
Neuroprotective Potentials of Berberine in Rotenone-Induced Parkinson’s Disease-like Motor Symptoms in Rats
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Drugs
2.3. Experimental Groups and Drug Treatment
2.4. Measurement of Body Weight
2.5. Open Field Test
2.6. Bar Catalepsy Test
2.7. Beam-Crossing Task
2.8. Rotarod Activity
2.9. Grip Strength Test
- 0—Fall off;
- 1—Hangs onto the wire using two forepaws;
- 2—Same as 1, but endeavors to climb on the wire;
- 3—Hangs onto the wire with two forepaws and one or both hind paws;
- 4—Hangs onto the wire with all forepaws plus the tail wrapped around the wire;
- 5—Escapes from the apparatus and falls onto the level surface.
2.10. Dissection and Homogenization
2.11. Estimation of Nitrite
2.12. Assessment of Lipid Peroxidative Indices
2.13. Measurement of Glutathione (GSH)
2.14. Measurement of Superoxide Dismutase (SOD) Activity
2.15. Measurement of Catalase (CAT) Activity
2.16. Measurement of Mitochondrial Function
2.17. Measurement of Neuroinflammatory Markers
2.18. Caspase-3 Colorimetric Assay
2.19. Determination of Protein
2.20. Statistical Analysis
3. Results
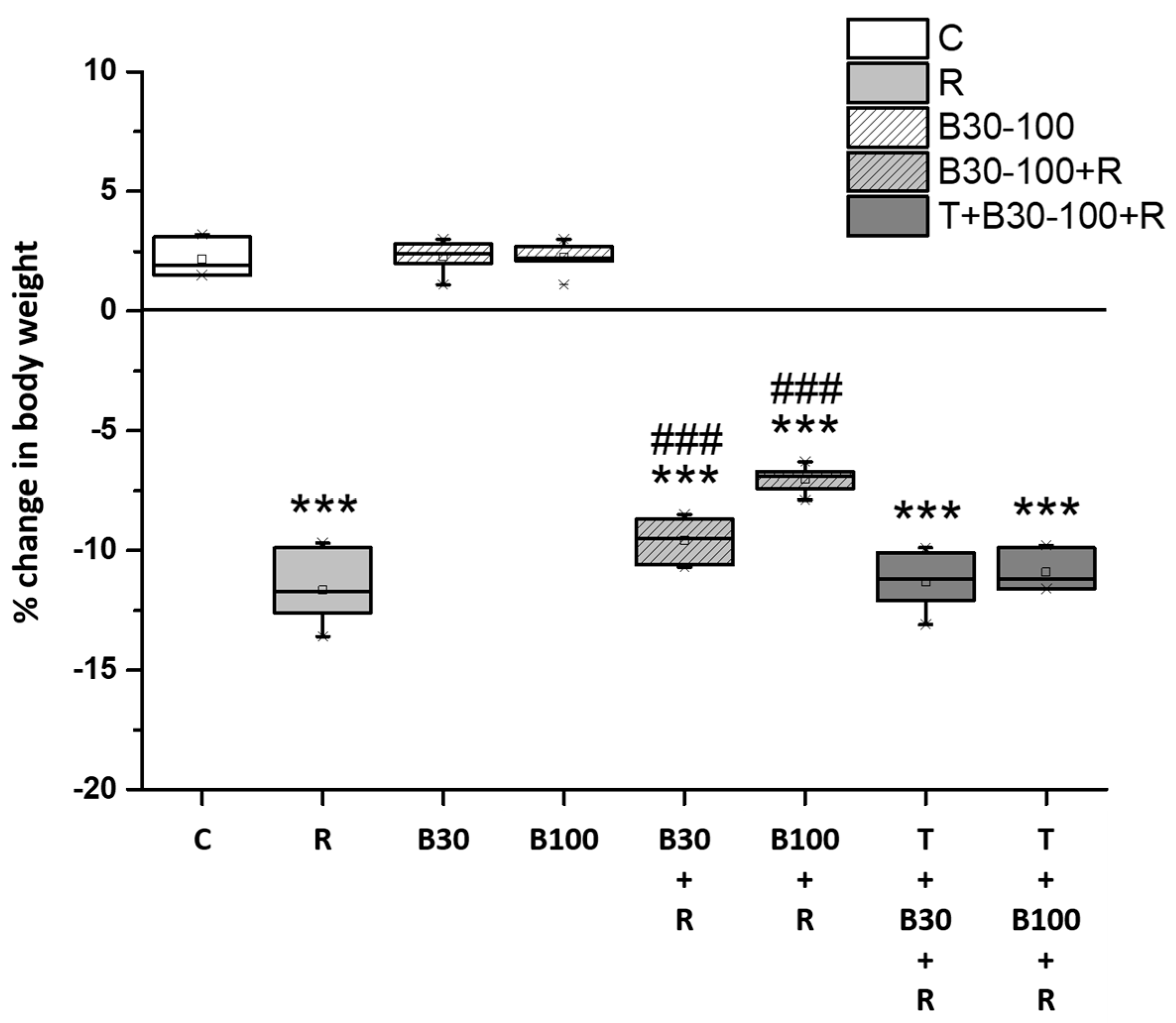
3.1. BBR Treatment Prevented RTN-Induced Decreases in Body Weight
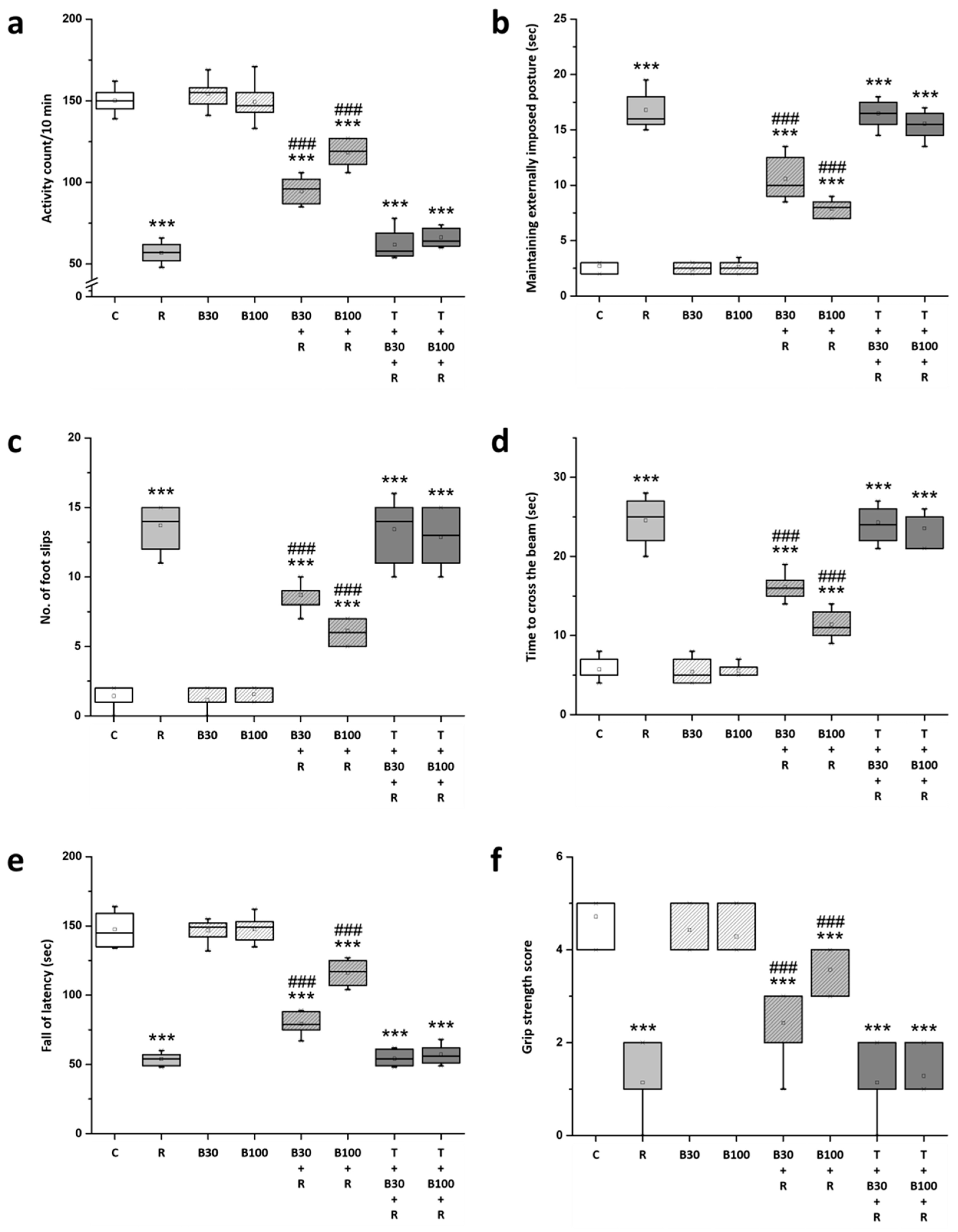
3.2. BBR Treatment Prevented RTN-Induced Motor Impairment
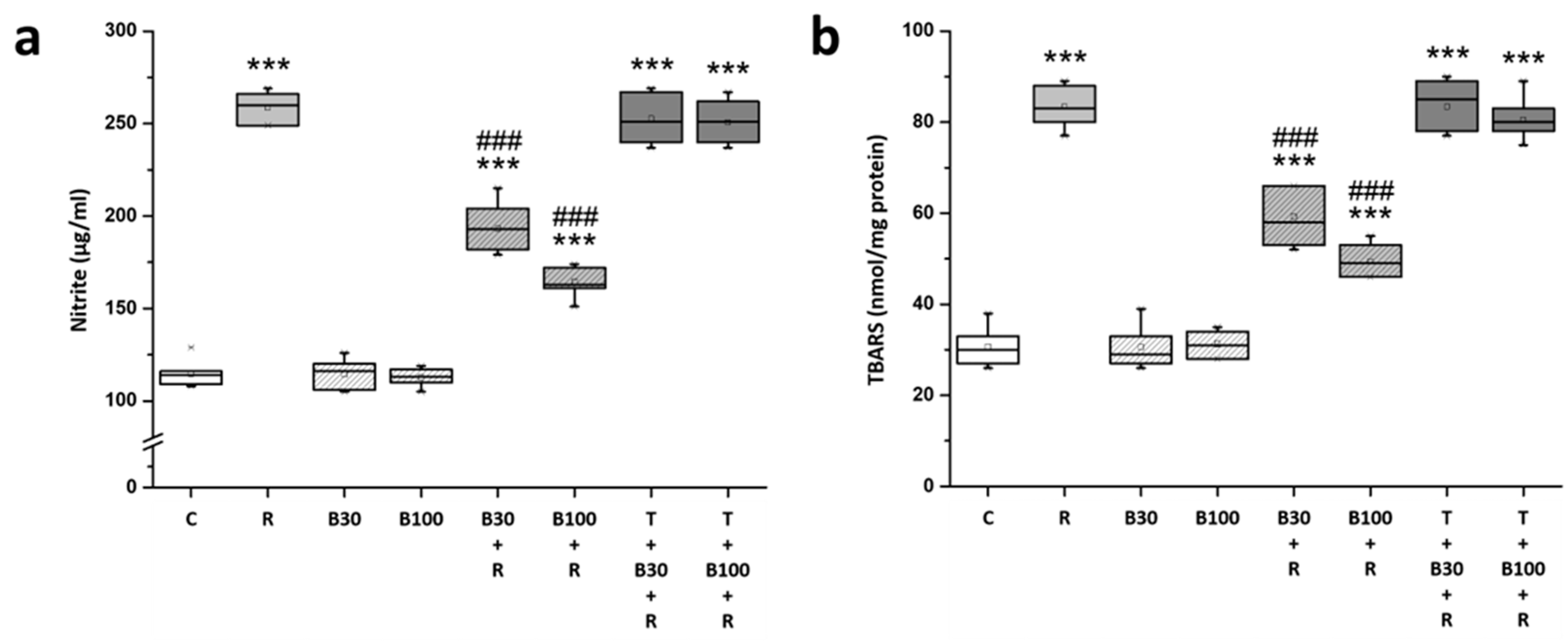
3.3. BBR Treatment Inhibited RTN-Induced Increases in Striatal Nitric Oxide and Lipid Peroxide Production
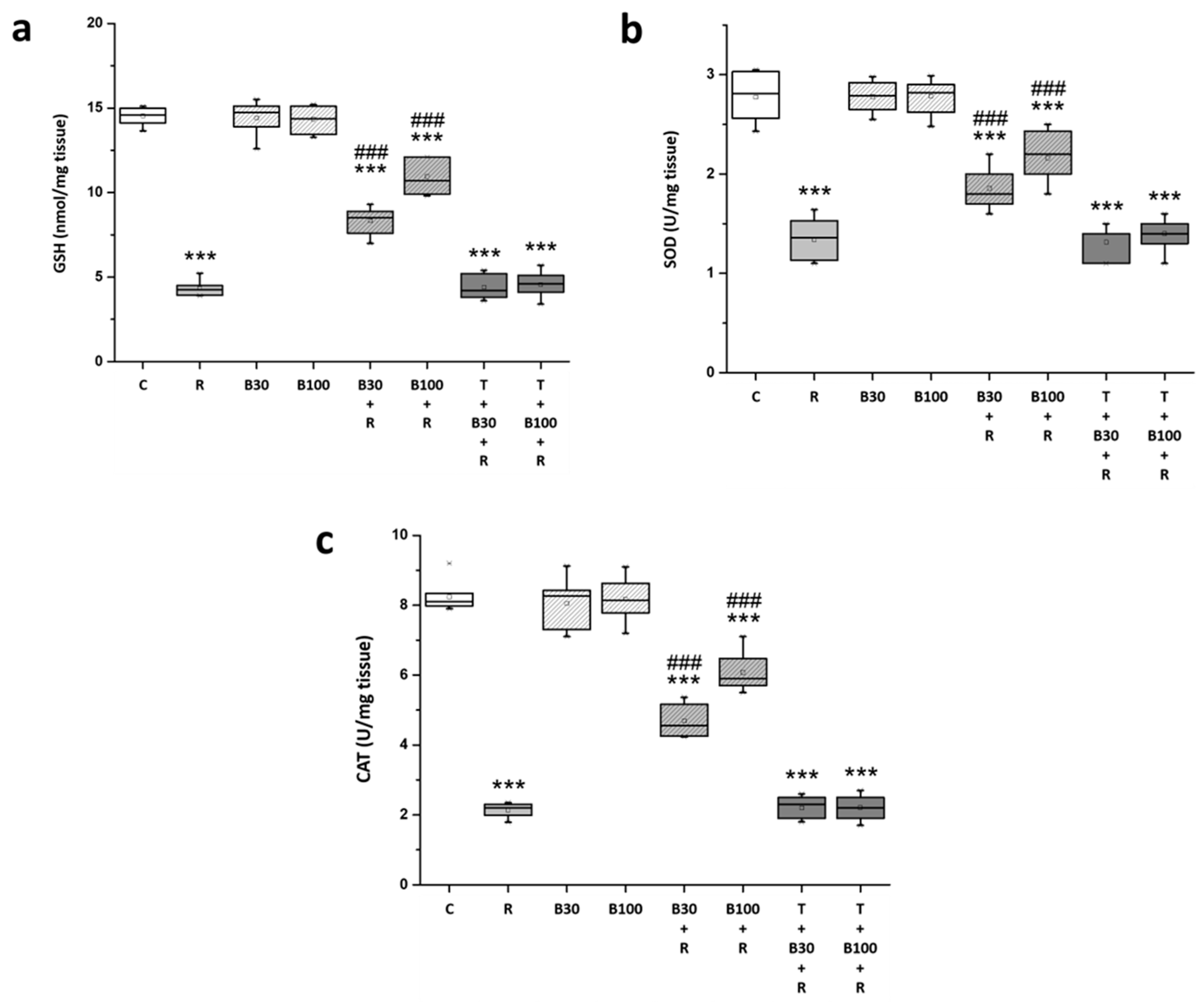
3.4. BBR Treatment Prevented RTN-Induced Decreases in Striatal Antioxidation Power
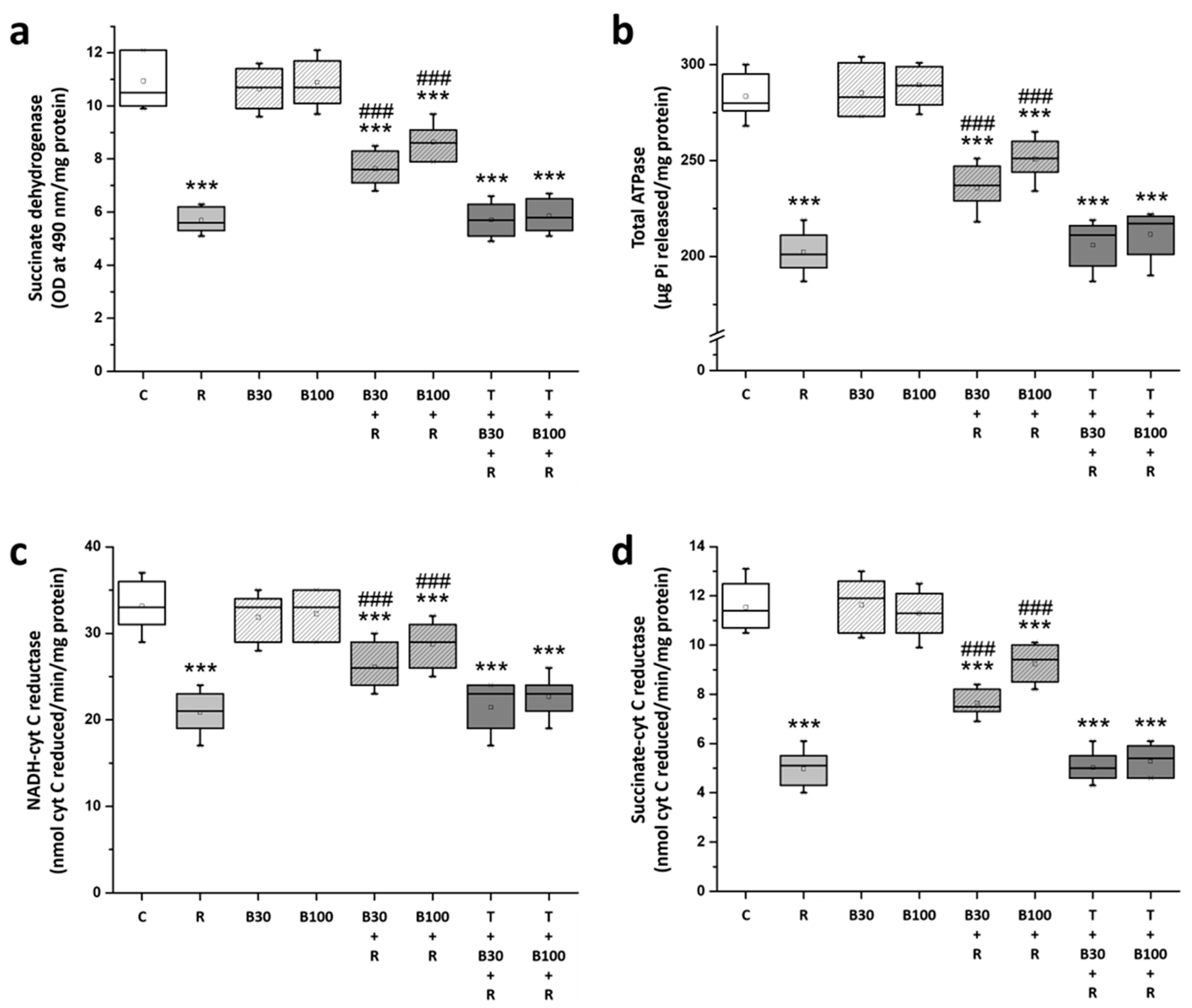
3.5. BBR Treatment Prevented RTN-Induced Striatal Mitochondrial Dysfunction
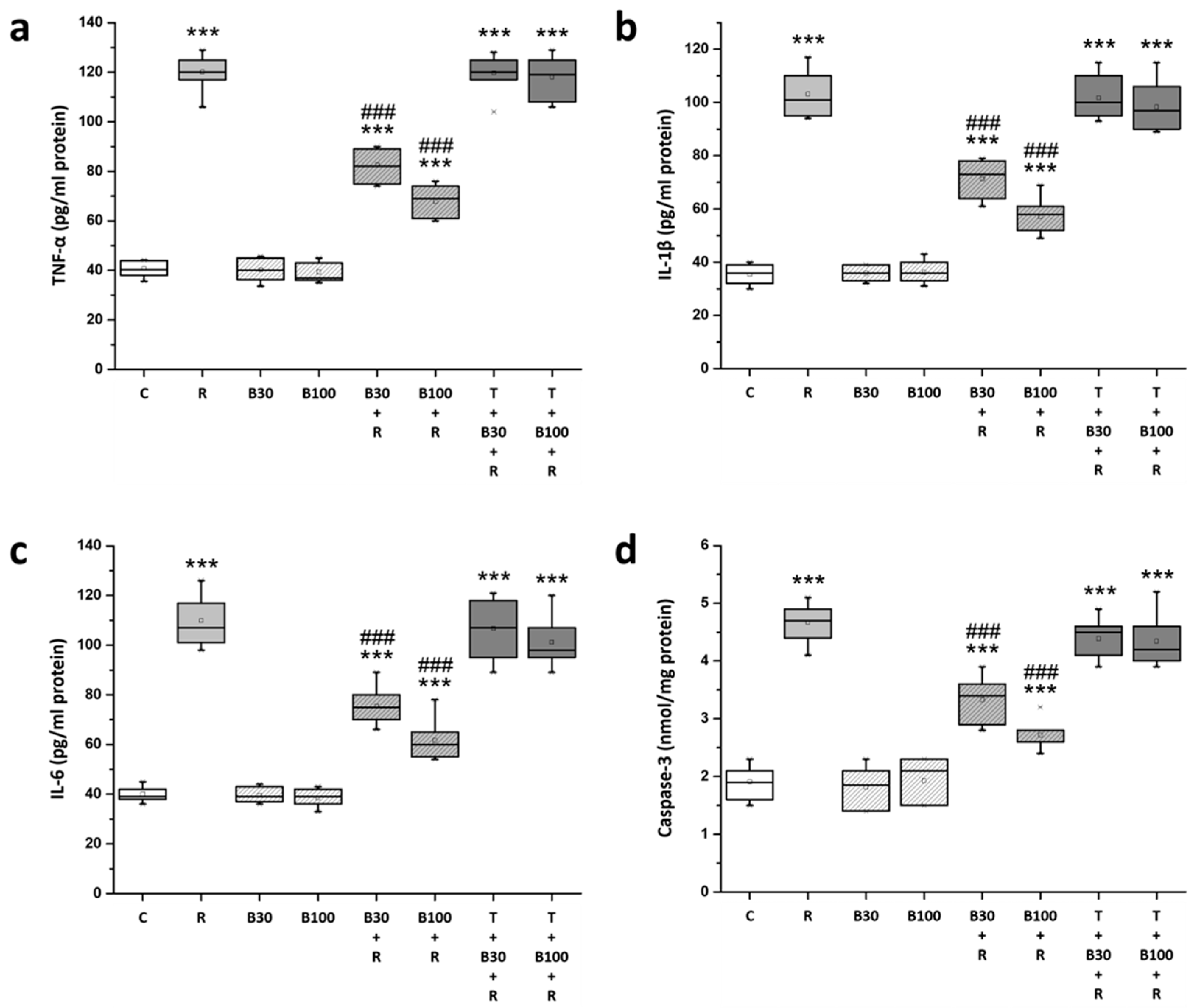
3.6. BBR Treatment Inhibited RTN-Induced Increases in Striatal Neuroinflammatory and Apoptotic Markers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ngo, K.J.; Paul, K.C.; Wong, D.; Kusters, C.D.J.; Bronstein, J.M.; Ritz, B.; Fogel, B.L. Lysosomal genes contribute to Parkinson’s disease near agriculture with high intensity pesticide use. NPJ Park. Dis. 2024, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.; Kaas, B. Parkinson’s disease: Risk factor modification and prevention. Semin. Neurol. 2022, 42, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.G.; Chan, B.J.L.; Koh, R.Y.; Ng, K.Y.; Chye, S.M. Prevention of Parkinson’s Disease: From Risk Factors to Early Interventions. CNS Neurol. Disord. Drug Targets 2024, 23, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Gutiérrez, M.T.; Serrano-García, N.; Orozco-Ibarra, M. Rotenone-induced model of parkinson’s disease: Beyond mitochondrial complex I inhibition. Mol. Neurobiol. 2023, 60, 1929–1948. [Google Scholar] [CrossRef] [PubMed]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease; Exon Publications: Brisbane, Australia, 2018; pp. 83–106. [Google Scholar]
- Lama, J.; Buhidma, Y.; Fletcher, E.J.; Duty, S. Animal models of Parkinson’s disease: A guide to selecting the optimal model for your research. Neuronal Signal. 2021, 5, NS20210026. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-N.; Wang, M.-H.; Soung, H.-S.; Chen, S.-M.; Fang, C.-H.; Lin, Y.-W.; Tseng, H.-C. L-theanine ameliorated rotenone-induced Parkinsonism-like symptoms in rats. Neurotox. Res. 2022, 40, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Garabadu, D.; Agrawal, N. Naringin exhibits neuroprotection against rotenone-induced neurotoxicity in experimental rodents. Neuromol. Med. 2020, 22, 314–330. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.-C.; Wang, M.-H.; Chang, K.-C.; Soung, H.-S.; Fang, C.-H.; Lin, Y.-W.; Li, K.-Y.; Yang, C.-C.; Tsai, C.-C. Protective effect of (−) epigallocatechin-3-gallate on rotenone-induced parkinsonism-like symptoms in rats. Neurotox. Res. 2020, 37, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Farombi, E.O.; Awogbindin, I.O.; Farombi, T.H.; Oladele, J.O.; Izomoh, E.R.; Aladelokun, O.B.; Ezekiel, I.O.; Adebambo, O.I.; Abah, V.O. Neuroprotective role of kolaviron in striatal redo-inflammation associated with rotenone model of Parkinson’s disease. Neurotoxicology 2019, 73, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Mansour, R.M.; Ahmed, M.A.; El-Sahar, A.E.; El Sayed, N.S. Montelukast attenuates rotenone-induced microglial activation/p38 MAPK expression in rats: Possible role of its antioxidant, anti-inflammatory and antiapoptotic effects. Toxicol. Appl. Pharmacol. 2018, 358, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kumar, P.; Deshmukh, R. Neuroprotective potential of spermidine against rotenone induced Parkinson’s disease in rats. Neurochem. Int. 2018, 116, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Zaitone, S.A.; Ahmed, E.; Elsherbiny, N.M.; Mehanna, E.T.; El-Kherbetawy, M.K.; ElSayed, M.H.; Alshareef, D.M.; Moustafa, Y.M. Caffeic acid improves locomotor activity and lessens inflammatory burden in a mouse model of rotenone-induced nigral neurodegeneration: Relevance to Parkinson’s disease therapy. Pharmacol. Rep. 2019, 71, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Gasmi, A.; Asghar, F.; Zafar, S.; Oliinyk, P.; Khavrona, O.; Lysiuk, R.; Peana, M.; Piscopo, S.; Antonyak, H.; Pen, J.J. Berberine: Pharmacological features in health, disease and aging. Curr. Med. Chem. 2024, 31, 1214–1234. [Google Scholar] [CrossRef] [PubMed]
- Imenshahidi, M.; Hosseinzadeh, H. Berberine and barberry (Berberis vulgaris): A clinical review. Phytother. Res. 2019, 33, 504–523. [Google Scholar] [CrossRef] [PubMed]
- Khoshandam, A.; Imenshahidi, M.; Hosseinzadeh, H. Pharmacokinetic of berberine, the main constituent of Berberis vulgaris L.: A comprehensive review. Phytother. Res. 2022, 36, 4063–4079. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, Q.; Ma, S.R.; Zhao, Z.X.; Pan, L.B.; Cong, L.; Han, P.; Peng, R.; Yu, H.; Lin, Y.; et al. Oral berberine improves brain dopa/dopamine levels to ameliorate Parkinson’s disease by regulating gut microbiota. Signal Transduct Target Ther. 2021, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Akbar, M.; Shabbir, A.; Rehman, K.; Akash, M.S.H.; Shah, M.A. Neuroprotective potential of berberine in modulating Alzheimer’s disease via multiple signaling pathways. J. Food Biochem. 2021, 45, e13936. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Fekri, H.S.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Therapeutic and biological activities of berberine: The involvement of Nrf2 signaling pathway. J. Cell. Biochem. 2020, 121, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wang, X.-C.; Fan, L.-L.; Zhu, Y.-H.; Wang, Q.; Chen, Y.-B. Berberine ameliorates lipopolysaccharide-induced cognitive impairment through SIRT1/NRF2/NF-κB signaling pathway in C57BL/6J Mice. Rejuvenation Res. 2022, 25, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Kang, C.; Che, S.; Su, J.; Sun, Q.; Ge, T.; Guo, Y.; Lv, J.; Sun, Z.; Yang, W. Berberine: A promising treatment for neurodegenerative diseases. Front. Pharmacol. 2022, 13, 845591. [Google Scholar] [CrossRef] [PubMed]
- Dadgostar, E.; Moghanlou, M.; Parvaresh, M.; Mohammadi, S.; Khandan, M.; Aschner, M.; Mirzaei, H.; Tamtaji, O.R. Can Berberine Serve as a New Therapy for Parkinson’s Disease? Neurotox. Res. 2022, 40, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Jia, Y.; Pan, D.; Ma, Z. Berberine alleviates rotenone-induced cytotoxicity by antioxidation and activation of PI3K/Akt signaling pathway in SH-SY5Y cells. Neuroreport 2020, 31, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Gendy, A.M.; Soubh, A.; Elnagar, M.R.; Hamza, E.; Ahmed, K.A.; Aglan, A.; El-Haddad, A.E.; Farag, M.A.; El-Sadek, H.M. New insights into the role of berberine against 3-nitropropionic acid-induced striatal neurotoxicity: Possible role of BDNF–TrkB–PI3K/Akt and NF-κB signaling. Food Chem. Toxicol. 2023, 175, 113721. [Google Scholar] [CrossRef] [PubMed]
- Kadir, A.; Singh, J.; Rahi, V.; Kumar, P. Berberine ameliorate haloperidol and 3-Nitropropionic acid-induced neurotoxicity in rats. Neurochem. Res. 2022, 47, 3285–3297. [Google Scholar] [CrossRef]
- Liu, D.-Q.; Chen, S.-P.; Sun, J.; Wang, X.-M.; Chen, N.; Zhou, Y.-Q.; Tian, Y.-K.; Ye, D.-W. Berberine protects against ischemia-reperfusion injury: A review of evidence from animal models and clinical studies. Pharmacol. Res. 2019, 148, 104385. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Shi, D.-D.; Li, W.; Cheng, D.; Zhang, Y.-D.; Zhang, S.; Tsoi, B.; Zhao, J.; Wang, Z.; Zhang, Z.-J. Berberine ameliorates depression-like behaviors in mice via inhibiting NLRP3 inflammasome-mediated neuroinflammation and preventing neuroplasticity disruption. J. Neuroinflamm. 2023, 20, 54. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, R.; Taab, Y.; Kiasalari, Z.; Afshin-Majd, S.; Baluchnejadmojarad, T.; Roghani, M. Berberine ameliorates intrahippocampal kainate-induced status epilepticus and consequent epileptogenic process in the rat: Underlying mechanisms. Biomed. Pharmacother. 2017, 87, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.-W.; Shaw, P.-C. Therapeutic efficacies of berberine against neurological disorders: An update of pharmacological effects and mechanisms. Cells 2022, 11, 796. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zou, Q.; Pu, Y.; Cai, Z.; Tang, Y. Berberine Rescues D-Ribose-Induced Alzheimer‘s Pathology via Promoting Mitophagy. Int. J. Mol. Sci. 2023, 24, 5896. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tian, Y.; Yan, F.; Zhao, F.; Wang, R.; Luo, Y.; Zheng, Y. Berberine protects against chronic cerebral hypoperfusion-induced cognitive impairment and hippocampal damage via regulation of the ERK/Nrf2 pathway. J. Chem. Neuroanat. 2022, 123, 102119. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.-N.; Cai, C.-Z.; Wu, M.-Y.; Su, H.-X.; Li, M.; Lu, J.-H. Neuroprotective effects of berberine in animal models of Alzheimer’s disease: A systematic review of pre-clinical studies. BMC Complement. Altern. Med. 2019, 19, 109. [Google Scholar] [CrossRef] [PubMed]
- Thangarajan, S.; Deivasigamani, A.; Natarajan, S.S.; Krishnan, P.; Mohanan, S.K. Neuroprotective activity of L-theanine on 3-nitropropionic acid-induced neurotoxicity in rat striatum. Int. J. Neurosci. 2014, 124, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kalonia, H.; Kumar, A. Novel protective mechanisms of antidepressants against 3-nitropropionic acid induced Huntington’s-like symptoms: A comparative study. J. Psychopharmacol. 2011, 25, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Tanabe, Y.; Fujii, Y.; Kikuta, T.; Shibata, H.; Shido, O. Chronic administration of docosahexaenoic acid ameliorates the impairment of spatial cognition learning ability in amyloid β–infused rats. J. Nutr. 2005, 135, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Misra, H.P.; Fridovich, I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [CrossRef] [PubMed]
- Beers, R.F.; Sizer, I.W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Moreadith, R.W.; Fiskum, G. Isolation of mitochondria from ascites tumor cells permeabilized with digitonin. Anal. Biochem. 1984, 137, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Gomez, C.; López-Cepero, J.M.; Boveris, A. Beneficial effects of moderate exercise on mice aging: Survival, behavior, oxidative stress, and mitochondrial electron transfer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R505–R511. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; McCoy, J.L.; Greenamyre, J.T. Phenothiazine normalizes the NADH/NAD+ ratio, maintains mitochondrial integrity and protects the nigrostriatal dopamine system in a chronic rotenone model of Parkinson’s disease. Redox Biol. 2019, 24, 101164. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Pace, L.; Bedse, G.; Michele Lavecchia, A.; De Marco, F.; Gaetani, S.; Serviddio, G. Glutamate and mitochondria: Two prominent players in the oxidative stress-induced neurodegeneration. Curr. Alzheimer Res. 2016, 13, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Resende, R.; Fernandes, T.; Pereira, A.C.; Marques, A.P.; Pereira, C.F. Endoplasmic reticulum-mitochondria contacts modulate reactive oxygen species-mediated signaling and oxidative stress in brain disorders: The key role of sigma-1 receptor. Antioxid. Redox Signal. 2022, 37, 758–780. [Google Scholar] [CrossRef] [PubMed]
- Fonnum, F.; Lock, E. The contributions of excitotoxicity, glutathione depletion and DNA repair in chemically induced injury to neurones: Exemplified with toxic effects on cerebellar granule cells. J. Neurochem. 2004, 88, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, R.; Lahti, A.; Kankaanranta, H.; Moilanen, E. Nitric oxide production and signaling in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2021, 41, 770–784. [Google Scholar] [CrossRef]
- Arab, H.H.; Safar, M.M.; Shahin, N.N. Targeting ROS-dependent AKT/GSK-3β/NF-κB and DJ-1/Nrf2 pathways by dapagliflozin attenuates neuronal injury and motor dysfunction in rotenone-induced Parkinson’s disease rat model. ACS Chem. Neurosci. 2021, 12, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.-H.; Wu, C.-W.; Lee, Y.-C.; Hung, C.-Y.; Chen, I.-C.; Wu, K.L. Nrf2 activation attenuates the early suppression of mitochondrial respiration due to the α-synuclein overexpression. Biomed. J. 2018, 41, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Moreira, S.; Fonseca, I.; Nunes, M.J.; Rosa, A.; Lemos, L.; Rodrigues, E.; Carvalho, A.N.; Outeiro, T.F.; Rodrigues, C.M.P.; Gama, M.J. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Exp. Neurol. 2017, 295, 77–87. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental | Treatment | n = 8 | |
|---|---|---|---|
| Group | ♂:♀ = 4:4 | ||
| 1. | C | control (normal saline; i.p. + distilled water; p.o. + DMSO; s.c.) for 21 days | |
| 2. | R | normal saline; i.p. + distilled water; p.o. + RTN (0.5 mg/kg; s.c.) for 21 days | |
| 3. | B30 | normal saline; i.p. + BBR (30 mg/kg; p.o.) + DMSO; s.c. for 21 days | |
| 4. | B100 | normal saline; i.p. + BBR (100 mg/kg; p.o.) + DMSO; s.c. for 21 days | |
| 5. | B30+R | normal saline; i.p. + BBR (30 mg/kg; p.o.) + RTN (0.5 mg/kg; s.c.) for 21 days | |
| 6. | B100+R | normal saline; i.p. + BBR (100 mg/kg; p.o.) + RTN (0.5 mg/kg; s.c.) for 21 days | |
| 7. | T+B30+R | TGN (10 mg/kg; i.p.) + BBR (30 mg/kg; p.o.) + RTN (0.5 mg/kg; s.c.) for 21 days | |
| 8. | T+B100+R | TGN (10 mg/kg; i.p.) + BBR (100 mg/kg; p.o.) + RTN (0.5 mg/kg; s.c.) for 21 days | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, H.-C.; Wang, M.-H.; Fang, C.-H.; Lin, Y.-W.; Soung, H.-S. Neuroprotective Potentials of Berberine in Rotenone-Induced Parkinson’s Disease-like Motor Symptoms in Rats. Brain Sci. 2024, 14, 596. https://doi.org/10.3390/brainsci14060596
Tseng H-C, Wang M-H, Fang C-H, Lin Y-W, Soung H-S. Neuroprotective Potentials of Berberine in Rotenone-Induced Parkinson’s Disease-like Motor Symptoms in Rats. Brain Sciences. 2024; 14(6):596. https://doi.org/10.3390/brainsci14060596
Chicago/Turabian StyleTseng, Hsiang-Chien, Mao-Hsien Wang, Chih-Hsiang Fang, Yi-Wen Lin, and Hung-Sheng Soung. 2024. "Neuroprotective Potentials of Berberine in Rotenone-Induced Parkinson’s Disease-like Motor Symptoms in Rats" Brain Sciences 14, no. 6: 596. https://doi.org/10.3390/brainsci14060596
APA StyleTseng, H.-C., Wang, M.-H., Fang, C.-H., Lin, Y.-W., & Soung, H.-S. (2024). Neuroprotective Potentials of Berberine in Rotenone-Induced Parkinson’s Disease-like Motor Symptoms in Rats. Brain Sciences, 14(6), 596. https://doi.org/10.3390/brainsci14060596