DNA Methylation in Autism Spectrum Disorders: Biomarker or Pharmacological Target?

 , and
, and 
Abstract
:1. Introduction
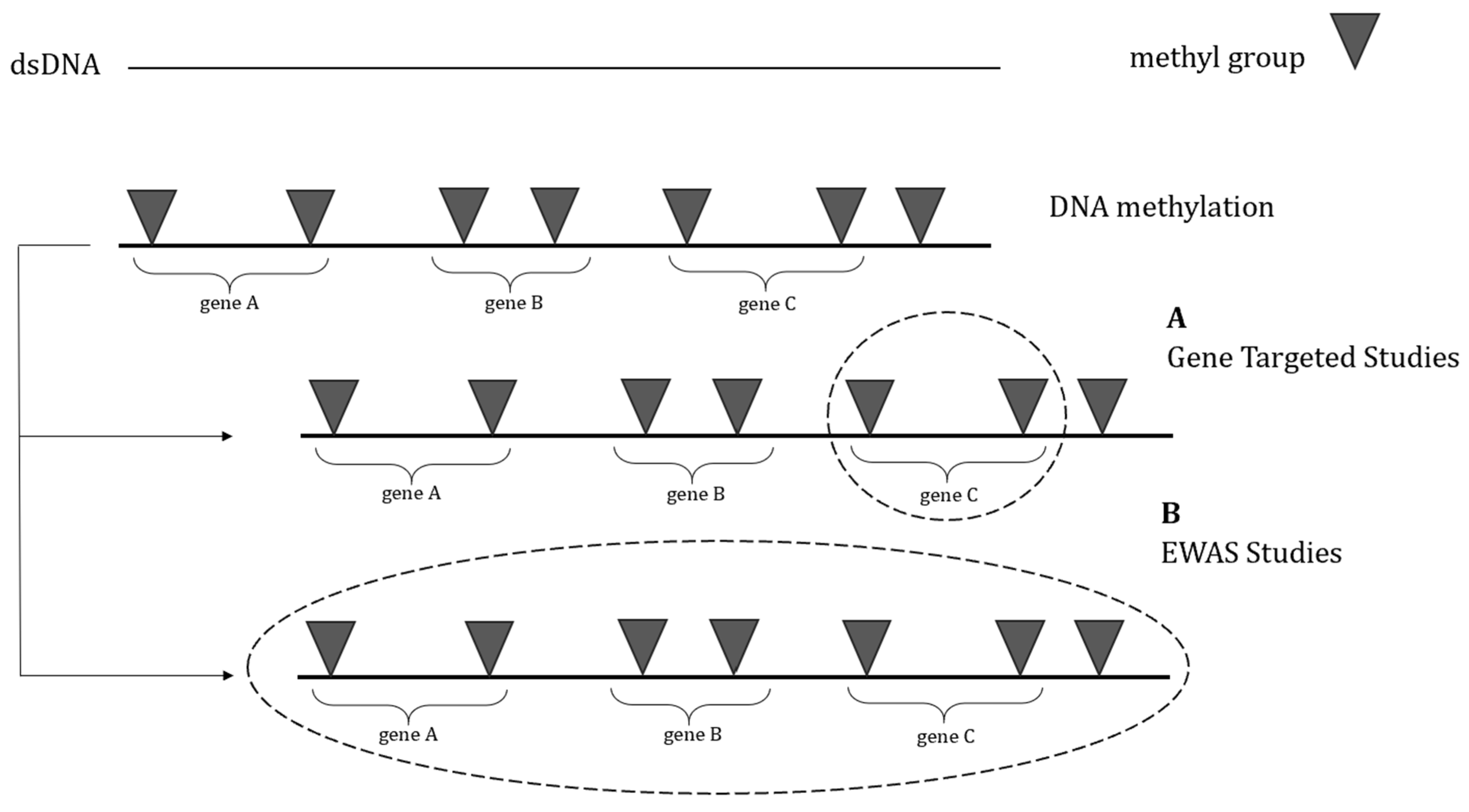
2. DNA Methylation
3. Alterations in DNA Methylation at Specific Genes Associated with Autism Spectrum Disorders
3.1. Hormones and Neurotransmitter Receptor Genes
3.1.1. Serotonin
HTR2A
HTR4
3.1.2. Oxytocin
3.1.3. Dopamine
3.1.4. GABA
3.1.5. Insulin
3.1.6. Estrogen
3.2. Genes Associated with Neural Matrix
3.2.1. RELN
3.2.2. SHANK3
3.2.3. ST8SIA2
3.3. Genes Associated with Gene Expression
3.3.1. MeCP2
3.3.2. NHIP
3.4. Genes Involved in Disorders Associated with ASD
3.4.1. FMR1
3.4.2. APOE
3.5. Genes Associated with Environmental Factors
3.5.1. WNT
3.5.2. KCC2
3.6. Other Genes
3.6.1. ACSF3
3.6.2. PPP2R2C
3.6.3. CYP2E1
4. Global DNA Methylation Profiling in Autism Spectrum Disorder
4.1. Clinical Evidence
4.1.1. EWASs on Post-Mortem Brain Tissues
4.1.2. EWASs on Peripheral Tissues
Studies on Monozygotic Twin Pairs
Studies on ASD Patients
4.2. Preclinical Evidence
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hirota, T.; King, B.H. Autism Spectrum Disorder: A Review. JAMA 2023, 329, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Kodak, T.; Bergmann, S. Autism Spectrum Disorder: Characteristics, Associated Behaviors, and Early Intervention. Pediatr. Clin. N. Am. 2020, 67, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Buescher, A.V.; Cidav, Z.; Knapp, M.; Mandell, D.S. Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatr. 2014, 168, 721–728. [Google Scholar] [CrossRef] [PubMed]
- King, B.H. Psychiatric comorbidities in neurodevelopmental disorders. Curr. Opin. Neurol. 2016, 29, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Spencer, D.; Yang, W.; Kelly, J.P.; Newschaffer, C.J.; Johnson, J.; Marshall, J.; Azocar, F.; Tabb, L.P.; Dennen, T. Injuries among children with autism spectrum disorder. Acad. Pediatr. 2014, 14, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Hirvikoski, T.; Mittendorfer-Rutz, E.; Boman, M.; Larsson, H.; Lichtenstein, P.; Bölte, S. Premature mortality in autism spectrum disorder. Br. J. Psychiatry 2016, 208, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Schendel, D.E.; Overgaard, M.; Christensen, J.; Hjort, L.; Jørgensen, M.; Vestergaard, M.; Parner, E.T. Association of Psychiatric and Neurologic Comorbidity with Mortality Among Persons with Autism Spectrum Disorder in a Danish Population. JAMA Pediatr. 2016, 170, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Lordan, R.; Storni, C.; De Benedictis, C.A. Autism Spectrum Disorders: Diagnosis and Treatment. In Autism Spectrum Disorders; Grabrucker, A.M., Ed.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar]
- Weuring, W.; Geerligs, J.; Koeleman, B.P. Gene therapies for monogenic autism spectrum disorders. Genes 2021, 12, 1667. [Google Scholar] [CrossRef]
- Antaki, D.; Guevara, J.; Maihofer, A.X.; Klein, M.; Gujral, M.; Grove, J.; Carey, C.E.; Hong, O.; Arranz, M.J.; Hervas, A. A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. Nat. Genet. 2022, 54, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, Y.; Matsumoto, J.; Miura, K.; Hasegawa, N.; Hashimoto, R. Genetics of autism spectrum disorders and future direction. J. Hum. Genet. 2023, 68, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Karimi, P.; Kamali, E.; Mousavi, S.M.; Karahmadi, M. Environmental factors influencing the risk of autism. J. Res. Med. Sci. 2017, 22, 27. [Google Scholar] [PubMed]
- Ladd-Acosta, C.; Fallin, M.D. The role of epigenetics in genetic and environmental epidemiology. Epigenomics 2016, 8, 271–283. [Google Scholar] [CrossRef]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Vorstman, J.A.S.; Staal, W.G.; van Daalen, E.; van Engeland, H.; Hochstenbach, P.F.R.; Franke, L. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol. Psychiatry 2006, 11, 18–28. [Google Scholar] [CrossRef]
- Bremer, A.; Giacobini, M.; Nordenskjöld, M.; Brøndum-Nielsen, K.; Mansouri, M.; Dahl, N.; Anderlid, B.; Schoumans, J. Screening for copy number alterations in loci associated with autism spectrum disorders by two-color multiplex ligation-dependent probe amplification. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, N.S.; Jain, J.M.N.; Chintakindi, K.P.; Singh, R.P.; Naik, U.; Akella, R.R.D. Aberrations in folate metabolic pathway and altered susceptibility to autism. Psychiatr. Genet. 2009, 19, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Solehdin, F.; Cohen, I.L.; Gonzalez, M.G.; Jenkins, E.C.; Lewis, M.E.S.; Holden, J.J.A. Population- and Family-Based Studies Associate the MTHFR Gene with Idiopathic Autism in Simplex Families. J. Autism Dev. Disord. 2011, 41, 938–944. [Google Scholar] [CrossRef]
- Smith, A.; Kaufman, F.; Sandy, M.S.; Cardenas, A. Cannabis Exposure During Critical Windows of Development: Epigenetic and Molecular Pathways Implicated in Neuropsychiatric Disease. Curr. Environ. Health Rep. 2020, 7, 325–342. [Google Scholar] [CrossRef]
- Abd-Nikfarjam, B.; Dolati-Somarin, A.; Baradaran Rahimi, V.; Askari, V.R. Cannabinoids in neuroinflammatory disorders: Focusing on multiple sclerosis, Parkinsons, and Alzheimers diseases. Biofactors 2023, 49, 560–583. [Google Scholar] [CrossRef] [PubMed]
- Askari, V.R.; Baradaran Rahimi, V.; Shafiee-Nick, R. Low Doses of β-Caryophyllene Reduced Clinical and Paraclinical Parameters of an Autoimmune Animal Model of Multiple Sclerosis: Investigating the Role of CB(2) Receptors in Inflammation by Lymphocytes and Microglial. Brain Sci. 2023, 13, 1092. [Google Scholar] [CrossRef] [PubMed]
- Askari, V.R.; Shafiee-Nick, R. Promising neuroprotective effects of β-caryophyllene against LPS-induced oligodendrocyte toxicity: A mechanistic study. Biochem. Pharmacol. 2019, 159, 154–171. [Google Scholar] [CrossRef] [PubMed]
- Askari, V.R.; Shafiee-Nick, R. The protective effects of β-caryophyllene on LPS-induced primary microglia M(1)/M(2) imbalance: A mechanistic evaluation. Life Sci. 2019, 219, 40–73. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.Q.V.; Miyake, K. Neurodevelopmental Disorders and Environmental Toxicants: Epigenetics as an Underlying Mechanism. Int. J. Genom. 2017, 2017, 7526592. [Google Scholar] [CrossRef]
- Keil, K.P.; Lein, P.J. DNA methylation: A mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ. Epigenetics 2016, 2, dvv012. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Schermelleh, L.; Walter, J.; Cardoso, M.C.; Leonhardt, H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. USA 2005, 102, 8905–8909. [Google Scholar] [CrossRef] [PubMed]
- Aapola, U.; Lyle, R.; Krohn, K.; Antonarakis, S.E.; Peterson, P. Isolation and initial characterization of the mouse Dnmt3l gene. Cytogenet. Cell Genet. 2001, 92, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Okano, M.; Lei, H.; Li, E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 2002, 129, 1983–1993. [Google Scholar] [CrossRef]
- Jia, D.; Jurkowska, R.Z.; Zhang, X.; Jeltsch, A.; Cheng, X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007, 449, 248–251. [Google Scholar] [CrossRef]
- Mayer, W.; Niveleau, A.; Walter, J.; Fundele, R.; Haaf, T. Demethylation of the zygotic paternal genome. Nature 2000, 403, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Paroush, Z.; Keshet, I.; Yisraeli, J.; Cedar, H. Dynamics of demethylation and activation of the alpha-actin gene in myoblasts. Cell 1990, 63, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Gujar, H.; Weisenberger, D.J.; Liang, G. The Roles of Human DNA Methyltransferases and Their Isoforms in Shaping the Epigenome. Genes 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Coulondre, C.; Miller, J.H.; Farabaugh, P.J.; Gilbert, W. Molecular basis of base substitution hotspots in Escherichia coli. Nature 1978, 274, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980, 8, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.; Taggart, M.; Frommer, M.; Miller, O.J.; Macleod, D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 1985, 40, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Binder, E.B. Epigenetic alterations in depression and antidepressant treatment. Dialogues Clin. Neurosci. 2014, 16, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, K.; Iovino, N.; Bogdanović, O. Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol. 2018, 19, 774–790. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8, 148. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Guhathakurta, S.; Singh, A.S.; Sinha, S.; Chatterjee, A.; Ahmed, S.; Ghosh, S.; Usha, R. Analysis of serotonin receptor 2A gene (HTR2A): Association study with autism spectrum disorder in the Indian population and investigation of the gene expression in peripheral blood leukocytes. Neurochem. Int. 2009, 55, 754–759. [Google Scholar] [CrossRef]
- Hranilovic, D.; Blazevic, S.; Stefulj, J.; Zill, P. DNA methylation analysis of HTR2A regulatory region in leukocytes of autistic subjects. Autism Res. 2016, 9, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ying, X.; Huang, L.; Zhao, Y.; Zhou, D.; Liu, J.; Zhong, J.; Huang, T.; Zhang, W.; Cheng, F. Association of human serotonin receptor 4 promoter methylation with autism spectrum disorder. Medicine 2020, 99, e18838. [Google Scholar] [CrossRef] [PubMed]
- Elagoz Yuksel, M.; Yuceturk, B.; Karatas, O.F.; Ozen, M.; Dogangun, B. The altered promoter methylation of oxytocin receptor gene in autism. J. Neurogenet. 2016, 30, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Andari, E.; Nishitani, S.; Kaundinya, G.; Caceres, G.A.; Morrier, M.J.; Ousley, O.; Smith, A.K.; Cubells, J.F.; Young, L.J. Epigenetic modification of the oxytocin receptor gene: Implications for autism symptom severity and brain functional connectivity. Neuropsychopharmacology 2020, 45, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Wieting, J.; Jahn, K.; Bleich, S.; Frieling, H.; Deest, M. A targeted long-read sequencing approach questions the association of OXTR methylation with high-functioning autism. Clin. Epigenetics 2023, 15, 195. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Song, C.; Hohmann, S.; Prokhorova, T.; Sheldrick-Michel, T.M.; Knöpfel, T. DNA Methylation Profiles of GAD1 in Human Cerebral Organoids of Autism Indicate Disrupted Epigenetic Regulation during Early Development. Int. J. Mol. Sci. 2022, 23, 9188. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Mordaunt, C.E.; Yasui, D.H.; Marathe, R.; Coulson, R.L.; Dunaway, K.W.; Jianu, J.M.; Walker, C.K.; Ozonoff, S.; Hertz-Picciotto, I. Placental DNA methylation levels at CYP2E1 and IRS2 are associated with child outcome in a prospective autism study. Hum. Mol. Genet. 2019, 28, 2659–2674. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liang, S.; Sun, Y.; Li, H.; Endo, F.; Nakao, M.; Saitoh, N.; Wu, L. Analysis of estrogen receptor β gene methylation in autistic males in a Chinese Han population. Metab. Brain Dis. 2017, 32, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Green, A.L.; Eid, A.; Zhan, L.; Zarbl, H.; Guo, G.L.; Richardson, J.R. Epigenetic regulation of the ontogenic expression of the dopamine transporter. Front. Genet. 2019, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Lintas, C.; Sacco, R.; Persico, A.M. Differential methylation at the RELN gene promoter in temporal cortex from autistic and typically developing post-puberal subjects. J. Neurodev. Disord. 2016, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, X.; Li, X.-L.; Towers, A.; Cao, X.; Wang, P.; Bowman, R.; Yang, H.; Goldstein, J.; Li, Y.-J. Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum. Mol. Genet. 2014, 23, 1563–1578. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, L.; Chai, X.; Liu, J. The association between ST8SIA2 gene and behavioral phenotypes in children with autism spectrum disorder. Front. Behav. Neurosci. 2022, 16, 929878. [Google Scholar] [CrossRef] [PubMed]
- Blumkin, E.; Levav-Rabkin, T.; Melamed, O.; Galron, D.; Golan, H.M. Gender-specific effect of Mthfr genotype and neonatal vigabatrin interaction on synaptic proteins in mouse cortex. Neuropsychopharmacology 2011, 36, 1714–1728. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Tian, Y.; Xu, C.; Wang, J.; Jin, Y. Prenatal and postnatal traffic pollution exposure, DNA methylation in Shank3 and MeCP2 promoter regions, H3K4me3 and H3K27me3 and sociability in rats’ offspring. Clin. Epigenetics 2021, 13, 180. [Google Scholar] [CrossRef]
- Nagarajan, R.; Hogart, A.; Gwye, Y.; Martin, M.R.; LaSalle, J.M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 2006, 1, 172–182. [Google Scholar] [CrossRef]
- Zhu, Y.; Gomez, J.A.; Laufer, B.I.; Mordaunt, C.E.; Mouat, J.S.; Soto, D.C.; Dennis, M.Y.; Benke, K.S.; Bakulski, K.M.; Dou, J. Placental methylome reveals a 22q13. 33 brain regulatory gene locus associated with autism. Genome Biol. 2022, 23, 46. [Google Scholar] [CrossRef] [PubMed]
- Jiraanont, P.; Kumar, M.; Tang, H.-T.; Espinal, G.; Hagerman, P.J.; Hagerman, R.J.; Chutabhakdikul, N.; Tassone, F. Size and methylation mosaicism in males with Fragile X syndrome. Expert. Rev. Mol. Diagn. 2017, 17, 1023–1032. [Google Scholar] [CrossRef]
- Budimirovic, D.B.; Schlageter, A.; Filipovic-Sadic, S.; Protic, D.D.; Bram, E.; Mahone, E.M.; Nicholson, K.; Culp, K.; Javanmardi, K.; Kemppainen, J. A genotype-phenotype study of high-resolution FMR1 nucleic acid and protein analyses in fragile X patients with neurobehavioral assessments. Brain Sci. 2020, 10, 694. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, Y.; Zhao, Y.; Yu, H.; Ying, X.; Zhou, D.; Zhong, J.; Zheng, Z.; Liu, J.; Pan, R. APOE hypermethylation is associated with autism spectrum disorder in a Chinese population. Exp. Ther. Med. 2018, 15, 4749–4754. [Google Scholar] [CrossRef]
- Lu, Z.; Liu, Z.; Mao, W.; Wang, X.; Zheng, X.; Chen, S.; Cao, B.; Huang, S.; Zhang, X.; Zhou, T. Locus-specific DNA methylation of Mecp2 promoter leads to autism-like phenotypes in mice. Cell Death Dis. 2020, 11, 85. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, L.; Zhu, X.; Cui, W.; Sun, Y.; Nishijo, H.; Peng, Y.; Li, R. Demethylation of Specific Wnt/β-Catenin Pathway Genes and its Upregulation in Rat Brain Induced by Prenatal Valproate Exposure. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2010, 293, 1947–1953. [Google Scholar] [CrossRef]
- Konopko, M.A.; Densmore, A.L.; Krueger, B.K. Sexually dimorphic epigenetic regulation of brain-derived neurotrophic factor in fetal brain in the valproic acid model of autism spectrum disorder. Dev. Neurosci. 2017, 39, 507–518. [Google Scholar] [CrossRef]
- Algothmi, K.; Alqurashi, A.; Alrofaidi, A.; Alharbi, M.; Farsi, R.; Alburae, N.; Ganash, M.; Azhari, S.; Basingab, F.; Almuhammadi, A.; et al. DNA Methylation Level of Transcription Factor Binding Site in the Promoter Region of Acyl-CoA Synthetase Family Member 3 (ACSF3) in Saudi Autistic Children. Pharmgenomics Pers. Med. 2022, 15, 131–142. [Google Scholar] [CrossRef]
- Kimura, R.; Nakata, M.; Funabiki, Y.; Suzuki, S.; Awaya, T.; Murai, T.; Hagiwara, M. An epigenetic biomarker for adult high-functioning autism spectrum disorder. Sci. Rep. 2019, 9, 13662. [Google Scholar] [CrossRef]
- Ju, L.S.; Yang, J.J.; Morey, T.E.; Gravenstein, N.; Seubert, C.N.; Resnick, J.L.; Zhang, J.Q.; Martynyuk, A.E. Role of epigenetic mechanisms in transmitting the effects of neonatal sevoflurane exposure to the next generation of male, but not female, rats. Br. J. Anaesth. 2018, 121, 406–416. [Google Scholar] [CrossRef]
- Bonnin, A.; Levitt, P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience 2011, 197, 1–7. [Google Scholar] [CrossRef]
- Cook, E.H.; Leventhal, B.L. The serotonin system in autism. Curr. Opin. Pediatr. 1996, 8, 348–354. [Google Scholar] [CrossRef]
- Vincent, J.B.; Noor, A.; Windpassinger, C.; Gianakopoulos, P.J.; Schwarzbraun, T.; Alfred, S.E.; Stachowiak, B.; Scherer, S.W.; Roberts, W.; Wagner, K. Characterization of a de novo translocation t (5; 18)(q33. 1; q12. 1) in an autistic boy identifies a breakpoint close to SH3TC2, ADRB2, and HTR4 on 5q, and within the desmocollin gene cluster on 18q. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 150, 817–826. [Google Scholar] [CrossRef]
- Williams, K.; Brignell, A.; Randall, M.; Silove, N.; Hazell, P. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst. Rev. 2013, 8, CD004677. [Google Scholar] [CrossRef]
- Spurlock, G.; Heils, A.; Holmans, P.; Williams, J.; D’Souza, U.; Cardno, A.; Murphy, K.; Jones, L.; Buckland, P.R.; McGuffin, P. A family based association study of T102C polymorphism in 5HT2A and schizophrenia plus identification of new polymorphisms in the promoter. Mol. Psychiatry 1998, 3, 42–49. [Google Scholar] [CrossRef]
- Warren, J.T.; Peacock, M.L.; Rodriguez, L.C.; Fink, J.K. An MspI polymorphism in the hyman serotonin receptor gene (HTR2): Detection by DGGE and RFLP analysis. Hum. Mol. Genet. 1993, 2, 338a. [Google Scholar] [CrossRef]
- Cho, I.H.; Yoo, H.J.; Park, M.; Lee, Y.S.; Kim, S.A. Family-based association study of 5-HTTLPR and the 5-HT2A receptor gene polymorphisms with autism spectrum disorder in Korean trios. Brain Res. 2007, 1139, 34–41. [Google Scholar] [CrossRef]
- Veenstra-VanderWeele, J.; Kim, S.J.; Lord, C.; Courchesne, R.; Akshoomoff, N.; Leventhal, B.L.; Courchesne, E.; Cook, E.H., Jr. Transmission disequilibrium studies of the serotonin 5-HT2A receptor gene (HTR2A) in autism. Am. J. Med. Genet. 2002, 114, 277–283. [Google Scholar] [CrossRef]
- Quintana, D.S.; Rokicki, J.; van der Meer, D.; Alnæs, D.; Kaufmann, T.; Córdova-Palomera, A.; Dieset, I.; Andreassen, O.A.; Westlye, L.T. Oxytocin pathway gene networks in the human brain. Nat. Commun. 2019, 10, 668. [Google Scholar] [CrossRef]
- Bethlehem, R.A.; Lombardo, M.V.; Lai, M.-C.; Auyeung, B.; Crockford, S.K.; Deakin, J.; Soubramanian, S.; Sule, A.; Kundu, P.; Voon, V. Intranasal oxytocin enhances intrinsic corticostriatal functional connectivity in women. Transl. Psychiatry 2017, 7, e1099. [Google Scholar] [CrossRef]
- Guastella, A.J.; Einfeld, S.L.; Gray, K.M.; Rinehart, N.J.; Tonge, B.J.; Lambert, T.J.; Hickie, I.B. Intranasal oxytocin improves emotion recognition for youth with autism spectrum disorders. Biol. Psychiatry 2010, 67, 692–694. [Google Scholar] [CrossRef]
- Hollander, E.; Novotny, S.; Hanratty, M.; Yaffe, R.; DeCaria, C.M.; Aronowitz, B.R.; Mosovich, S. Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharmacology 2003, 28, 193–198. [Google Scholar] [CrossRef]
- Kosaka, H.; Munesue, T.; Ishitobi, M.; Asano, M.; Omori, M.; Sato, M.; Tomoda, A.; Wada, Y. Long-term oxytocin administration improves social behaviors in a girl with autistic disorder. BMC Psychiatry 2012, 12, 110. [Google Scholar] [CrossRef]
- Moerkerke, M.; Daniels, N.; Tibermont, L.; Tang, T.; Evenepoel, M.; Van der Donck, S.; Debbaut, E.; Prinsen, J.; Chubar, V.; Claes, S.; et al. Chronic oxytocin administration stimulates the oxytocinergic system in children with autism. Nat. Commun. 2024, 15, 58. [Google Scholar] [CrossRef]
- Siecinski, S.K.; Giamberardino, S.N.; Spanos, M.; Hauser, A.C.; Gibson, J.R.; Chandrasekhar, T.; Trelles, M.D.P.; Rockhill, C.M.; Palumbo, M.L.; Cundiff, A.W.; et al. Genetic and epigenetic signatures associated with plasma oxytocin levels in children and adolescents with autism spectrum disorder. Autism Res. 2023, 16, 502–523. [Google Scholar] [CrossRef]
- Evenepoel, M.; Moerkerke, M.; Daniels, N.; Chubar, V.; Claes, S.; Turner, J.; Vanaudenaerde, B.; Willems, L.; Verhaeghe, J.; Prinsen, J.; et al. Endogenous oxytocin levels in children with autism: Associations with cortisol levels and oxytocin receptor gene methylation. Transl. Psychiatry 2023, 13, 235. [Google Scholar] [CrossRef]
- LoParo, D.; Waldman, I. The oxytocin receptor gene (OXTR) is associated with autism spectrum disorder: A meta-analysis. Mol. Psychiatry 2015, 20, 640–646. [Google Scholar] [CrossRef]
- Gorentla, B.K.; Vaughan, R.A. Differential effects of dopamine and psychoactive drugs on dopamine transporter phosphorylation and regulation. Neuropharmacology 2005, 49, 759–768. [Google Scholar] [CrossRef]
- Spencer, T.J.; Biederman, J.; Faraone, S.V.; Madras, B.K.; Bonab, A.A.; Dougherty, D.D.; Batchelder, H.; Clarke, A.; Fischman, A.J. Functional genomics of attention-deficit/hyperactivity disorder (ADHD) risk alleles on dopamine transporter binding in ADHD and healthy control subjects. Biol. Psychiatry 2013, 74, 84–89. [Google Scholar] [CrossRef]
- Nakamura, K.; Sekine, Y.; Ouchi, Y.; Tsujii, M.; Yoshikawa, E.; Futatsubashi, M.; Tsuchiya, K.J.; Sugihara, G.; Iwata, Y.; Suzuki, K. Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch. Gen. Psychiatry 2010, 67, 59–68. [Google Scholar] [CrossRef]
- Rodríguez-Traver, E.; Solís, O.; Díaz-Guerra, E.; Ortiz, Ó.; Vergaño-Vera, E.; Méndez-Gómez, H.R.; García-Sanz, P.; Moratalla, R.; Vicario-Abejón, C. Role of Nurr1 in the generation and differentiation of dopaminergic neurons from stem cells. Neurotox. Res. 2016, 30, 14–31. [Google Scholar] [CrossRef]
- Schmitt, K.C.; Reith, M.E. Regulation of the dopamine transporter: Aspects relevant to psychostimulant drugs of abuse. Ann. N. Y. Acad. Sci. 2010, 1187, 316–340. [Google Scholar] [CrossRef]
- Zhou, S.; Yu, Y. Synaptic EI balance underlies efficient neural coding. Front. Neurosci. 2018, 12, 46. [Google Scholar] [CrossRef]
- Askari, V.R.; Baradaran Rahimi, V.; Ghorbani, A.; Rakhshandeh, H. Hypnotic Effect of Ocimum basilicum on Pentobarbital-Induced Sleep in Mice. Iran. Red. Crescent Med. J. 2016, 18, e24261. [Google Scholar] [CrossRef]
- Baradaran Rahimi, V.; Askari, V.R.; Tajani, A.S.; Hosseini, A.; Rakhshandeh, H. Evaluation of the Sleep-Prolonging Effect of Lagenaria vulgaris and Cucurbita pepo Extracts on Pentobarbital-Induced Sleep and Possible Mechanisms of Action. Medicina 2018, 54, 55. [Google Scholar] [CrossRef]
- Sohal, V.S.; Rubenstein, J.L. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Hegarty, J.P.; Weber, D.J.; Cirstea, C.M.; Beversdorf, D.Q. Cerebro-cerebellar functional connectivity is associated with cerebellar excitation–inhibition balance in autism spectrum disorder. J. Autism Dev. Disord. 2018, 48, 3460–3473. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, S.K.; Kang, W.S.; Park, J.K.; Kim, Y.J.; Nam, M.; Kim, J.W.; Chung, J.-H. Association between IRS 1 Gene Polymorphism and Autism Spectrum Disorder: A Pilot Case-Control Study in Korean Males. Int. J. Mol. Sci. 2016, 17, 1227. [Google Scholar] [CrossRef]
- Baron-Cohen, S.; Lombardo, M.V.; Auyeung, B.; Ashwin, E.; Chakrabarti, B.; Knickmeyer, R. Why are autism spectrum conditions more prevalent in males? PLoS Biol. 2011, 9, e1001081. [Google Scholar] [CrossRef]
- Östlund, H.; Keller, E.; Hurd, Y.L. Estrogen receptor gene expression in relation to neuropsychiatric disorders. Ann. N. Y. Acad. Sci. 2003, 1007, 54–63. [Google Scholar] [CrossRef]
- ter Horst, G.J. Estrogen in the limbic system. Vitam. Horm. 2010, 82, 319–338. [Google Scholar]
- Crider, A.; Thakkar, R.; Ahmed, A.O.; Pillai, A. Dysregulation of estrogen receptor beta (ERβ), aromatase (CYP19A1), and ER co-activators in the middle frontal gyrus of autism spectrum disorder subjects. Mol. Autism 2014, 5, 46. [Google Scholar] [CrossRef]
- Wei, H.; Liang, F.; Meng, G.; Nie, Z.; Zhou, R.; Cheng, W.; Wu, X.; Feng, Y.; Wang, Y. Redox/methylation mediated abnormal DNA methylation as regulators of ambient fine particulate matter-induced neurodevelopment related impairment in human neuronal cells. Sci. Rep. 2016, 6, 33402. [Google Scholar] [CrossRef]
- Shih, P.-Y.; Fang, Y.-L.; Shankar, S.; Lee, S.-P.; Hu, H.-T.; Chen, H.; Wang, T.-F.; Hsia, K.-C.; Hsueh, Y.-P. Phase separation and zinc-induced transition modulate synaptic distribution and association of autism-linked CTTNBP2 and SHANK3. Nat. Commun. 2022, 13, 2664. [Google Scholar] [CrossRef]
- Wang, Y.; Chiola, S.; Yang, G.; Russell, C.; Armstrong, C.J.; Wu, Y.; Spampanato, J.; Tarboton, P.; Ullah, H.A.; Edgar, N.U. Modeling human telencephalic development and autism-associated SHANK3 deficiency using organoids generated from single neural rosettes. Nat. Commun. 2022, 13, 5688. [Google Scholar] [CrossRef]
- Li, K.; Liang, X.; Xie, X.; Tian, L.; Yan, J.; Lin, B.; Liu, H.; Lai, W.; Liu, X.; Xi, Z. Role of SHANK3 in concentrated ambient PM2. 5 exposure induced autism-like phenotype. Heliyon 2023, 9, e14328. [Google Scholar] [CrossRef]
- Singleton, M.K.; Gonzales, M.L.; Leung, K.N.; Yasui, D.H.; Schroeder, D.I.; Dunaway, K.; LaSalle, J.M. MeCP2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiol. Dis. 2011, 43, 190–200. [Google Scholar] [CrossRef]
- Nguyen, M.V.; Du, F.; Felice, C.A.; Shan, X.; Nigam, A.; Mandel, G.; Robinson, J.K.; Ballas, N. MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 2012, 32, 10021–10034. [Google Scholar] [CrossRef]
- Moretti, P.; Zoghbi, H.Y. MeCP2 dysfunction in Rett syndrome and related disorders. Curr. Opin. Genet. Dev. 2006, 16, 276–281. [Google Scholar] [CrossRef]
- Wen, Z.; Cheng, T.L.; Li, G.Z.; Sun, S.B.; Yu, S.Y.; Zhang, Y.; Du, Y.S.; Qiu, Z. Identification of autism-related MECP2 mutations by whole-exome sequencing and functional validation. Mol. Autism 2017, 8, 43. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.; Fu, Y.-H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Foraker, J.; Millard, S.P.; Leong, L.; Thomson, Z.; Chen, S.; Keene, C.D.; Bekris, L.M.; Yu, C.-E. The APOE gene is differentially methylated in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 48, 745–755. [Google Scholar] [CrossRef]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Hung, C.; Fujisawa, Y.; Sakaguchi, D.; Angelastro, J.; Omanska-Klusek, A.; Schoenfeld, R.; Giulivi, C. Mitochondrial dysfunction in Pten haplo-insufficient mice with social deficits and repetitive behavior: Interplay between Pten and p53. PLoS ONE 2012, 7, e42504. [Google Scholar] [CrossRef]
- Wisner, K.L.; Leckman-Westin, E.; Finnerty, M.; Essock, S.M. Valproate prescription prevalence among women of childbearing age. Psychiatr. Serv. 2011, 62, 218–220. [Google Scholar] [CrossRef]
- Gerard, E.E.; Meador, K.J. An update on maternal use of antiepileptic medications in pregnancy and neurodevelopment outcomes. J. Pediatr. Genet. 2015, 4, 94–110. [Google Scholar]
- Mychasiuk, R.; Richards, S.; Nakahashi, A.; Kolb, B.; Gibb, R. Effects of rat prenatal exposure to valproic acid on behaviour and neuro-anatomy. Dev. Neurosci. 2012, 34, 268–276. [Google Scholar] [CrossRef]
- Schneider, T.; Przewłocki, R. Behavioral alterations in rats prenatally exposed to valproic acid: Animal model of autism. Neuropsychopharmacology 2005, 30, 80–89. [Google Scholar] [CrossRef]
- Huberman Samuel, M.; Meiri, G.; Dinstein, I.; Flusser, H.; Michaelovski, A.; Bashiri, A.; Menashe, I. Exposure to general anesthesia may contribute to the association between cesarean delivery and autism spectrum disorder. J. Autism Dev. Disord. 2019, 49, 3127–3135. [Google Scholar] [CrossRef]
- Laporta, M.L.; Sprung, J.; Fejedelem, C.A.; Henning, D.T.; Weaver, A.L.; Hanson, A.C.; Schroeder, D.R.; Myers, S.M.; Voigt, R.G.; Weingarten, T.N. Association between exposure of children to general anesthesia and autism spectrum disorder. J. Autism Dev. Disord. 2021, 52, 4301–4310. [Google Scholar] [CrossRef]
- Ladd-Acosta, C.; Hansen, K.D.; Briem, E.; Fallin, M.D.; Kaufmann, W.E.; Feinberg, A.P. Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry 2014, 19, 862–871. [Google Scholar] [CrossRef]
- Nardone, S.; Sams, D.S.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef]
- Nardone, S.; Sams, D.S.; Zito, A.; Reuveni, E.; Elliott, E. Dysregulation of Cortical Neuron DNA Methylation Profile in Autism Spectrum Disorder. Cereb. Cortex 2017, 27, 5739–5754. [Google Scholar] [CrossRef]
- Corley, M.J.; Vargas-Maya, N.; Pang, A.P.S.; Lum-Jones, A.; Li, D.; Khadka, V.; Sultana, R.; Blanchard, D.C.; Maunakea, A.K. Epigenetic Delay in the Neurodevelopmental Trajectory of DNA Methylation States in Autism Spectrum Disorders. Front. Genet. 2019, 10, 907. [Google Scholar] [CrossRef]
- Wong, C.C.Y.; Smith, R.G.; Hannon, E.; Ramaswami, G.; Parikshak, N.N.; Assary, E.; Troakes, C.; Poschmann, J.; Schalkwyk, L.C.; Sun, W.; et al. Genome-wide DNA methylation profiling identifies convergent molecular signatures associated with idiopathic and syndromic autism in post-mortem human brain tissue. Hum. Mol. Genet. 2019, 28, 2201–2211. [Google Scholar] [CrossRef]
- Takahashi, E.; Allan, N.; Peres, R.; Ortug, A.; van der Kouwe, A.J.W.; Valli, B.; Ethier, E.; Levman, J.; Baumer, N.; Tsujimura, K.; et al. Integration of structural MRI and epigenetic analyses hint at linked cellular defects of the subventricular zone and insular cortex in autism: Findings from a case study. Front. Neurosci. 2022, 16, 1023665. [Google Scholar] [CrossRef]
- Wong, C.C.; Meaburn, E.L.; Ronald, A.; Price, T.S.; Jeffries, A.R.; Schalkwyk, L.C.; Plomin, R.; Mill, J. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry 2014, 19, 495–503. [Google Scholar] [CrossRef]
- Liang, S.; Li, Z.; Wang, Y.; Li, X.; Yang, X.; Zhan, X.; Huang, Y.; Gao, Z.; Zhang, M.; Sun, C.; et al. Genome-Wide DNA Methylation Analysis Reveals Epigenetic Pattern of SH2B1 in Chinese Monozygotic Twins Discordant for Autism Spectrum Disorder. Front. Neurosci. 2019, 13, 712. [Google Scholar] [CrossRef]
- Saffari, A.; Arno, M.; Nasser, E.; Ronald, A.; Wong, C.C.Y.; Schalkwyk, L.C.; Mill, J.; Dudbridge, F.; Meaburn, E.L. RNA sequencing of identical twins discordant for autism reveals blood-based signatures implicating immune and transcriptional dysregulation. Mol. Autism 2019, 10, 38. [Google Scholar] [CrossRef]
- Hannon, E.; Schendel, D.; Ladd-Acosta, C.; Grove, J.; Hansen, C.S.; Andrews, S.V.; Hougaard, D.M.; Bresnahan, M.; Mors, O.; Hollegaard, M.V.; et al. Elevated polygenic burden for autism is associated with differential DNA methylation at birth. Genome Med. 2018, 10, 19. [Google Scholar] [CrossRef]
- Mordaunt, C.E.; Jianu, J.M.; Laufer, B.I.; Zhu, Y.; Hwang, H.; Dunaway, K.W.; Bakulski, K.M.; Feinberg, J.I.; Volk, H.E.; Lyall, K.; et al. Cord blood DNA methylome in newborns later diagnosed with autism spectrum disorder reflects early dysregulation of neurodevelopmental and X-linked genes. Genome Med. 2020, 12, 88. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Radhakrishna, U. Placental DNA methylation changes and the early prediction of autism in full-term newborns. PLoS ONE 2021, 16, e0253340. [Google Scholar] [CrossRef]
- Mordaunt, C.E.; Mouat, J.S.; Schmidt, R.J.; LaSalle, J.M. Comethyl: A network-based methylome approach to investigate the multivariate nature of health and disease. Brief. Bioinform. 2022, 23, bbab554. [Google Scholar] [CrossRef]
- Jasoliya, M.; Gu, J.; AlOlaby, R.R.; Durbin-Johnson, B.; Chedin, F.; Tassone, F. Profiling Genome-Wide DNA Methylation in Children with Autism Spectrum Disorder and in Children with Fragile X Syndrome. Genes 2022, 13, 1795. [Google Scholar] [CrossRef]
- Papale, L.A.; Zhang, Q.; Li, S.; Chen, K.; Keleş, S.; Alisch, R.S. Genome-wide disruption of 5-hydroxymethylcytosine in a mouse model of autism. Hum. Mol. Genet. 2015, 24, 7121–7131. [Google Scholar] [CrossRef]
- Muehlmann, A.M.; Bliznyuk, N.; Duerr, I.; Yang, T.P.; Lewis, M.H. Early exposure to a methyl donor supplemented diet and the development of repetitive motor behavior in a mouse model. Dev. Psychobiol. 2020, 62, 77–87. [Google Scholar] [CrossRef]
- Stoccoro, A.; Conti, E.; Scaffei, E.; Calderoni, S.; Coppedè, F.; Migliore, L.; Battini, R. DNA Methylation Biomarkers for Young Children with Idiopathic Autism Spectrum Disorder: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 9138. [Google Scholar] [CrossRef]
- Gascon, E.; Lynch, K.; Ruan, H.; Almeida, S.; Verheyden, J.M.; Seeley, W.W.; Dickson, D.W.; Petrucelli, L.; Sun, D.; Jiao, J.; et al. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 2014, 20, 1444–1451. [Google Scholar] [CrossRef]
- Yang, Y.; Shu, X.; Liu, D.; Shang, Y.; Wu, Y.; Pei, L.; Xu, X.; Tian, Q.; Zhang, J.; Qian, K.; et al. EPAC null mutation impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron 2012, 73, 774–788. [Google Scholar] [CrossRef]

{kind=link}
| Clinical Studies | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | Protein | Genomic Region | Groups of Subjects | Tissue | ASD Diagnosis | N of Subjects (in Each Group) | Age of Subjects (in Each Group) | Main DNA Methylation Finding | Ref. |
| HTR2A | Promoter | ASD, HC | Blood | DSM-4 | 90 + 101 | 5.84 ± 2.96 y | No association was proved | [44] | |
| Promoter | ASC, HC | Blood | DSM-4 | 90 + 66 | 4–45 y 19–60 y | Different methylation patterns | [45] | ||
| HTR4 | Promoter | ASD, HC | Blood | DSM-5 | 61 + 66 | 4.02 ± 2.83 y 5.76 ± 0.72 y | ↓ in ASD and females | [46] | |
| OXTR | Oxytocin receptor | Promoter | ASD, TD | Blood | DSM-5-TR, CARS | 27 + 39 | 39.27± 16.95 m 43.69 ± 20.51 m | ↓ in ASD | [47] |
| Oxytocin receptor | Promoter | ASD, neurotypical group | Saliva | DSM-5 ASD | 35 + 64 | 27.02 ± 5.34 y 28.22 ± 6.96 y | ↑ in the intron 1 area in ASD | [48] | |
| Oxytocin receptor | Promoter | HFA, HC | Blood | German guideline | 20 + 20 | 30.5 ± 7.8 y 30.7 ± 7.8 y | Not reported a single significant group-dependent methylated site | [49] | |
| GAD1 | Glutamic acid decarboxylase | Regulatory | ASD, control | Brain | DSM-5 | 4 + 5 | 25.9 ± 0.8 y 26.1 ± 0.8 y | More diverse in cerebral organoids from ASD subjects | [50] |
| IRS2 | Insulin receptor substrate 2 | Exon 1, Intron 1 | ASD, TD | MARBLES placenta | DSM-5 | 20 + 21 | - | ↑ in ASD | [51] |
| ESR2 | ESR2 | Proximal promoter region and an untranslated exon | ASD, HC | Blood | DSM-4 | 54 + 54 | 4.24 ± 0.98 y 4.37 ± 0.80 y | Different methylation patterns | [52] |
| RELN | Reelin | Promoter | ASD, HC | Brain | - | 6 + 6 | 21.0 ± 2.9 y 22.0 ± 1.8 y | Different methylation patterns | [54] |
| SHANK3 | SH3 and multiple ankyrin repeat domains 3 | Promoter | ASD, control | Brain | DSM-5 and ADI-R | 54 + 43 | - | ↑ SHANK3 CGIs | [55] |
| ST8SIA2 | ST8 alpha-N-acetyl-neuraminide | Chr. 15: 92,984,625/Chr. 15: 92998561 | ASD, TD | Blood | DSM-5, ADI-R, ADOS | 69 + 76 | 4.47 ± 1.23 y 4.59 ± 1.19 y | ↑ in ASD | [56] |
| MeCP2 | Methyl CpG binding protein 2 | Promoter | ASD, control | Brain | Not mentioned | 9 + 9 | 5–56 y 9–56 y | ↑ in ASD | [59] |
| NHIP | Neuronal hypoxia-inducible, placenta associated | 22q13.33 | ASD, TD | EARLI/MARBLES placenta | ADOS/ADI-R/MSEL | MARBLES: 46 + 46 EARLI: 16 + 31 | - | ↓ in ASD | [60] |
| FMR1 | FMRP | Promoter | FXS, control | Blood | ADOS | 12 + 5 | 1–28 y 1–28 y | ↑ in 4 cases (3 of which were ASD) | [61] |
| Gene | FXS + ASD, HC | Blood | DSM-5 | 18 | Male: 14.4 ±11.9 y Female: 14.7 ± 10.9 y | FMRP levels correlate with FMR1 gene methylation | [62] | ||
| APOE | APOE | Promoter | ASD, HC | Blood | DSM-4 | 62 + 73 | - | ↑ in ASD | [63] |
| ACSF3 | Malonyl-CoA synthetase | Promoter | ASD, control | Blood | DSM-5 | 19 +18 | 6–12 y 3–11 y | No DNA in the binding site of SP1 within the ACSF3 promoter | [67] |
| PPP2R2C | PP2A | Promoter | ASD, HC | Blood | DSM-5, ADOS ASSQ-R | 29 + 29 | - | Hypermethylation and gene downregulation | [68] |
| CYP2E1 | Cytochrome P450 Family 2 Subfamily E Member 1 | Intron 1, Exon 2 | ASD, TD | MARBLES placenta | DSM-5 | 20 + 21 | - | ↓ in ASD | [51] |
| Preclinical Studies | |||||||||
| Gene | Protein | Genomic Region | Animal/Strain | Animal Model | Tissue | Age of subjects | Main DNA Methylation Finding | Ref. | |
| DAT | DAT | Promoter | Long-Evans rat | Midbrain and striatum | 10 w | ↑ in midbrain and striatum | [53] | ||
| RELN | Reelin | Promoter | Balb/cAnNCrlBR mice | Cerebral cortex | 4 d | Unmethylated | [57] | ||
| SHANK3 | SH3 and multiple ankyrin repeat domains 3 | Promoter | Wistar rats | PND exposure to pollution | Brain | 25 d | ↑ in exposed rats | [58] | |
| MeCP2 | MeCP2 | TSS | Adult mice | Hippocampus | 3–18 w | ↑ in hippocampus | [64] | ||
| methyl CpG binding protein 2 | Promoter | Wistar rats | PND exposure to pollution | Brain | 25 d | ↑ in exposed rats | [58] | ||
| WNT | WNT1/WNT2 | Promoter | Rat | Rat VPA ASD model | Frontal cortex and hippocampus | Offspring | Increased expression | [65] | |
| KCC2 | Potassium (K+)/chloride (Cl−) symporter | Promoter | Sprague Dawley rats | Parental exposure to sevoflurane | Hypothalamus, hippocampus | 5 d | ↑ only in male offspring | [69] | |
| Clinical Studies | |||||
|---|---|---|---|---|---|
| Subjects | ASD Diagnosis | Tissue | N of Subjects | Methods | Refs. |
| Autism cases vs. unrelated controls | ADI-R, ADOS | Post-mortem brain tissue (dorsolateral prefrontal cortex, temporal cortex, and cerebellum) | 19 autism cases, 21 unrelated controls | Infinium HumanMethylation450 BeadChip, bump hunting approach, and a permutation-based multiple testing correction method | [120] |
| autism cases vs. controls | ADI-R | Two cortical regions, prefrontal cortex (BA10), and anterior cingulate gyrus (BA24) | 13 autism cases, 12 controls | DNA was converted with sodium bisulfite and probed with the Illumina 450 K methylation array | [121] |
| ASD vs. controls | ADI-R | Anterior PFC, BA10, frontal cortex BA9 and BA8 | 16 male ASD and 15 male controls | 450 K BeadArray, targeted next-generation bisulfite sequencing | [122] |
| ASD vs. TD | ADI-R | Subventricular zone of the lateral ventricles | 17 ASD, 17 TD | ELISA, Illumina 450k Array-Based DNA Methylation Analyses | [123] |
| ASD vs. non-psychiatric control donors | Not mentioned | Post-mortem tissues samples [PFC, TC, and CBL] | 43 ASD, 38 controls | Illumina Infinium HumanMethylation450 BeadChip array of bisulfite converted DNA | [124] |
| Case study ASD | ADI-R, ADOS | Post-mortem brain samples, SVZ, and insular cortex | 7 cases | Whole-genome Bisulfite Sequencing | [125] |
| MZ twins discordant | Not mentioned | Blood | 50 MZ twin pairs | Illumina Infinium HumanMethylation27 BeadChip array of bisulfite-converted DNA | [126] |
| ASD Monozygotic Twins | DSM-5, ADOS | Blood | 5 pairs of ASD-discordant MZ twins, 4 pairs of ASD-concordant MZ twins, and 30 pairs of sporadic patients | Illumina Infinium Human Methylation 450BeadChip array of bisulfite converted DNA | [127] |
| MZ twins; concordant ASC vs. discordant ASC vs. control | ADI-R, ADOS | Blood | 6 concordant ASCs, 6 discordant ASCs, and 11 control pairs (total N = 46) | Illumina 27 K DNA methylation dataset; the edgeR package for differential expression analyses | [128] |
| ASD cases vs. matched controls | DPCRR, DNPR | Blood | 1316 (equal numbers of ASD cases and matched controls) | mQTL | [129] |
| MARBLES ASD vs. TD | ADOS, ADI-R, MSEL, DSM-5 | Placenta | 20 ASD, 21 TD | Illumina HiSeq 2000 array of bisulfite converted DNA | [51] |
| TD vs. ASD | ADOS, MSEL | Umbilical cord blood samples | 56 TD, 50 ASD | Whole-genome bisulfite sequencing | [130] |
| Autism cases vs. controls | DSM-IV | Placental tissue | 14 cases, 10 control | Illumina HumanMethylation450 BeadChip (450K) | [131] |
| TD vs. ASD | ADOS, MSEL | Male cord blood samples | 39 TD, 35 ASD | Develop the R package Comethyl | [132] |
| ASD vs. TD (MARBLES and EARLI) | ADOS, ADI-R, MSEL, DSM-5 | Placenta | 46 ASD, 46 TD | WGBS, Illumina HiSeq X array of bisulfite converted DNA | [60] |
| ASD, FXSA, TD | ADOS, DSM-V | Peripheral blood | 23 ASD, 23 FXSA, 11 TD | Data processing and analysis were performed in R (version 4.0.2) using minfi, limma, DMRcate, ChAMP, and methylCC packages | [133] |
| Preclinical Studies | |||||
| Animal/Strain | Animal Model | Tissue | Method | Refs. | |
| Mice | autism mouse model (Cntnap2−/−) | striatum | Bioconductor package edgeR | [134] | |
| Mice | C58 mice | cerebellum, striatum, and cortex | MethylFlash Methylated DNA Quantification Kit (Epigentek) protocol | [135] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gholamalizadeh, H.; Amiri-Shahri, M.; Rasouli, F.; Ansari, A.; Baradaran Rahimi, V.; Reza Askari, V. DNA Methylation in Autism Spectrum Disorders: Biomarker or Pharmacological Target? Brain Sci. 2024, 14, 737. https://doi.org/10.3390/brainsci14080737
Gholamalizadeh H, Amiri-Shahri M, Rasouli F, Ansari A, Baradaran Rahimi V, Reza Askari V. DNA Methylation in Autism Spectrum Disorders: Biomarker or Pharmacological Target? Brain Sciences. 2024; 14(8):737. https://doi.org/10.3390/brainsci14080737
Chicago/Turabian StyleGholamalizadeh, Hanieh, Maedeh Amiri-Shahri, Fatemeh Rasouli, Arina Ansari, Vafa Baradaran Rahimi, and Vahid Reza Askari. 2024. "DNA Methylation in Autism Spectrum Disorders: Biomarker or Pharmacological Target?" Brain Sciences 14, no. 8: 737. https://doi.org/10.3390/brainsci14080737