disease: Clinical Practice in 2021. JPAD-J. Prev. Alzheimers Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, S.; Carrillo, M.C.; Farias, S.T.; Goldberg, T.E.; Hendrix, J.A.; Jaeger, J.; Knopman, D.S.; Langbaum, J.B.; Park, D.C.; Ropacki, M.T.; et al. Measuring cognition and function in the preclinical stage of Alzheimer’s disease. Alzheimers Dement. 2018, 4, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Jian, B.; Liu, Z. Transmembrane Protein 175, a Lysosomal Ion Channel Related to Parkinson’s Disease. Biomolecules 2023, 13, 802. [Google Scholar] [CrossRef] [PubMed]
- Cramb, K.M.L.; Beccano-Kelly, D.; Cragg, S.J.; Wade-Martins, R. Impaired dopamine release in Parkinson’s disease. Brain 2023, 146, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022, 21, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.X.; Tian, Y.; Wang, Z.T.; Ma, Y.H.; Tan, L.; Yu, J.T. The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. JPAD-J. Prev. Alzheimers Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Grabher, B.J. Effects of Alzheimer Disease on Patients and Their Family. J. Nucl. Med. Technol. 2018, 46, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Mechanisms of Disease Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 328460. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bernstein, M.A.; Fox, N.C.; Thompson, P.; Alexander, G.; Harvey, D.; Borowski, B.; Britson, P.J.; Whitwell, J.L.; Ward, C.; et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J. Magn. Reson. Imaging 2008, 27, 685–691. [Google Scholar] [CrossRef]
- Hulstaert, F.; Blennow, K.; Ivanoiu, A.; Schoonderwaldt, H.C.; Riemenschneider, M.; De Deyn, P.P.; Bancher, C.; Cras, P.; Wiltfang, J.; Mehta, P.D.; et al. Improved discrimination of AD patients using β-amyloid((1-42)) and tau levels in CSF. Neurology 1999, 52, 1555–1562. [Google Scholar] [CrossRef]
- Balasundaram, A.; Srinivasan, S.; Prasad, A.; Malik, J.; Kumar, A. Hippocampus Segmentation-Based Alzheimer’s Disease Diagnosis and Classification of MRI Images. Arab. J. Sci. Eng. 2023, 48, 10249–10265. [Google Scholar] [CrossRef] [PubMed]
- Esumi, S.; Ushio, S.; Zamami, Y. Polypharmacy in Older Adults with Alzheimer’s Disease. Medicina 2022, 58, 1445. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.X.; Yu, F.; Lyu, Y.; Lu, X.F. Promising candidates from drug clinical trials: Implications for clinical treatment of Alzheimer’s disease in China. Front. Neurol 2022, 13, 1034243. [Google Scholar] [CrossRef] [PubMed]
- Alhazmi, H.A.; Albratty, M. An update on the novel and approved drugs for Alzheimer disease. Saudi Pharm. J. 2022, 30, 1755–1764. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef]
- Kamal, A.; Almenar-Queralt, A.; LeBlanc, J.F.; Roberts, E.A.; Goldstein, L.S.B. Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature 2001, 414, 643–648. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Tan, J.Z.A.; Gleeson, P.A. The trans-Golgi network is a major site for α-secretase processing of amyloid precursor protein in primary neurons. J. Biol. Chem. 2019, 294, 1618–1631. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.M.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Morkuniene, R.; Cizas, P.; Jankeviciute, S.; Petrolis, R.; Arandarcikaite, O.; Krisciukaitis, A.; Borutaite, V. Small Aβ1-42 oligomer-induced membrane depolarization of neuronal and microglial cells: Role of N-methyl-D-aspartate receptors. J. Neurosci. Res. 2015, 93, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Novak, M.; Edwards, P.C.; Klug, A.; Tichelaar, W.; Crowther, R.A. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 2020, 143, 2803–2817. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W.J. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Williamson, R.; Scales, T.; Clark, B.R.; Gibb, G.; Reynolds, C.H.; Kellie, S.; Bird, I.N.; Varndell, I.M.; Sheppard, P.W.; Everall, I.; et al. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-β peptide exposure: Involvement of src family protein kinases. J. Neurosci. 2002, 22, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Byers, H.L.; Wray, S.; Leung, K.Y.; Saxton, M.J.; Seereeram, A.; Reynolds, C.H.; Ward, M.A.; Anderton, B.H. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J. Biol. Chem. 2007, 282, 23645–23654. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.; Hanger, D.P.; Miller, C.C.J.; Lovestone, S. The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol 2013, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Grundke-Iqbal, I.; Iqbal, K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res. Mol. Brain Res. 1996, 38, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Tanimukai, H.; Grundke-Iqbal, I.; Iqbal, K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am. J. Pathol. 2005, 166, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Shen, T.; Tran, H.; Nichols, B.; et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 2017, 9, eaag0481. [Google Scholar] [CrossRef] [PubMed]
- Simic, G.; Leko, M.B.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milosevic, N.; Bazadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 464423. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.D.; Li, B.; Grundke-Iqbal, I.; Iqbal, K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc. Natl. Acad. Sci. USA 2006, 103, 8864–8869. [Google Scholar] [CrossRef]
- Goshima, Y.; Hida, T.; Gotoh, T. Computational Analysis of Axonal Transport: A Novel Assessment of Neurotoxicity, Neuronal Development and Functions. Int. J. Mol. Sci. 2012, 13, 3414–3430. [Google Scholar] [CrossRef]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of Tau Pathology by the Microglial Fractalkine Receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht-Talboo, J.; Landreth, G.E. Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 2005, 25, 8240–8249. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015, 6, 6176. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Jekabsone, A.; Mander, P.K.; Tickler, A.; Sharpe, M.; Brown, G.C. Fibrillar beta-amyloid peptide Aβ1-40 activates microglial proliferation via stimulating TNF-α release and H2O2 derived from NADPH oxidase: A cell culture study. J. Neuroinflammation 2006, 3, 24. [Google Scholar] [CrossRef]
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discovery 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.L.; Liu, Y.F.; Wang, X.Y.; Ma, H.B.; He, J.S.; Zhang, Y.; Yu, C.H.; Guan, W.J.; Ma, Y.H. The effects of amyloid-β42 oligomer on the proliferation and activation of astrocytes in vitro. In Vitr. Cell. Dev. Biol.-Anim. 2011, 47, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Maphis, N.; Xu, G.X.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Larke, L.E.C.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E.; Cuello, A.C.; Fisher, A. Reimagining cholinergic therapy for Alzheimer’s disease. Brain 2022, 145, 2250–2275. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Chen, Z.R.; Huang, J.B.; Yang, S.L.; Hong, F.F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Ferreira, S.T. Inflammation, Defective Insulin Signaling, and Mitochondrial Dysfunction as Common Molecular Denominators Connecting Type 2 Diabetes to Alzheimer Disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Vani, T.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Lacor, P.N.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Insulin Receptor Dysfunction Impairs Cellular Clearance of Neurotoxic Oligomeric Aβ. J. Biol. Chem. 2009, 284, 18742–18753. [Google Scholar] [CrossRef] [PubMed]
- Petra, A.I.; Panagiotidou, S.; Hatziagelaki, E.; Stewart, J.M.; Conti, P.; Theoharides, T.C. Gut-Microbiota-Brain Axis and Its Effect on Neuropsychiatric Disorders With Suspected Immune Dysregulation. Clin. Ther. 2015, 37, 984–995. [Google Scholar] [CrossRef]
- Pistollato, F.; Sumalla Cano, S.; Elio, I.; Masias Vergara, M.; Giampieri, F.; Battino, M. Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 2016, 74, 624–634. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Daulatzai, M.A. Chronic Functional Bowel Syndrome Enhances Gut-Brain Axis Dysfunction, Neuroinflammation, Cognitive Impairment, and Vulnerability to Dementia. Neurochem. Res. 2014, 39, 624–644. [Google Scholar] [CrossRef]
- Naseer, M.I.; Bibi, F.; Alqahtani, M.H.; Chaudhary, A.G.; Azhar, E.I.; Kamal, M.A.; Yasir, M. Role of Gut Microbiota in Obesity, Type 2 Diabetes and Alzheimer’s Disease. Cns Neurol. Disord.-Drug Targets 2014, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.D.C.; Diaz-Cintra, S.; et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Portet, F.; Dauvilliers, Y.; Campion, D.; Raux, G.; Hauw, J.J.; Lyon-Caen, O.; Camu, W.; Touchon, J. Very early onset AD with a de novo mutation in the presenilin 1 gene (Met 233 Leu). Neurology 2003, 61, 1136–1137. [Google Scholar] [CrossRef] [PubMed]
- Snider, B.J.; Norton, J.; Coats, M.A.; Chakraverty, S.; Hou, C.E.; Jervis, R.; Lendon, C.L.; Goate, A.M.; McKeel, D.W.; Morris, J.C. Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch. Neurol. 2005, 62, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Marks, A.R. Targeting ryanodine receptors to treat human diseases. J. Clin. Investig. 2023, 133, e162891. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.H.; Shineman, D.; Müller, M.; Cárdenas, C.; Mei, L.J.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.Y.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 57. [Google Scholar] [CrossRef]
- Gibson, G.E.; Chen, H.L.; Xu, H.; Qiu, L.H.; Xu, Z.S.; Denton, T.T.; Shi, Q.L. Deficits in the mitochondrial enzyme α-ketoglutarate dehydrogenase lead to Alzheimer’s disease-like calcium dysregulation. Neurobiol. Aging 2012, 33, 1121-e13. [Google Scholar] [CrossRef]
- Keller, J.N.; Guo, Q.; Holtsberg, F.W.; Bruce-Keller, A.J.; Mattson, M.P. Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 4439–4450. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.Y.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.C.; Zhong, C.J. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef] [PubMed]
- Lezza, A.M.; Boffoli, D.; Scacco, S.; Cantatore, P.; Gadaleta, M.N. Correlation between mitochondrial DNA 4977-bp deletion and respiratory chain enzyme activities in aging human skeletal muscles. Biochem. Biophys. Res. Commun. 1994, 205, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Han, S.H.; Son, S.M.; Hong, H.S.; Choi, Y.J.; Byun, J.; Mook-Jung, I. Mitochondria-Specific Accumulation of Amyloid β Induces Mitochondrial Dysfunction Leading to Apoptotic Cell Death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Calingasan, N.Y.; Yu, F.M.; Mauck, W.M.; Toidze, M.; Almeida, C.G.; Takahashi, R.H.; Carlson, G.A.; Beal, M.F.; Lin, M.T.; et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J. Neurochem. 2004, 89, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. SOD1 (Copper/Zinc Superoxide Dismutase) Deficiency Drives Amyloid β Protein Oligomerization and Memory Loss in Mouse Model of Alzheimer Disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Höltta, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Vanderstichele, H.; Bibl, M.; Engelborghs, S.; Le Bastard, N.; Lewczuk, P.; Molinuevo, J.L.; Parnetti, L.; Perret-Liaudet, A.; Shaw, L.M.; Teunissen, C.; et al. Standardization of preanalytical aspects of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagnosis: A consensus paper from the Alzheimer’s Biomarkers Standardization Initiative. Alzheimers Dement. 2012, 8, 65–73. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Mattsson, N.; Hansson, O.; Alzheimer’s Dis, N. Cerebrospinal fluid analysis detects cerebral amyloid-β accumulation earlier than positron emission tomography. Brain 2016, 139, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical diagnosis of Alzheimer’s disease: Recommendations of the International Working Group. Lancet Neurol. 2021, 20, 484–496. [Google Scholar] [CrossRef]
- Simonsen, A.H.; Herukka, S.K.; Andreasen, N.; Baldeiras, I.; Bjerke, M.; Blennow, K.; Engelborghs, S.; Frisoni, G.B.; Gabryelewicz, T.; Galluzzi, S.; et al. Recommendations for CSF AD biomarkers in the diagnostic evaluation of dementia. Alzheimers Dement. 2017, 13, 274–284. [Google Scholar] [CrossRef]
- Shoji, M.; Matsubara, E.; Kanai, M.; Watanabe, M.; Nakamura, T.; Tomidokoro, Y.; Shizuka, M.; Wakabayashi, K.; Igeta, Y.; Ikeda, Y.; et al. Combination assay of CSF tau, A beta 1-40 and A beta 1-42(43) as a biochemical marker of Alzheimer’s disease. J. Neurol. Sci. 1998, 158, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Lewczuk, P.; Esselmann, H.; Otto, M.; Maler, J.M.; Henkel, A.W.; Henkel, M.K.; Eikenberg, O.; Antz, C.; Krause, W.R.; Reulbach, U.; et al. Neurochemical diagnosis of Alzheimer’s dementia by CSF Aβ42, Aβ42/Aβ40 ratio and total tau. Neurobiol. Aging 2004, 25, 273–281. [Google Scholar] [CrossRef]
- Janelidze, S.; Zetterberg, H.; Mattsson, N.; Palmqvist, S.; Vanderstichele, H.; Lindberg, O.; van Westen, D.; Stomrud, E.; Minthon, L.; Blennow, K.; et al. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: Better diagnostic markers of Alzheimer disease. Ann. Clin. Transl. Neurol. 2016, 3, 154–165. [Google Scholar] [CrossRef]
- Chételat, G.; Arbizu, J.; Barthel, H.; Garibotto, V.; Law, I.; Morbelli, S.; van de Giessen, E.; Agosta, F.; Barkhof, F.; Brooks, D.J.; et al. Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. Lancet Neurol. 2020, 19, 951–962. [Google Scholar] [CrossRef]
- Shaffer, J.L.; Petrella, J.R.; Sheldon, F.C.; Choudhury, K.R.; Calhoun, V.D.; Edward Coleman, R.; Murali Doraiswamy, P. Predicting cognitive decline in subjects at risk for Alzheimer disease by using combined cerebrospinal fluid, MR imaging, and PET biomarkers. Radiology 2013, 266, 583–591. [Google Scholar] [CrossRef]
- Bergeron, D.; Gorno-Tempini, M.L.; Rabinovici, G.D.; Santos-Santos, M.A.; Seeley, W.; Miller, B.L.; Pijnenburg, Y.; Keulen, M.A.; Groot, C.; van Berckel, B.N.M.; et al. Prevalence of amyloid-β pathology in distinct variants of primary progressive aphasia. Ann. Neurol. 2018, 84, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, T.A.; Leuzy, A.; Therriault, J.; Chamoun, M.; Lussier, F.; Tissot, C.; Strandberg, O.; Palmqvist, S.; Stomrud, E.; Ferreira, P.C.L.; et al. Discriminative accuracy of the A/T/N scheme to identify cognitive impairment due to Alzheimer’s disease. Alzheimers Dement 2023, 15, e12390. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Boccardi, M.; Barkhof, F.; Blennow, K.; Cappa, S.; Chiotis, K.; Demonet, J.F.; Garibotto, V.; Giannakopoulos, P.; Gietl, A.; et al. Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol. 2017, 16, 661–676. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, B.C.; Wolk, D.A.; Alzheimers Dis Neuroimaging, I. MRI cortical thickness biomarker predicts AD-like CSF and cognitive decline in normal adults. Neurology 2012, 78, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Buckner, R.L.; Sepulcre, J.; Talukdar, T.; Krienen, F.M.; Liu, H.S.; Hedden, T.; Andrews-Hanna, J.R.; Sperling, R.A.; Johnson, K.A. Cortical Hubs Revealed by Intrinsic Functional Connectivity: Mapping, Assessment of Stability, and Relation to Alzheimer’s Disease. J. Neurosci. 2009, 29, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.G.; Wu, X.; Wang, Z.Q.; Zhang, N.; Dong, H.Q.; Yao, L.; Li, K.C. Impairment and compensation coexist in amnestic MCI default mode network. Neuroimage 2010, 50, 48–55. [Google Scholar] [CrossRef]
- Risacher, S.L.; Saykin, A.J.; West, J.D.; Shen, L.; Firpi, H.A.; McDonald, B.C.; ADNI. Baseline MRI Predictors of Conversion from MCI to Probable AD in the ADNI Cohort. Curr. Alzheimer Res. 2009, 6, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Leys, D.; Barkhof, F.; Huglo, D.; Weinstein, H.C.; Vermersch, P.; Kuiper, M.; Steinling, M.; Wolters, E.C.; Valk, J. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: Diagnostic value and neuropsychological correlates. J. Neurol. Neurosurg. Psychiatry 1992, 55, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Frühbeis, C.; Fröhlich, D.; Krämer-Albers, E.-M. Emerging Roles of Exosomes in Neuron–Glia Communication. Front Physiol. 2012, 3, 119. [Google Scholar] [CrossRef]
- Sinha, M.S.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma Exosomal miRNAs in Persons with and without Alzheimer Disease: Altered Expression and Prospects for Biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.B.; Vanderstichele, H.; Figurski, M.; Aisen, P.S.; Petersen, R.C.; Weiner, M.W.; Jack, C.R.; Jagust, W.; Decarli, C.; Toga, A.W.; et al. Factors affecting Aβ plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol. 2011, 122, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Lövheim, H.; Elgh, F.; Johansson, A.; Zetterberg, H.; Blennow, K.; Hallmans, G.; Eriksson, S. Plasma concentrations of free amyloid β cannot predict the development of Alzheimer’s disease. Alzheimers Dement. 2017, 13, 778–782. [Google Scholar] [CrossRef] [PubMed]
- van Oijen, M.; Hofman, A.; Soares, H.D.; Koudstaal, P.J.; Breteler, M.M.B. Plasma Aβ1-40 and Aβ1-42 and the risk of dementia: A prospective case-cohort study. Lancet Neurol. 2006, 5, 655–660. [Google Scholar] [CrossRef]
- Xu, W.H.; Kawarabayashi, T.; Matsubara, E.; Deguchi, K.; Murakami, T.; Harigaya, Y.; Ikeda, M.; Amari, M.; Kuwano, R.; Abe, K.; et al. Plasma antibodies to Aβ40 and Aβ42 in patients with Alzheimer’s disease and normal controls. Brain Res. 2008, 1219, 169–179. [Google Scholar] [CrossRef]
- Janelidze, S.; Stomrud, E.; Palmqvist, S.; Zetterberg, H.; van Westen, D.; Jeromin, A.; Song, L.; Hanlon, D.; Tan Hehir, C.A.; Baker, D.; et al. Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci. Rep. 2016, 6, 26801. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Stocker, H.; Nabers, A.; Perna, L.; Möllers, T.; Rujescu, D.; Hartmann, A.; Holleczek, B.; Schöttker, B.; Gerwert, K.; Brenner, H. Prediction of Alzheimer’s disease diagnosis within 14 years through Aβ misfolding in blood plasma compared to APOE4 status, and other risk factors. Alzheimers Dement. 2020, 16, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L.S.; Xiong, C.J.; et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019, 93, E1647–E1659. [Google Scholar] [CrossRef]
- Ashton, N.J.; Brum, W.S.; Di Molfetta, G.; Benedet, A.L.; Arslan, B.; Jonaitis, E.; Langhough, R.E.; Cody, K.; Wilson, R.; Carlsson, C.M.; et al. Diagnostic Accuracy of a Plasma Phosphorylated Tau 217 Immunoassay for Alzheimer Disease Pathology. JAMA Neurol. 2024, 81, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 70–76. [Google Scholar] [CrossRef]
- Laske, C.; Stransky, E.; Leyhe, T.; Eschweiler, G.W.; Wittorf, A.; Richartz, E.; Bartels, M.; Buchkremer, G.; Schott, K. Stage-dependent BDNF serum concentrations in Alzheimer’s disease. J. Neural Transm. 2006, 113, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Laske, C.; Stransky, E.; Leyhe, T.; Koehler, N.; Schott, K. P3–340: Decrease of BDNF serum concentration from MCI to early Alzheimer’s disease. Alzheimer’s Dement. 2006, 2, S475. [Google Scholar] [CrossRef]
- Maruszak, A.; Silajdzic, E.; Lee, H.; Murphy, T.; Liu, B.; Shi, L.; de Lucia, C.; Douiri, A.; Salta, E.; Nevado, A.J.; et al. Predicting progression to Alzheimer’s disease with human hippocampal progenitors exposed to serum. Brain 2023, 146, 2045–2058. [Google Scholar] [CrossRef]
- Zhao, Z.; Chuah, J.H.; Lai, K.W.; Chow, C.O.; Gochoo, M.; Dhanalakshmi, S.; Wang, N.; Bao, W.; Wu, X. Conventional machine learning and deep learning in Alzheimer’s disease diagnosis using neuroimaging: A review. Front. Comput. Neurosc. 2023, 17, 1038636. [Google Scholar] [CrossRef]
- Hansson, O.; Lehmann, S.; Otto, M.; Zetterberg, H.; Lewczuk, P. Advantages and disadvantages of the use of the CSF Amyloid (A) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimer’s Res. Ther. 2019, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; Dekosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.L.; Thal, L.J.; Gamzu, E.R.; Davis, C.S.; Woolson, R.F.; Gracon, S.I.; Drachman, D.A.; Schneider, L.S.; Whitehouse, P.J.; Hoover, T.M. A double-blind, placebo-controlled multicenter study of tacrine for Alzheimer’s disease. The Tacrine Collaborative Study Group. NEJM 1992, 327, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Aguilera, O.M.; Ismaili, L.; Iriepa, I.; Diez-Iriepa, D.; Chabchoub, F.; Marco-Contelles, J.; Pérez, M. Tacrines as Therapeutic Agents for Alzheimer’s Disease. V. Recent Developments. Chem. Rec. 2021, 21, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Min, S.L.S.; Liew, S.Y.; Chear, N.J.Y.; Goh, B.H.; Tan, W.N.; Khaw, K.Y. Plant Terpenoids as the Promising Source of Cholinesterase Inhibitors for Anti-AD Therapy. Biology 2022, 11, 307. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.B.; Zimmerman, H.J.; Knapp, M.J.; Gracon, S.I.; Lewis, K.W. Hepatotoxic Effects of Tacrine Administration in Patients With Alzheimer’s Disease. JAMA 1994, 271, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Cacabelos, R.; Oset-Gasque, M.J.; Samadi, A.; Marco-Contelles, J. Novel tacrine-related drugs as potential candidates for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2013, 23, 1916–1922. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Ogura, H.; Arai, Y.; Iimura, Y.; Yamanishi, Y. Research and development of donepezil hydrochloride, a new type of acetylcholinesterase inhibitor. Jpn. J. Pharmacol. 2002, 89, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef]
- Rogers, S.L.; Farlow, M.R.; Doody, R.S.; Mohs, R.; Friedhoff, L.T.; Donepezil Study Group. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology 1998, 50, 136–145. [Google Scholar] [CrossRef]
- Miculas, D.C.; Negru, P.A.; Bungau, S.G.; Behl, T.; ul Hassan, S.S.; Tit, D.M. Pharmacotherapy Evolution in Alzheimer’s Disease: Current Framework and Relevant Directions. Cells 2023, 12, 131. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.K.; Grossberg, G.T. Rivastigmine for Alzheimer’s disease. Expert Rev. Neurother. 2005, 5, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Jann, M.W. Rivastigmine, a new-generation cholinesterase inhibitor for the treatment of Alzheimer’s disease. Pharmacotherapy 2000, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Moreno, T.; González-Acedo, A.; Rivas-Domínguez, A.; García-Morales, V.; García-Cozar, F.J.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics 2022, 14, 1117. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.P.; Chang, L.R.; Song, Y.Z.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.D.; Pugh, S.; Laack, N.N.; Wefel, J.S.; Khuntia, D.; Meyers, C.; Choucair, A.; Fox, S.; Suh, J.H.; Roberge, D.; et al. Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: A randomized, double-blind, placebo-controlled trial. Neuro-oncology 2013, 15, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef]
- Vaz, M.; Silva, V.; Monteiro, C.; Silvestre, S. Role of Aducanumab in the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Clin. Interv. Aging 2022, 17, 797–810. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, J.; Williams, L.; Stella, H.; Leitermann, K.; Mikulskis, A.; O’Gorman, J.; Sevigny, J. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement 2016, 2, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Rezai, A.R.; D’Haese, P.-F.; Finomore, V.; Carpenter, J.; Ranjan, M.; Wilhelmsen, K.; Mehta, R.I.; Wang, P.; Najib, U.; Vieira Ligo Teixeira, C.; et al. Ultrasound Blood-Brain Barrier Opening and Aducanumab in Alzheimer’s Disease. N. Engl. J. Med. 2024, 390, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Chowdhury, N.S. Novel anti-amyloid-beta (Aβ) monoclonal antibody lecanemab for Alzheimer’s disease: A systematic review. Int. J. Immunopathol. Pharmacol. 2023, 37, 03946320231209839. [Google Scholar] [CrossRef] [PubMed]
- Monfared, A.A.T.; Tafazzoli, A.; Ye, W.C.; Chavan, A.; Zhang, Q.W. Long-Term Health Outcomes of Lecanemab in Patients with Early Alzheimer’s Disease Using Simulation Modeling. Neurol. Ther. 2022, 11, 863–880. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Apostolova, L.; Rabinovici, G.D.; Atri, A.; Aisen, P.; Greenberg, S.; Hendrix, S.; Selkoe, D.; Weiner, M.; Petersen, R.C.; et al. Lecanemab: Appropriate Use Recommendations. JPAD-J. Prev. Alzheimers Dis. 2023, 10, 362–377. [Google Scholar] [CrossRef]
- Xiao, S.F.; Chan, P.; Wang, T.; Hong, Z.; Wang, S.Z.; Kuang, W.H.; He, J.C.; Pan, X.P.; Zhou, Y.Y.; Ji, Y.; et al. A 36-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate Alzheimer’s dementia. Alzheimer’s Res. Ther. 2021, 13, 62. [Google Scholar] [CrossRef]
- Wang, X.Y.; Sun, G.Q.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.Q.; Chu, X.K.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Lu, J.J.; Pan, Q.Q.; Zhou, J.Q.; Weng, Y.; Chen, K.L.; Shi, L.; Zhu, G.X.; Chen, C.L.; Li, L.; Geng, M.Y.; et al. Pharmacokinetics, distribution, and excretion of sodium oligomannate, a recently approved anti-Alzheimer’s disease drug in China. J. Pharm. Anal. 2022, 12, 145–155. [Google Scholar] [CrossRef]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef]
- Price, B.R.; Sudduth, T.L.; Weekman, E.M.; Johnson, S.; Hawthorne, D.; Woolums, A.; Wilcock, D.M. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J. Neuroinflammation 2020, 17, 238. [Google Scholar] [CrossRef] [PubMed]
- Atwal, J.K.; Chen, Y.M.; Chiu, C.; Mortensen, D.L.; Meilandt, W.J.; Liu, Y.C.; Heise, C.E.; Hoyte, K.; Luk, W.; Lu, Y.M.; et al. A Therapeutic Antibody Targeting BACE1 Inhibits Amyloid-β Production in Vivo. Sci. Transl. Med. 2011, 3, 84ra43. [Google Scholar] [CrossRef] [PubMed]
- Pachón-Angona, I.; Maj, M.; Wnorowski, A.; Martin, H.; Józwiak, K.; Ismaili, L. Synthesis of new Hantzsch adducts showing Ca2+ channel blockade capacity, cholinesterase inhibition and antioxidant power. Future Med. Chem. 2021, 13, 1717–1729. [Google Scholar] [CrossRef] [PubMed]
- Nimmagadda, A.; Shi, Y.; Cai, J.F. γ-AApeptides as a New Strategy for Therapeutic Development. Curr. Med. Chem. 2019, 26, 2313–2329. [Google Scholar] [CrossRef] [PubMed]
- Vellas, B.; Sol, O.; Snyder, P.J.; Ousset, P.J.; Haddad, R.; Maurin, M.; Lemarié, J.C.; Désiré, L.; Pando, M.P.; Grp, E.H.T.S. EHT0202 in Alzheimer’s Disease: A 3-Month, Randomized, Placebo-Controlled, Double-Blind Study. Curr. Alzheimer Res. 2011, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N. Alzheimer’s Disease Drug Development Pipeline 2020. JPAD-J. Prev. Alzheimers Dis. 2020, 7, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.Q.; Vassar, R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Muntimadugu, E.; Dhommati, R.; Jain, A.; Gopala, V.; Challa, S.; Shaheen, M.; Khan, W. Intranasal delivery of nanoparticle encapsulated tarenflurbil: A potential brain targeting strategy for Alzheimer’s disease. Eur. J. Pharm. Sci. 2016, 92, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Soeda, Y.; Takashima, A. New Insights Into Drug Discovery Targeting Tau Protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Kellar, D.; Register, T.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal insulin modulates cerebrospinal fluid markers of neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A randomized trial. Sci. Rep. 2022, 12, 1346. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.E.; Loughrey, D.; Lawlor, B.A.; Robertson, I.H.; Walsh, C.; Brennan, S. The impact of cognitive training and mental stimulation on cognitive and everyday functioning of healthy older adults: A systematic review and meta-analysis. Ageing Res. Rev. 2014, 15, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.S.; de Leon, C.F.M.; Barnes, L.L.; Schneider, J.A.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Participation in cognitively stimulating activities and risk of incident Alzheimer disease. JAMA-J. Am. Med. Assoc. 2002, 287, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Abrahamson, K. How Exercise Influences Cognitive Performance When Mild Cognitive Impairment Exists A Literature Review. J. Psychosoc. Nurs. Ment. Health Serv. 2016, 54, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Buch, E.; Weber, C.; Cohen, L.G.; Braun, C.; Dimyan, M.A.; Ard, T.; Mellinger, J.; Caria, A.; Soekadar, S.; Fourkas, A.; et al. Think to move: A neuromagnetic brain-computer interface (BCI) system for chronic stroke. Stroke 2008, 39, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Wolpaw, J.R.; McFarland, D.J. Control of a two-dimensional movement signal by a noninvasive brain-computer interface in humans. Proc. Natl. Acad. Sci. USA 2004, 101, 17849–17854. [Google Scholar] [CrossRef]
- Little, S.; Pogosyan, A.; Neal, S.; Zavala, B.; Zrinzo, L.; Hariz, M.; Foltynie, T.; Limousin, P.; Ashkan, K.; FitzGerald, J.; et al. Adaptive Deep Brain Stimulation in Advanced Parkinson Disease. Ann. Neurol. 2013, 74, 449–457. [Google Scholar] [CrossRef]

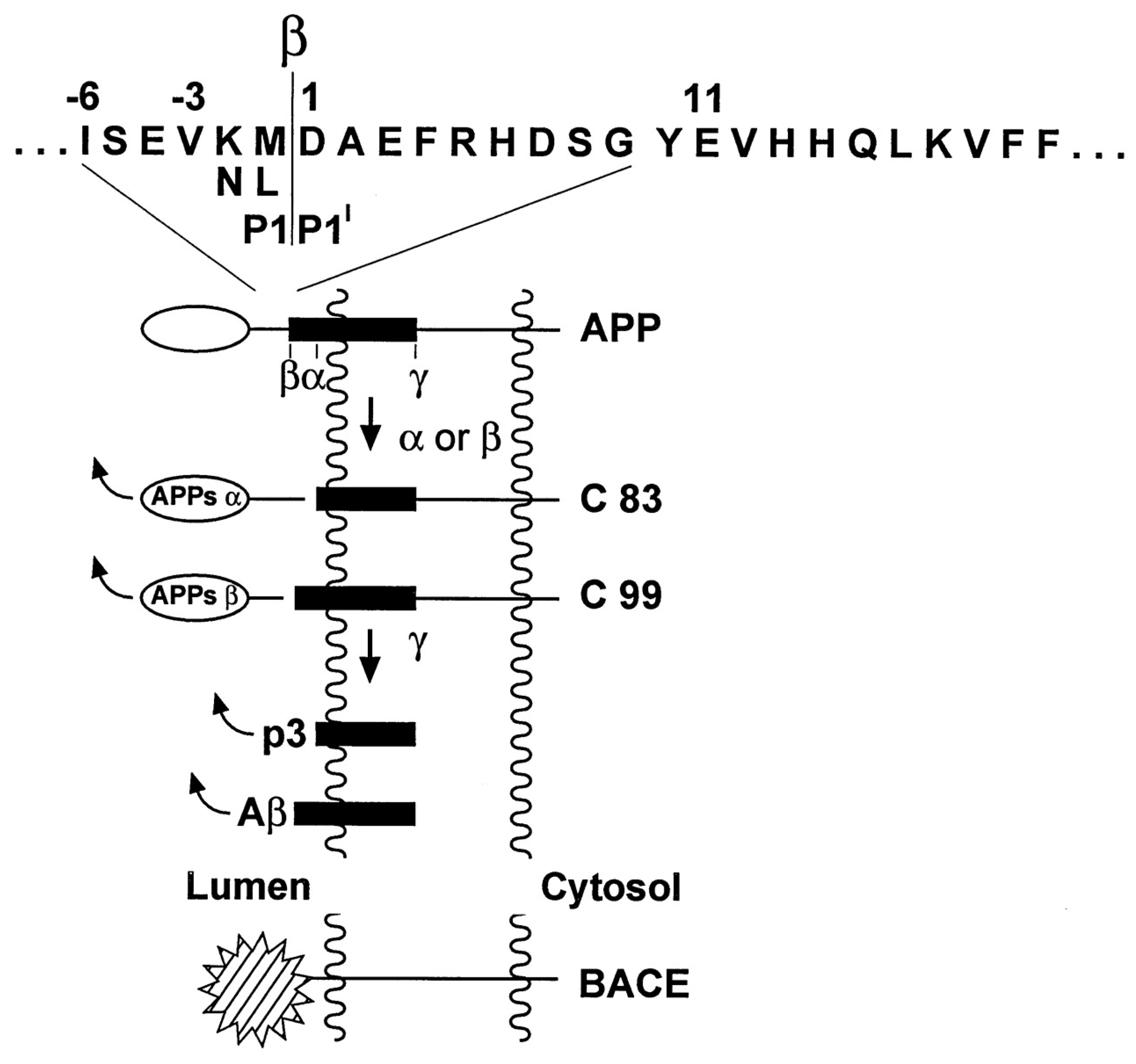

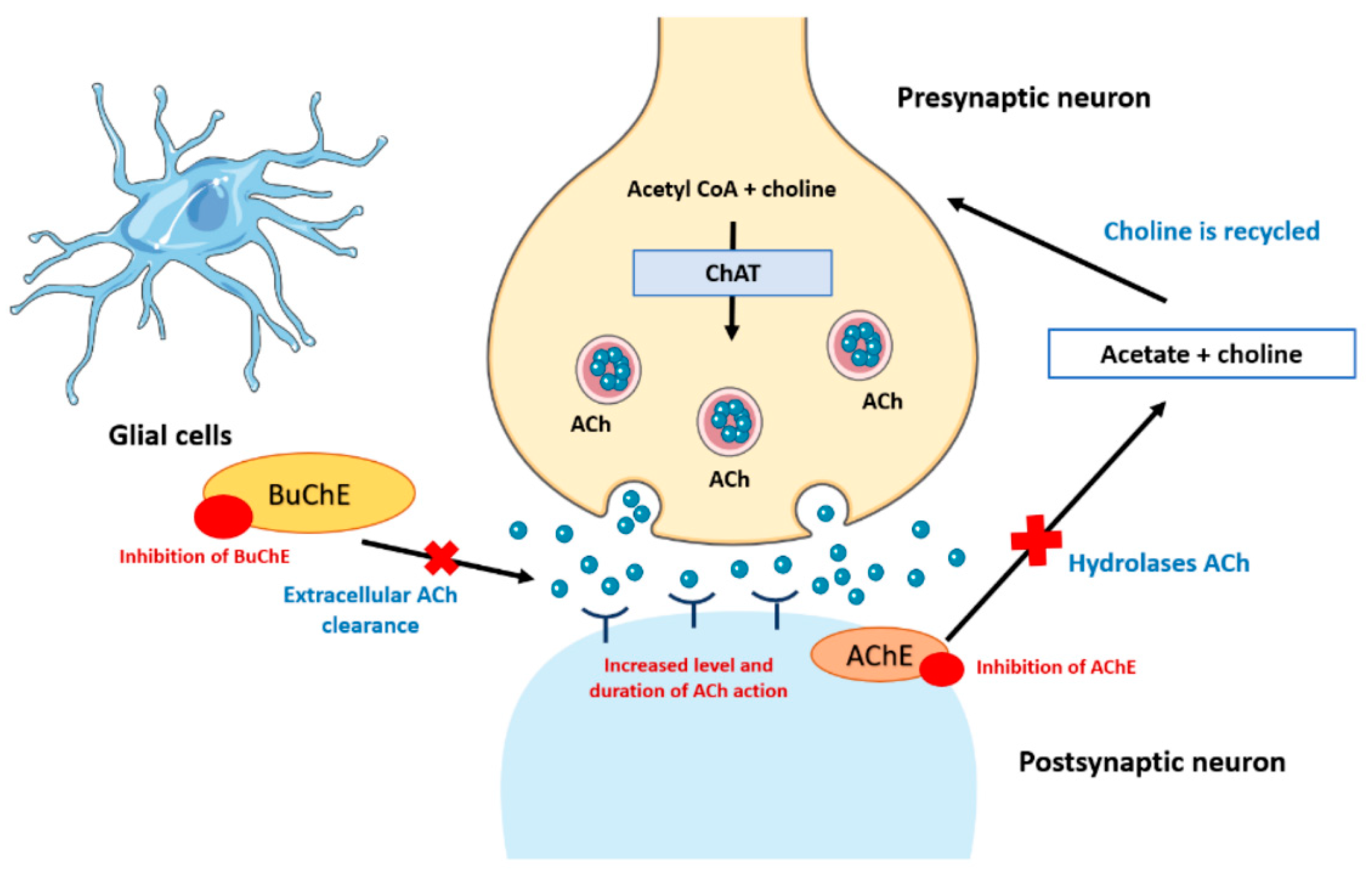


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Marker | Advantage | Disadvantage | References |
|---|---|---|---|---|
| CSF molecular diagnosis | Aβ42 T-Tau P-Tau | accurate | invasive, high rate of misclassification | [102,108,141] |
| PET | glucose metabolism Aβ, Tau protein | noninvasive, sensitive | expensive, confused with other diseases | [111,112] |
| MRI | medial temporal atrophy | noninvasive | expensive, confused with other diseases | [1,142] |
| blood tests | plasma exosomes plasma Aβ40, Aβ42, p-Tau serum blood–brain barrier | minimally invasive, diversity of markers | relatively less accurate | [120,122,129,139] |
| artificial intelligence diagnosis | a deep-learning model based on MRI and PET | efficient | immaturity of technology | [140] |
| Name of Drug | Type of Drug | Side Effect | Approved by | References |
|---|---|---|---|---|
| Tacrine | acetylcholinesterase inhibitor | strong hepatotoxicity | FDA | [146] |
| Donepezil | acetylcholinesterase inhibitor | insomnia, nausea, loss of appetite, muscle cramps, muscle weakness | FDA | [150] |
| Rivastigmine | acetylcholinesterase inhibitor | Relatively low side effects focusing on the gastrointestinal area | FDA | [154] |
| Galantamine | acetylcholinesterase inhibitor | convulsions, severe nausea, stomach cramps, vomiting, irregular breathing, confusion, muscle weakness | FDA | [149] |
| Memantine | glutamate receptor antagonist | dizziness, headache, and confusion | FDA | [158] |
| Aducanumab | Aβ monoclonal antibody | cerebral edema, sulcus effusion cerebral hemorrhage | FDA | [160] |
| Lecanemab | Aβ monoclonal antibody | minor cerebral hemorrhage rare macrohemorrhage | FDA | [166] |
| Sodium Oligomannate | the drug targeting the brain-gut axis | Relatively low side effects | China-FDA | [168,169] |
| Mechanism of Action | Phase of Clinical Trials | References | |
|---|---|---|---|
| (1) | Aβ aggregation inhibitors | in phase 2 | [174] |
| (2) | α-secretase modulators | most are in phase 2 | [175,176] |
| (3) | β-secretase inhibitors | most are in phase 1 and 2, the farthest and is in phase 2/3 | [171,177] |
| (4) | γ-secretase inhibitors | failed in phase 3 | [178] |
| (5) | inhibitors of Tau hyperphosphorylation | failed in phase 2 | [31] |
| (6) | Tau protein aggregation inhibitors | failed in phase 3 | [179] |
| (7) | drugs that enhance Tau clearance | in phase 1 | [179,180] |
| (8) | intranasal insulin | in phase 2 | [181] |
| (9) | TREM2-activating antibodies | preclinical | [171] |
| (10) | Ca2+ channel inhibitors | preclinical | [173] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Qiu, L. Research Progress on the Pathogenesis, Diagnosis, and Drug Therapy of Alzheimer’s Disease. Brain Sci. 2024, 14, 590. https://doi.org/10.3390/brainsci14060590
Yang Y, Qiu L. Research Progress on the Pathogenesis, Diagnosis, and Drug Therapy of Alzheimer’s Disease. Brain Sciences. 2024; 14(6):590. https://doi.org/10.3390/brainsci14060590
Chicago/Turabian StyleYang, Yixuan, and Lina Qiu. 2024. "Research Progress on the Pathogenesis, Diagnosis, and Drug Therapy of Alzheimer’s Disease" Brain Sciences 14, no. 6: 590. https://doi.org/10.3390/brainsci14060590