
Three Cases of Spinocerebellar Ataxia Type 2 (SCA2) and Pediatric Literature Review: Do Not Forget Trinucleotide Repeat Disorders in Childhood-Onset Progressive Ataxia

 , , , , , ,
, , , , , , 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
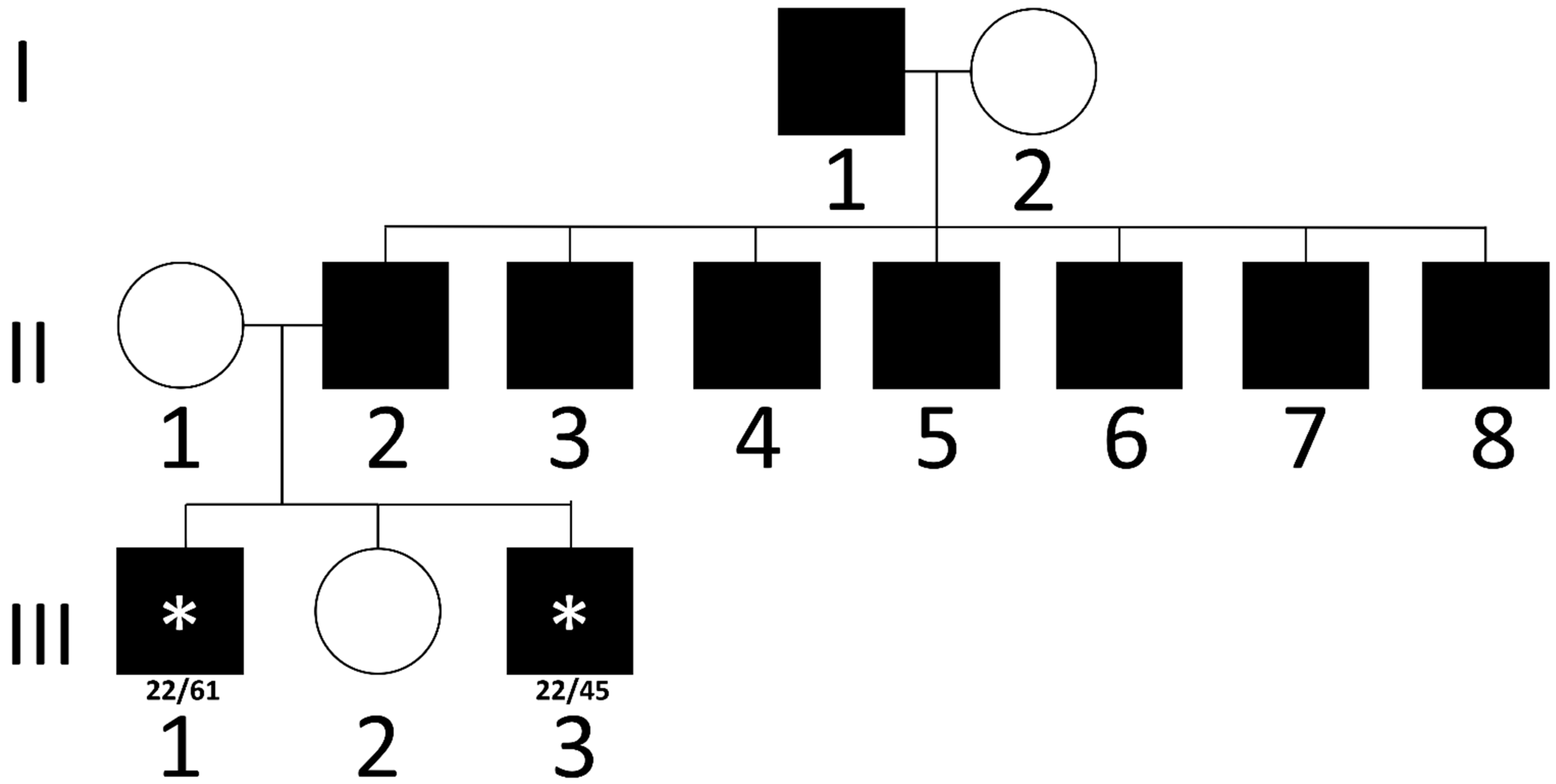
3.1. Case Reports
3.2. The Literature Review
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Subramony, S.H.; Burns, M.; Kugelmann, E.L.; Zingariello, C.D. Inherited Ataxias in Children. Pediatr. Neurol. 2022, 131, 54–62. [Google Scholar] [CrossRef]
- Fogel, B.L. Childhood Cerebellar Ataxia. J. Child Neurol. 2012, 27, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Raslan, I.R.; Barsottini, O.G.; Pedroso, J.L. A Proposed Clinical Classification and a Diagnostic Approach for Congenital Ataxias. Neurol. Clin. Pract. 2021, 11, e328–e336. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Pérez, L.C.; Rodríguez-Labrada, R.; Fernandez-Ruiz, J. Spinocerebellar Ataxia Type 2: Clinicogenetic Aspects, Mechanistic Insights, and Management Approaches. Front. Neurol. 2017, 8, 472. [Google Scholar] [CrossRef]
- Durr, A. Autosomal Dominant Cerebellar Ataxias: Polyglutamine Expansions and Beyond. Lancet Neurol. 2010, 9, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Koenig, M. Progressive Cerebellar Atrophy: Hereditary Ataxias and Disorders with Spinocerebellar Degeneration. Handb. Clin. Neurol. 2013, 113, 1869–1878. [Google Scholar] [CrossRef]
- Fogel, B.L.; Lee, H.; Deignan, J.L.; Strom, S.P.; Kantarci, S.; Wang, X.; Quintero-Rivera, F.; Vilain, E.; Grody, W.W.; Perlman, S.; et al. Exome Sequencing in the Clinical Diagnosis of Sporadic or Familial Cerebellar Ataxia. JAMA Neurol. 2014, 71, 1237–1246. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Rüb, U.; Auburger, G. Spinocerebellar Ataxia 2 (SCA2). Cerebellum 2008, 7, 115–124. [Google Scholar] [CrossRef]
- Sánchez-Corona, J.; Ramirez-Garcia, S.A.; Castañeda-Cisneros, G.; Andrea Gutiérrez-Rubio, S.; Volpini, V.; Sánchez-Garcia, D.M.; Elías García-Ortiz, J.; García-Cruz, D. A Clinical Report of the Massive CAG Repeat Expansion in Spinocerebellar Ataxia Type 2: Severe Onset in a Mexican Child and Review Previous Cases. Genet. Mol. Biol. 2020, 43, e20190325. [Google Scholar] [CrossRef]
- Avelino, M.A.; Pedroso, J.L.; Orlacchio, A.; Barsottini, O.G.P.; Masruha, M.R. Neonatal SCA2 Presenting With Choreic Movements and Dystonia With Dystonic Jerks, Retinitis, Seizures, and Hypotonia. Mov. Disord. Clin. Pract. 2014, 1, 252–254. [Google Scholar] [CrossRef]
- Singh, A.; Faruq, M.; Mukerji, M.; Dwivedi, M.K.; Pruthi, S.; Kapoor, S. Infantile Onset Spinocerebellar Ataxia 2 (SCA2): A Clinical Report with Review of Previous Cases. J. Child Neurol. 2014, 29, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Vinther-Jensen, T.; Ek, J.; Duno, M.; Skovby, F.; Hjermind, L.E.; Nielsen, J.E.; Nielsen, T.T. Germ-Line CAG Repeat Instability Causes Extreme CAG Repeat Expansion with Infantile-Onset Spinocerebellar Ataxia Type 2. Eur. J. Hum. Genet. 2013, 21, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Di Fabio, R.; Santorelli, F.; Bertini, E.; Balestri, M.; Cursi, L.; Tessa, A.; Pierelli, F.; Casali, C. Infantile Childhood Onset of Spinocerebellar Ataxia Type 2. Cerebellum 2012, 11, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Paciorkowski, A.R.; Shafrir, Y.; Hrivnak, J.; Patterson, M.C.; Tennison, M.B.; Clark, H.B.; Gomez, C.M. Massive Expansion of SCA2 with Autonomic Dysfunction, Retinitis Pigmentosa, and Infantile Spasms. Neurology 2011, 77, 1055–1060. [Google Scholar] [CrossRef]
- Mao, R.; Aylsworth, A.S.; Potter, N.; Wilson, W.G.; Breningstall, G.; Wick, M.J.; Babovic-Vuksanovic, D.; Nance, M.; Patterson, M.C.; Gomez, C.M.; et al. Childhood-Onset Ataxia: Testing for Large CAG-Repeats in SCA2 and SCA7. Am. J. Med. Genet. 2002, 110, 338–345. [Google Scholar] [CrossRef]
- Babovic-Vuksanovic, D.; Snow, K.; Patterson, M.C.; Michels, V.V. Spinocerebellar Ataxia Type 2 (SCA 2) in an Infant with Extreme CAG Repeat Expansion. Am. J. Med. Genet. 1998, 79, 383–387. [Google Scholar] [CrossRef]
- Abdel-Aleem, A.; Zaki, M.S. Spinocerebellar Ataxia Type 2 (SCA2) in an Egyptian Family Presenting with Polyphagia and Marked CAG Expansion in Infancy. J. Neurol. 2008, 255, 413–419. [Google Scholar] [CrossRef]
- Dirik, E.; Yiş, U.; Başak, N.; Soydan, E.; Hüdaoǧlu, O.; Özgönül, F. Spinocerebellar Ataxia Type 2 in a Turkish Family. J. Child Neurol. 2007, 22, 891–894. [Google Scholar] [CrossRef]
- Moretti, P.; Blazo, M.; Garcia, L.; Armstrong, D.; Lewis, R.A.; Roa, B.; Scaglia, F. Spinocerebellar Ataxia Type 2 (SCA2) Presenting with Ophthalmoplegia and Developmental Delay in Infancy. Am. J. Med. Genet. A 2004, 124A, 392–396. [Google Scholar] [CrossRef]
- Almaguer-Mederos, L.; Falcón, N.; Almira, Y.; Zaldivar, Y.; Almarales, D.; Góngora, E.; Herrera, M.; Batallán, K.; Armiñán, R.; Manresa, M.; et al. Estimation of the Age at Onset in Spinocerebellar Ataxia Type 2 Cuban Patients by Survival Analysis. Clin. Genet. 2010, 78, 169–174. [Google Scholar] [CrossRef]
- Marcelo, A.; Afonso, I.T.; Afonso-Reis, R.; Brito, D.V.C.; Costa, R.G.; Rosa, A.; Alves-Cruzeiro, J.; Ferreira, B.; Henriques, C.; Nobre, R.J.; et al. Autophagy in Spinocerebellar Ataxia Type 2, a Dysregulated Pathway, and a Target for Therapy. Cell Death Dis. 2021, 12, 1117. [Google Scholar] [CrossRef]
- Miyatake, S.; Koshimizu, E.; Fujita, A.; Doi, H.; Okubo, M.; Wada, T.; Hamanaka, K.; Ueda, N.; Kishida, H.; Minase, G.; et al. Rapid and Comprehensive Diagnostic Method for Repeat Expansion Diseases Using Nanopore Sequencing. npj Genom. Med. 2022, 7, 1–15. [Google Scholar] [CrossRef]
- Available online: https://www.ern-rnd.eu/wp-content/uploads/2020/10/ERN-RND-Diagnostic-Flowchart-for-early-Ataxias_final.pdf (accessed on 30 November 2024).



{kind=link}
{kind=link}
{kind=link}
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | Subject 1 | Subject 2 | Subject 3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Repeat number | 884 | 104 | 320 | 124 | 92 | 300 | 750 | 500 | >200 | 220 | 200 | 500 | 350 | 400 | 230 | 220 | 69–75 | 70 | 62 | 58 | 61 | 45 |
| Inheritance (allele) | Maternal (49) | Paternal (n/a) | Paternal (47) | Paternal (45) | Paternal (51) | Paternal (43) | Paternal (40) | Paternal (40) | Maternal (45) | Maternal (43) | Paternal (42) | Maternal (45) | Paternal (40) | Paternal (43) | Paternal (40) | Paternal (43) | Paternal (39) | Paternal (40) | n/a | Paternal (37) | Paternal (n/a) | Paternal (n/a) |
| Age of onset | neonatal | neonatal | 6 months | 6 months | 4 months | 2 months | infantile | 3 months | 3 months | 2 weeks | 6 months | 10 months | 3 months | 10 months | 11 months | neonatal | 2 years | 5 years | 2 months | 8 years | 6 years | 6 months |
| Signs or symptoms at onset | Nystagmus, dysphagia | Hypotonia, dysphagia | Hypotonia, developmental regression | Hypotonia, eye movement abnormalities, myoclonus | Developmental delay, hypotonia, dyskinesia | Hypotonia | Developmental delay, microcephaly | Developmental regression | Focal seizures | Apnea episodes | Hypotonia | Hypotonia, developmental delay | Hypotonia, seizures | Hypotonia, developmental delay | Hypotonia, developmental delay | Hypotonia, apnoea episodes | Ataxia, progressive movement disorder | Ataxia, developmental delay | Eye movement abnormalities | Learning disability, cerebellar ataxia | Limb tremor | Febrile seizures, mild developmental delay |
| Developmental delay | y | y | y | y | y | y | y | y | y | y | y | y | y | y | y | |||||||
| Developmental regression | y | y | y | y | y | y | y | y | ||||||||||||||
| Ataxia | y | y | y | y | y | y | y | |||||||||||||||
| Eye movement abnormalities | y | y | y | y | y | y | y | y | y | y | ||||||||||||
| Spasticity | y | y | ||||||||||||||||||||
| Mov. Disorder | y | y | y | y | y | y | ||||||||||||||||
| Seizures | y | y | y | y | y | y | y | |||||||||||||||
| EEG abnormalities | y | y | y | y | y | y | y | y | y | y | ||||||||||||
| Cerebellar atrophy (MRI) | y | y | y | y | y | y | y | y | y | y | y | y | y | y | y | y | y | |||||
| Neuropathy | y | y | y | |||||||||||||||||||
| Pigmentary retinitis | y | y | y | y | y | y | y | y | y | y | y | y | ||||||||||
| Microcephaly | y | y | y | y | y | |||||||||||||||||
| Dysphagia | y | y | y | y | y | y | y | y | y | y | ||||||||||||
| Comorbidities | Dysmorphic features, PEG placement 1 year, GERD | Autonomic disfunction, optic nerve atrophy | Autonomic instability | Optic nerve atrophy | Visual impairment | Vasomotor instability, polyphagy, obesity | Incontinence, drooling | Incontinence, drooling | Borderline intellectual functioning | Lower limb contractures, scoliosis, respiratory insufficiency with tracheostomy, death at 18 years | ||||||||||||
| Reference | [9] | [10] | [11] | [12] | [13] | [14] | [14] | [14] | [14] | [14] | [14] | [15] | [15] | [15] | [15] | [16] | [17] | [18] | [19] | This work | This work | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sartorelli, J.; Pomponi, M.G.; Garone, G.; Vasco, G.; Cumbo, F.; Colona, V.L.; D’Amico, A.; Bertini, E.; Nicita, F. Three Cases of Spinocerebellar Ataxia Type 2 (SCA2) and Pediatric Literature Review: Do Not Forget Trinucleotide Repeat Disorders in Childhood-Onset Progressive Ataxia. Brain Sci. 2025, 15, 156. https://doi.org/10.3390/brainsci15020156
Sartorelli J, Pomponi MG, Garone G, Vasco G, Cumbo F, Colona VL, D’Amico A, Bertini E, Nicita F. Three Cases of Spinocerebellar Ataxia Type 2 (SCA2) and Pediatric Literature Review: Do Not Forget Trinucleotide Repeat Disorders in Childhood-Onset Progressive Ataxia. Brain Sciences. 2025; 15(2):156. https://doi.org/10.3390/brainsci15020156
Chicago/Turabian StyleSartorelli, Jacopo, Maria Grazia Pomponi, Giacomo Garone, Gessica Vasco, Francesca Cumbo, Vito Luigi Colona, Adele D’Amico, Enrico Bertini, and Francesco Nicita. 2025. "Three Cases of Spinocerebellar Ataxia Type 2 (SCA2) and Pediatric Literature Review: Do Not Forget Trinucleotide Repeat Disorders in Childhood-Onset Progressive Ataxia" Brain Sciences 15, no. 2: 156. https://doi.org/10.3390/brainsci15020156
APA StyleSartorelli, J., Pomponi, M. G., Garone, G., Vasco, G., Cumbo, F., Colona, V. L., D’Amico, A., Bertini, E., & Nicita, F. (2025). Three Cases of Spinocerebellar Ataxia Type 2 (SCA2) and Pediatric Literature Review: Do Not Forget Trinucleotide Repeat Disorders in Childhood-Onset Progressive Ataxia. Brain Sciences, 15(2), 156. https://doi.org/10.3390/brainsci15020156