Emerging Medications for Treatment-Resistant Depression: A Review with Perspective on Mechanisms and Challenges


Abstract
1. Introduction
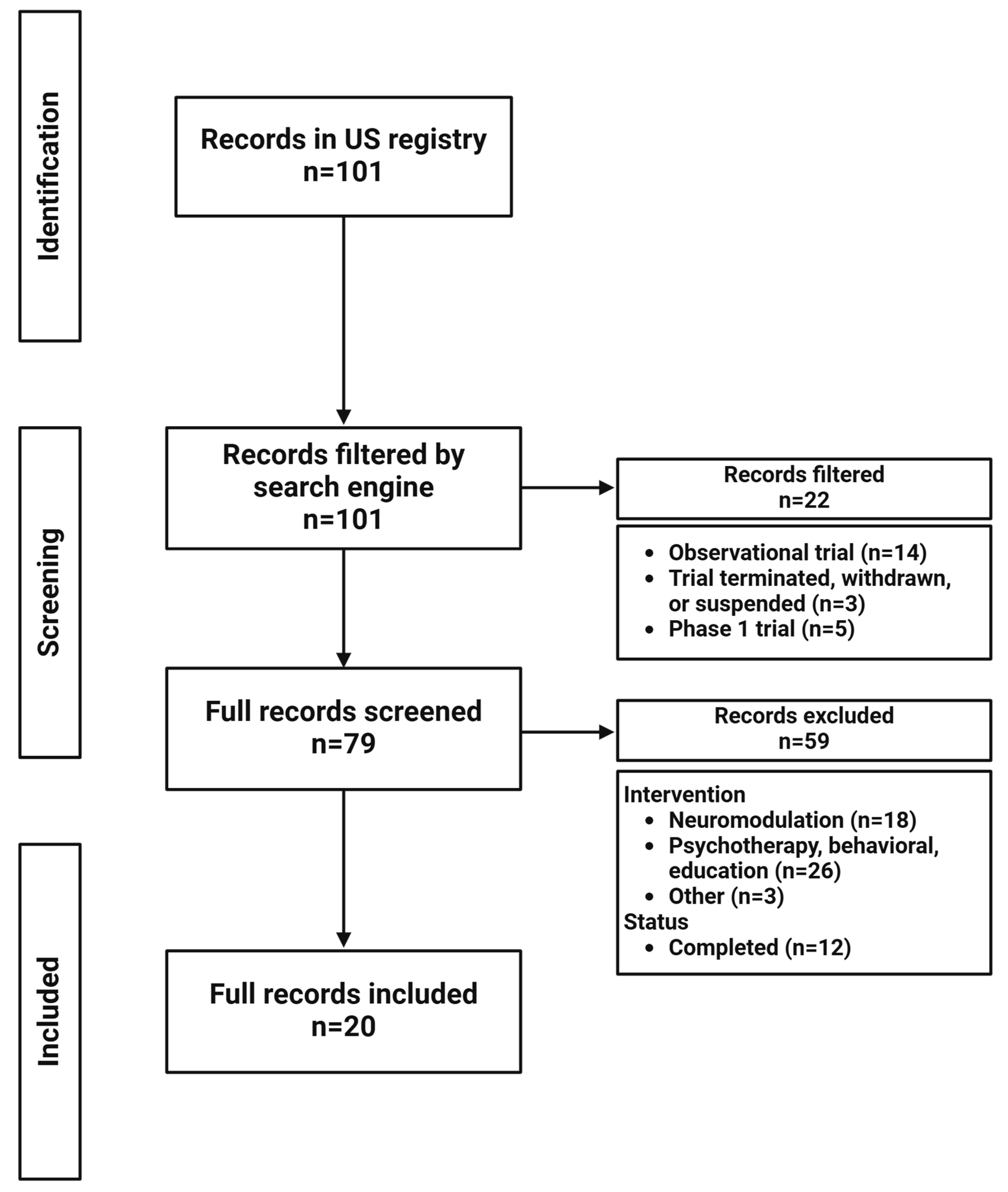
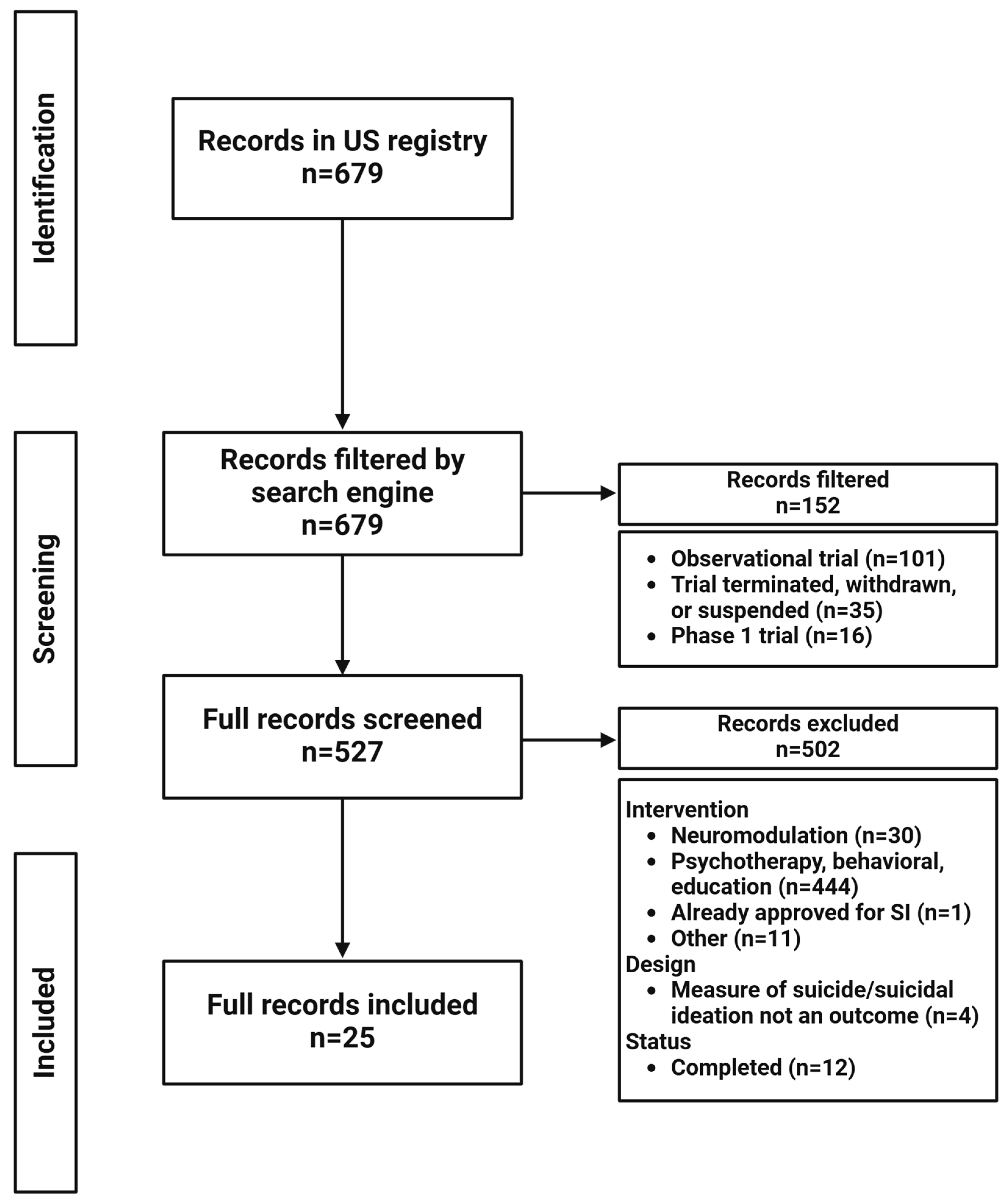
2. Materials and Methods
3. Results
3.1. Glutamatergic Signaling Modulators
3.2. Non-Glutamatergic Drugs
3.2.1. GABAergic Modulators
3.2.2. Monoaminergic Modulators
3.2.3. Psychedelics
3.2.4. Anti-Inflammatory and Immune Modulators
3.2.5. Opioid Receptor Modulators
3.2.6. Additional Mechanisms
3.3. Transdiagnostic Approaches to Treatment and Application of Biomarkers
3.3.1. Anhedonia
3.3.2. Suicidality
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| SSRI | selective serotonin reuptake inhibitor |
| SNRI | serotonin norepinephrine reuptake inhibitor |
| MDD | major depressive disorder |
| TCA | tricyclic antidepressant |
| MAOI | monoamine oxidase inhibitor |
| ADM | antidepressant medication |
| SRI | serotonin reuptake inhibitor |
| TRD | treatment-resistant depression |
| FDA | Food and Drug Administration |
| XR | extended release |
| MDE | major depressive episode |
| RCT | randomized controlled trial |
| PTSD | post-traumatic stress disorder |
| GABA | gamma-aminobutyric acid |
| NMDAR | N-methyl-D-aspartate receptor |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor |
| TrkB | tropomyosin receptor kinase B |
| eEF2 | eukaryotic elongation factor 2 |
| mTORC1 | mammalian target of rapamycin complex 1 |
| DCS | d-cycloserine |
| mGluR | metabotropic glutamate receptor |
| HNK | 2R,6R-hydroxynorketamine |
| 5-HT | 5-hydroxytryptamine, serotonin |
| PPD | post-partum depression |
| NDRI | norepinephrine and dopamine reuptake inhibitor |
| SERT | serotonin transporter |
| NET | norepinephrine transporter |
| DAT | dopamine transporter |
| DMT | dimethyltryptamine |
| PAT | psychedelic-assisted therapy |
| 5-MeO-DMT | 5-methoxy-dimethyltryptamine |
| CRP | C-reactive protein |
| TNF | tumor necrosis factor |
| IL | interleukin |
| IFN | interferon |
| EPA | eicosapentaeonic acid |
| HPA | hypothalamic–pituitary–adrenal |
| MAP2 | microtubule associated protein type-2 |
| rTMS | repetitive transcranial magnetic stimulation |
| CB | cannabinoid |
References
- Kazdin, A.E.; Wu, C.-S.; Hwang, I.; Puac-Polanco, V.; Sampson, N.A.; Al-Hamzawi, A.; Alonso, J.; Andrade, L.H.; Benjet, C.; Caldas-de-Almeida, J.-M. Antidepressant use in low-middle-and high-income countries: A World Mental Health Surveys report. Psychol. Med. 2021, 53, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Brody, D.J.; Gu, Q. Antidepressant Use Among Adults: United States, 2015–2018; US Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2020.
- Luo, Y.; Kataoka, Y.; Ostinelli, E.G.; Cipriani, A.; Furukawa, T.A. National Prescription Patterns of Antidepressants in the Treatment of Adults With Major Depression in the US Between 1996 and 2015: A Population Representative Survey Based Analysis. Front. Psychiatry 2020, 11, 35. [Google Scholar] [CrossRef]
- Gabriel, F.C.; de Melo, D.O.; Fráguas, R.; Leite-Santos, N.C.; Mantovani da Silva, R.A.; Ribeiro, E. Pharmacological treatment of depression: A systematic review comparing clinical practice guideline recommendations. PLoS ONE 2020, 15, e0231700. [Google Scholar] [CrossRef]
- Lee, E.H.; Han, P.L. Reciprocal interactions across and within multiple levels of monoamine and cortico-limbic systems in stress-induced depression: A systematic review. Neurosci. Biobehav. Rev. 2019, 101, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Sinyor, M.; Levitt, A.J.; Cheung, A.H.; Schaffer, A.; Kiss, A.; Dowlati, Y.; Lanctôt, K.L. Does inclusion of a placebo arm influence response to active antidepressant treatment in randomized controlled trials? Results from pooled and meta-analyses. J. Clin. Psychiatry 2010, 71, 4632. [Google Scholar] [CrossRef]
- Overall, J. Methodologic issues in the epidemiology of treatment resistant depression. Pharmacopsychiatry 1974, 7, 129–138. [Google Scholar] [CrossRef]
- Zhou, X.; Keitner, G.I.; Qin, B.; Ravindran, A.V.; Bauer, M.; Del Giovane, C.; Zhao, J.; Liu, Y.; Fang, Y.; Zhang, Y.; et al. Atypical Antipsychotic Augmentation for Treatment-Resistant Depression: A Systematic Review and Network Meta-Analysis. Int. J. Neuropsychopharmacol. 2015, 18, pyv060. [Google Scholar] [CrossRef]
- Al Shirawi, M.I.; Edgar, N.E.; Kennedy, S.H. Brexpiprazole in the Treatment of Major Depressive Disorder. Clin. Med. Insights Ther. 2017, 9, 1179559X17731801. [Google Scholar] [CrossRef]
- Luan, S.; Wan, H.; Zhang, L.; Zhao, H. Efficacy, acceptability, and safety of adjunctive aripiprazole in treatment-resistant depression: A meta-analysis of randomized controlled trials. Neuropsychiatr. Dis. Treat. 2018, 14, 467–477. [Google Scholar] [CrossRef]
- Sachs, G.S.; Yeung, P.P.; Rekeda, L.; Khan, A.; Adams, J.L.; Fava, M. Adjunctive Cariprazine for the Treatment of Patients With Major Depressive Disorder: A Randomized, Double-Blind, Placebo-Controlled Phase 3 Study. Am. J. Psychiatry 2023, 180, 241–251. [Google Scholar] [CrossRef]
- Li, G.; Zhang, L.; DiBernardo, A.; Wang, G.; Sheehan, J.J.; Lee, K.; Reutfors, J.; Zhang, Q. A retrospective analysis to estimate the healthcare resource utilization and cost associated with treatment-resistant depression in commercially insured US patients. PLoS ONE 2020, 15, e0238843. [Google Scholar] [CrossRef] [PubMed]
- Rybak, Y.E.; Lai, K.S.; Ramasubbu, R.; Vila-Rodriguez, F.; Blumberger, D.M.; Chan, P.; Delva, N.; Giacobbe, P.; Gosselin, C.; Kennedy, S.H. Treatment-resistant major depressive disorder: Canadian expert consensus on definition and assessment. Depress. Anxiety 2021, 38, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Sforzini, L.; Worrell, C.; Kose, M.; Anderson, I.M.; Aouizerate, B.; Arolt, V.; Bauer, M.; Baune, B.T.; Blier, P.; Cleare, A.J.; et al. A Delphi-method-based consensus guideline for definition of treatment-resistant depression for clinical trials. Mol. Psychiatry 2022, 27, 1286–1299. [Google Scholar] [CrossRef]
- Trivedi, M.H.; Rush, A.J.; Wisniewski, S.R.; Nierenberg, A.A.; Warden, D.; Ritz, L.; Norquist, G.; Howland, R.H.; Lebowitz, B.; McGrath, P.J. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR* D: Implications for clinical practice. Am. J. Psychiatry 2006, 163, 28–40. [Google Scholar] [CrossRef]
- Souery, D.; Pitchot, W. Switching antidepressants in patients with treatment-resistant depression. In Managing Treatment-Resistant Depression: Road to Novel Therapeutics; Quevedo, J., Riva-Posse, P., Bobo, W.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 167–174. [Google Scholar]
- Weitz, E.S.; Hollon, S.D.; Twisk, J.; Van Straten, A.; Huibers, M.J.; David, D.; DeRubeis, R.J.; Dimidjian, S.; Dunlop, B.W.; Cristea, I.A. Baseline depression severity as moderator of depression outcomes between cognitive behavioral therapy vs pharmacotherapy: An individual patient data meta-analysis. JAMA Psychiatry 2015, 72, 1102–1109. [Google Scholar] [CrossRef]
- Conway, C.R.; George, M.S.; Sackeim, H.A. Toward an evidence-based, operational definition of treatment-resistant depression: When enough is enough. JAMA Psychiatry 2017, 74, 9–10. [Google Scholar] [CrossRef]
- Dunlop, B.W.; Kaye, J.L.; Youngner, C.; Rothbaum, B. Assessing treatment-resistant posttraumatic stress disorder: The Emory treatment resistance interview for PTSD (E-TRIP). Behav. Sci. 2014, 4, 511–527. [Google Scholar] [CrossRef]
- Dunlop, B.W.C.; Choi, K.S.; Rajendra, J.K.; Nemeroff, C.B.; Craighead, W.E.; Mayberg, H.S. Shared and unique changes in brain connectivity among depressed patients remitting with pharmacotherapy versus psychotherapy. Am. J. Psychiatry, 2023; in press. [Google Scholar]
- Leykin, Y.; Amsterdam, J.D.; DeRubeis, R.J.; Gallop, R.; Shelton, R.C.; Hollon, S.D. Progressive resistance to a selective serotonin reuptake inhibitor but not to cognitive therapy in the treatment of major depression. J. Consult. Clin. Psychol. 2007, 75, 267. [Google Scholar] [CrossRef]
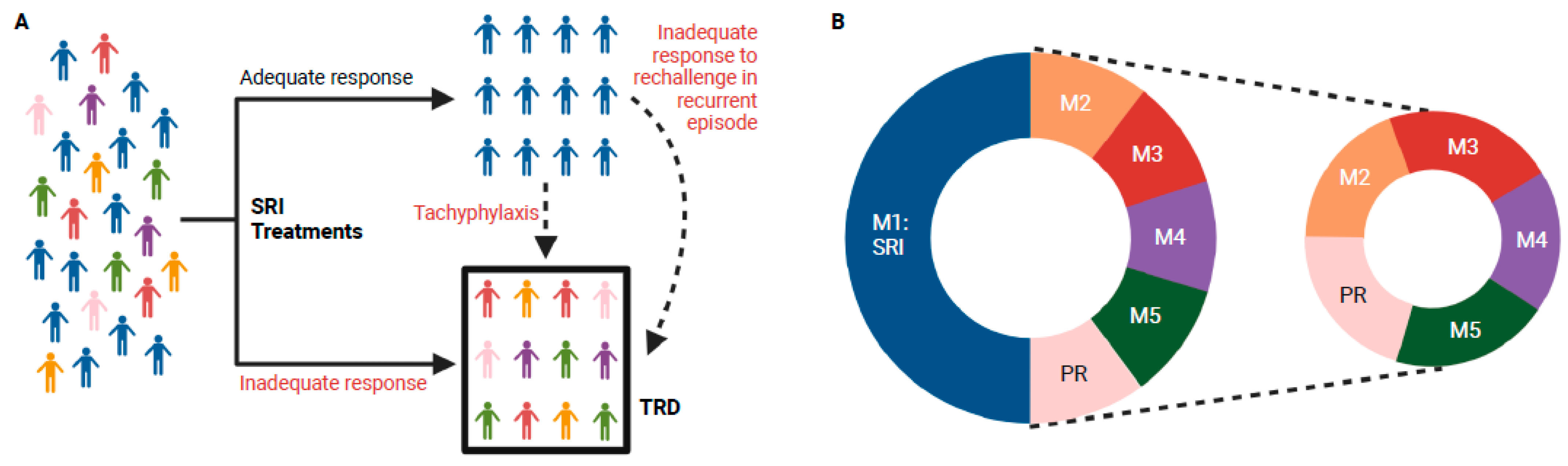
- Amsterdam, J.D.; Williams, D.; Michelson, D.; Adler, L.A.; Dunner, D.L.; Nierenberg, A.A.; Reimherr, F.W.; Schatzberg, A.F. Tachyphylaxis after repeated antidepressant drug exposure in patients with recurrent major depressive disorder. Neuropsychobiology 2009, 59, 227–233. [Google Scholar] [CrossRef]
- Fava, G.A.; Cosci, F.; Guidi, J.; Rafanelli, C. The deceptive manifestations of treatment resistance in depression: A new look at the problem. Psychother. Psychosom. 2020, 89, 265–273. [Google Scholar] [CrossRef]
- Gaynes, B.N.; Lux, L.; Gartlehner, G.; Asher, G.; Forman-Hoffman, V.; Green, J.; Boland, E.; Weber, R.P.; Randolph, C.; Bann, C. Defining treatment-resistant depression. Depress. Anxiety 2020, 37, 134–145. [Google Scholar] [CrossRef]
- Fekadu, A.; Donocik, J.G.; Cleare, A.J. Standardisation framework for the Maudsley staging method for treatment resistance in depression. BMC Psychiatry 2018, 18, 100. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, B.W.; Medeiros Da Frota Ribeiro, C. Randomized Controlled Trials and the Efficacy of Psychotropic Medications. In NeuroPsychopharmacotherapy; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–56. [Google Scholar]
- Nierenberg, A.; Amsterdam, J. Treatment-resistant depression: Definition and treatment approaches. J. Clin. Psychiatry 1990, 51, 39–47; discussion 48. [Google Scholar] [PubMed]
- McCann, D.J.; Petry, N.M.; Bresell, A.; Isacsson, E.; Wilson, E.; Alexander, R.C. Medication nonadherence, “professional subjects,” and apparent placebo responders: Overlapping challenges for medications development. J. Clin. Psychopharmacol. 2015, 35, 566. [Google Scholar] [CrossRef] [PubMed]
- Devine, E.G.; Waters, M.E.; Putnam, M.; Surprise, C.; O’Malley, K.; Richambault, C.; Fishman, R.L.; Knapp, C.M.; Patterson, E.H.; Sarid-Segal, O. Concealment and fabrication by experienced research subjects. Clin. Trials 2013, 10, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.D.; Razza, L.B.; Weissman, C.R.; Karbi, J.; Vine, T.; Mulsant, L.S.; Brunoni, A.R.; Husain, M.I.; Mulsant, B.H.; Blumberger, D.M. Magnitude of the placebo response across treatment modalities used for treatment-resistant depression in adults: A systematic review and meta-analysis. JAMA Netw. Open 2021, 4, e2125531. [Google Scholar] [CrossRef] [PubMed]
- Marder, S.R.; Laughren, T.; Romano, S.J. Why Are Innovative Drugs Failing in Phase III? Am. J. Psychiatry 2017, 174, 829–831. [Google Scholar] [CrossRef]
- Albert, P.R.; Benkelfat, C. The neurobiology of depression–revisiting the serotonin hypothesis. II. Genetic, epigenetic and clinical studies. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120535. [Google Scholar] [CrossRef]
- Fusar-Poli, P.; Solmi, M.; Brondino, N.; Davies, C.; Chae, C.; Politi, P.; Borgwardt, S.; Lawrie, S.M.; Parnas, J.; McGuire, P. Transdiagnostic psychiatry: A systematic review. World Psychiatry 2019, 18, 192–207. [Google Scholar] [CrossRef]
- Dalgleish, T.; Black, M.; Johnston, D.; Bevan, A. Transdiagnostic approaches to mental health problems: Current status and future directions. J. Consult. Clin. Psychol. 2020, 88, 179–195. [Google Scholar] [CrossRef]
- Kist, J.D.; Vrijsen, J.N.; Mulders, P.C.R.; van Eijndhoven, P.F.P.; Tendolkar, I.; Collard, R.M. Transdiagnostic psychiatry: Symptom profiles and their direct and indirect relationship with well-being. J. Psychiatr. Res. 2023, 161, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Maj, M. Why the clinical utility of diagnostic categories in psychiatry is intrinsically limited and how we can use new approaches to complement them. World Psychiatry 2018, 17, 121–122. [Google Scholar] [CrossRef]
- Rogan, T.; Wilkinson, S.T. The Role of Psychotherapy in the Management of Treatment-Resistant Depression. Psychiatr. Clin. N. Am. 2023, 46, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Conway, C.R.; Hristidis, V.C.; Gott, B.M.; Willie, J. Vagus Nerve Stimulation for Treatment-Resistant Depression: Current Status of This Novel Treatment. Psychiatr. Ann. 2022, 52, 272–282. [Google Scholar] [CrossRef]
- Müller, H.H.O.; Moeller, S.; Lücke, C.; Lam, A.P.; Braun, N.; Philipsen, A. Vagus Nerve Stimulation (VNS) and Other Augmentation Strategies for Therapy-Resistant Depression (TRD): Review of the Evidence and Clinical Advice for Use. Front. Neurosci. 2018, 12, 239. [Google Scholar] [CrossRef]
- Adu, M.K.; Shalaby, R.; Chue, P.; Agyapong, V.I.O. Repetitive Transcranial Magnetic Stimulation for the Treatment of Resistant Depression: A Scoping Review. Behav. Sci. 2022, 12, 195. [Google Scholar] [CrossRef]
- Dandekar, M.P.; Fenoy, A.J.; Carvalho, A.F.; Soares, J.C.; Quevedo, J. Deep brain stimulation for treatment-resistant depression: An integrative review of preclinical and clinical findings and translational implications. Mol. Psychiatry 2018, 23, 1094–1112. [Google Scholar] [CrossRef]
- Wu, Y.; Mo, J.; Sui, L.; Zhang, J.; Hu, W.; Zhang, C.; Wang, Y.; Liu, C.; Zhao, B.; Wang, X.; et al. Deep Brain Stimulation in Treatment-Resistant Depression: A Systematic Review and Meta-Analysis on Efficacy and Safety. Front. Neurosci. 2021, 15, 655412. [Google Scholar] [CrossRef]
- Pittenger, C.; Duman, R.S. Stress, Depression, and Neuroplasticity: A Convergence of Mechanisms. Neuropsychopharmacology 2008, 33, 88–109. [Google Scholar] [CrossRef]
- D’Sa, C.; Duman, R.S. Antidepressants and neuroplasticity. Bipolar Disord. 2002, 4, 183–194. [Google Scholar] [CrossRef]
- Aleksandrova, L.R.; Phillips, A.G. Neuroplasticity as a convergent mechanism of ketamine and classical psychedelics. Trends Pharmacol. Sci. 2021, 42, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Barsaglini, A.; Sartori, G.; Benetti, S.; Pettersson-Yeo, W.; Mechelli, A. The effects of psychotherapy on brain function: A systematic and critical review. Prog. Neurobiol. 2014, 114, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lyden, H.; Espinoza, R.; Pirnia, T.; Clark, K.; Joshi, S.; Leaver, A.; Woods, R.; Narr, K. Electroconvulsive therapy mediates neuroplasticity of white matter microstructure in major depression. Transl. Psychiatry 2014, 4, e380. [Google Scholar] [CrossRef]
- Pirnia, T.; Joshi, S.; Leaver, A.; Vasavada, M.; Njau, S.; Woods, R.; Espinoza, R.; Narr, K. Electroconvulsive therapy and structural neuroplasticity in neocortical, limbic and paralimbic cortex. Transl. Psychiatry 2016, 6, e832. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol. Psychiatry 2000, 48, 766–777. [Google Scholar] [CrossRef]
- Koolschijn, P.C.M.; van Haren, N.E.; Lensvelt-Mulders, G.J.; Hulshoff Pol, H.E.; Kahn, R.S. Brain volume abnormalities in major depressive disorder: A meta-analysis of magnetic resonance imaging studies. Hum. Brain Mapp. 2009, 30, 3719–3735. [Google Scholar] [CrossRef]
- Videbech, P.; Ravnkilde, B. Hippocampal volume and depression: A meta-analysis of MRI studies. Am. J. Psychiatry 2004, 161, 1957–1966. [Google Scholar] [CrossRef]
- Belleau, E.L.; Treadway, M.T.; Pizzagalli, D.A. The Impact of Stress and Major Depressive Disorder on Hippocampal and Medial Prefrontal Cortex Morphology. Biol. Psychiatry 2019, 85, 443–453. [Google Scholar] [CrossRef]
- David, D.J.; Samuels, B.A.; Rainer, Q.; Wang, J.W.; Marsteller, D.; Mendez, I.; Drew, M.; Craig, D.A.; Guiard, B.P.; Guilloux, J.P.; et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 2009, 62, 479–493. [Google Scholar] [CrossRef]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000, 20, 9104–9110. [Google Scholar] [CrossRef]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Malberg, J.E.; Hen, R.; Madsen, T.M. Adult Neurogenesis and Antidepressant Treatment: The Surprise Finding by Ron Duman and the Field 20 Years Later. Biol. Psychiatry 2021, 90, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Franjic, D.; Skarica, M.; Ma, S.; Arellano, J.I.; Tebbenkamp, A.T.N.; Choi, J.; Xu, C.; Li, Q.; Morozov, Y.M.; Andrijevic, D.; et al. Transcriptomic taxonomy and neurogenic trajectories of adult human, macaque, and pig hippocampal and entorhinal cells. Neuron 2022, 110, 452–469.e414. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Sousa, A.M.M.; Gao, T.; Skarica, M.; Li, M.; Santpere, G.; Esteller-Cucala, P.; Juan, D.; Ferrández-Peral, L.; Gulden, F.O.; et al. Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 2018, 362, aat8077. [Google Scholar] [CrossRef]
- Sousa, A.M.M.; Zhu, Y.; Raghanti, M.A.; Kitchen, R.R.; Onorati, M.; Tebbenkamp, A.T.N.; Stutz, B.; Meyer, K.A.; Li, M.; Kawasawa, Y.I.; et al. Molecular and cellular reorganization of neural circuits in the human lineage. Science 2017, 358, 1027–1032. [Google Scholar] [CrossRef]
- Williams, N.R.; Heifets, B.D.; Blasey, C.; Sudheimer, K.; Pannu, J.; Pankow, H.; Hawkins, J.; Birnbaum, J.; Lyons, D.M.; Rodriguez, C.I.; et al. Attenuation of Antidepressant Effects of Ketamine by Opioid Receptor Antagonism. Am. J. Psychiatry 2018, 175, 1205–1215. [Google Scholar] [CrossRef]
- Zarate, C.A., Jr.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef]
- Zheng, W.; Cai, D.-B.; Xiang, Y.-Q.; Jiang, W.-L.; Sim, K.; Ungvari, G.S.; Huang, X.; Huang, X.-X.; Ning, Y.-P.; Xiang, Y.-T. Adjunctive intranasal esketamine for major depressive disorder: A systematic review of randomized double-blind controlled-placebo studies. J. Affect. Disord. 2020, 265, 63–70. [Google Scholar] [CrossRef]
- Sanacora, G.; Treccani, G.; Popoli, M. Towards a glutamate hypothesis of depression: An emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 2012, 62, 63–77. [Google Scholar] [CrossRef]
- Auer, D.P.; Pütz, B.; Kraft, E.; Lipinski, B.; Schill, J.; Holsboer, F. Reduced glutamate in the anterior cingulate cortex in depression: An in vivo proton magnetic resonance spectroscopy study. Biol. Psychiatry 2000, 47, 305–313. [Google Scholar] [CrossRef]
- Hasler, G.; van der Veen, J.W.; Tumonis, T.; Meyers, N.; Shen, J.; Drevets, W.C. Reduced prefrontal glutamate/glutamine and γ-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 2007, 64, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Zink, M.; Vollmayr, B.; Gebicke-Haerter, P.J.; Henn, F.A. Reduced expression of glutamate transporters vGluT1, EAAT2 and EAAT4 in learned helpless rats, an animal model of depression. Neuropharmacology 2010, 58, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Choudary, P.V.; Molnar, M.; Evans, S.J.; Tomita, H.; Li, J.Z.; Vawter, M.P.; Myers, R.M.; Bunney, W.E.; Akil, H.; Watson, S.J.; et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl. Acad. Sci. USA 2005, 102, 15653–15658. [Google Scholar] [CrossRef] [PubMed]
- Banasr, M.; Duman, R.S. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol. Psychiatry 2008, 64, 863–870. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Riggs, L.M.; Gould, T.D. Ketamine and the future of rapid-acting antidepressants. Annu. Rev. Clin. Psychol. 2021, 17, 207–231. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef]
- Sanacora, G.; Smith, M.A.; Pathak, S.; Su, H.L.; Boeijinga, P.H.; McCarthy, D.J.; Quirk, M.C. Lanicemine: A low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol. Psychiatry 2014, 19, 978–985. [Google Scholar] [CrossRef]
- Fogaça, M.V.; Wu, M.; Li, C.; Li, X.-Y.; Picciotto, M.R.; Duman, R.S. Inhibition of GABA interneurons in the mPFC is sufficient and necessary for rapid antidepressant responses. Mol. Psychiatry 2021, 26, 3277–3291. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, Y.; Sang, K.; Dong, Y.; Ni, Z.; Ma, S.; Hu, H. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 2018, 554, 317–322. [Google Scholar] [CrossRef]
- Jourdi, H.; Hsu, Y.T.; Zhou, M.; Qin, Q.; Bi, X.; Baudry, M. Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J. Neurosci. 2009, 29, 8688–8697. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Georgiou, P.; Fischell, J.; Elmer, G.I.; Alkondon, M.; Yuan, P.; Pribut, H.J.; Singh, N.S. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016, 533, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.P.; Traynelis, S.F.; Siffert, J.; Pope, L.E.; Matsumoto, R.R. Pharmacology of dextromethorphan: Relevance to dextromethorphan/quinidine (Nuedexta®) clinical use. Pharmacol. Ther. 2016, 164, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Iosifescu, D.V.; Jones, A.; O’Gorman, C.; Streicher, C.; Feliz, S.; Fava, M.; Tabuteau, H. Efficacy and Safety of AXS-05 (Dextromethorphan-Bupropion) in Patients With Major Depressive Disorder: A Phase 3 Randomized Clinical Trial (GEMINI). J. Clin. Psychiatry 2022, 83, 41226. [Google Scholar] [CrossRef] [PubMed]
- Tabuteau, H.; Jones, A.; Anderson, A.; Jacobson, M.; Iosifescu, D.V. Effect of AXS-05 (Dextromethorphan-Bupropion) in Major Depressive Disorder: A Randomized Double-Blind Controlled Trial. Am. J. Psychiatry 2022, 179, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Axsome Therapeutics, Inc. Axsome Therapeutics Announces Topline Results of the STRIDE-1 Phase 3 Trial in Treatment Resistant Depression and Expert Call to Discuss Clinical Implications. Available online: https://www.globenewswire.com/news-release/2020/03/30/2008163/0/en/Axsome-Therapeutics-Announces-Topline-Results-of-the-STRIDE-1-Phase-3-Trial-in-Treatment-Resistant-Depression-and-Expert-Call-to-Discuss-Clinical-Implications.html (accessed on 22 December 2024).
- Nemeroff, C.B. Back to the future: Esmethadone, the (maybe) nonopiate opiate, and depression. Am. J. Psychiatry 2022, 179, 83–84. [Google Scholar] [CrossRef] [PubMed]
- De Martin, S.; Gabbia, D.; Folli, F.; Bifari, F.; Fiorina, P.; Ferri, N.; Stahl, S.; Inturrisi, C.E.; Pappagallo, M.; Traversa, S.; et al. REL-1017 (Esmethadone) Increases Circulating BDNF Levels in Healthy Subjects of a Phase 1 Clinical Study. Front. Pharmacol. 2021, 12, 671859. [Google Scholar] [CrossRef] [PubMed]
- Fava, M.; Stahl, S.; Pani, L.; De Martin, S.; Pappagallo, M.; Guidetti, C.; Alimonti, A.; Bettini, E.; Mangano, R.M.; Wessel, T.; et al. REL-1017 (Esmethadone) as Adjunctive Treatment in Patients With Major Depressive Disorder: A Phase 2a Randomized Double-Blind Trial. Am. J. Psychiatry 2022, 179, 122–131. [Google Scholar] [CrossRef]
- Relmada Therapeutics Relmada Therapeutics Reports That Data Monitoring Committee (DMC) Assessment Indicates That the Phase 3 Reliance II Trial is Futile at its Interim Analysis and is Unlikely to Meet the Primary Efficacy Endpoint with Statistical Significance. Available online: https://www.relmada.com/for-investors/news/detail/306/relmada-therapeutics-reports-that-data-monitoring-committee (accessed on 22 December 2024).
- Nagele, P.; Duma, A.; Kopec, M.; Gebara, M.A.; Parsoei, A.; Walker, M.; Janski, A.; Panagopoulos, V.N.; Cristancho, P.; Miller, J.P.; et al. Nitrous Oxide for Treatment-Resistant Major Depression: A Proof-of-Concept Trial. Biol. Psychiatry 2015, 78, 10–18. [Google Scholar] [CrossRef]
- Nagele, P.; Palanca Ben, J.; Gott, B.; Brown, F.; Barnes, L.; Nguyen, T.; Xiong, W.; Salloum Naji, C.; Espejo Gemma, D.; Lessov-Schlaggar Christina, N.; et al. A phase 2 trial of inhaled nitrous oxide for treatment-resistant major depression. Sci. Transl. Med. 2021, 13, eabe1376. [Google Scholar] [CrossRef]
- Nagele, P.; Zorumski, C.F.; Conway, C. Exploring Nitrous Oxide as Treatment of Mood Disorders: Basic Concepts. J. Clin. Psychopharmacol. 2018, 38, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Gideons, E.S.; Kavalali, E.T.; Monteggia, L.M. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc. Natl. Acad. Sci. USA 2014, 111, 8649–8654. [Google Scholar] [CrossRef] [PubMed]
- Miller, O.H.; Yang, L.; Wang, C.-C.; Hargroder, E.A.; Zhang, Y.; Delpire, E.; Hall, B.J. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. eLife 2014, 3, e03581. [Google Scholar] [CrossRef] [PubMed]
- Henter, I.D.; Park, L.T.; Zarate, C.A. Novel Glutamatergic Modulators for the Treatment of Mood Disorders: Current Status. CNS Drugs 2021, 35, 527–543. [Google Scholar] [CrossRef]
- Kew, J.N. Positive and negative allosteric modulation of metabotropic glutamate receptors: Emerging therapeutic potential. Pharmacol. Ther. 2004, 104, 233–244. [Google Scholar] [CrossRef]
- Ghaemi, N.; Sverdlov, A.; Shelton, R.; Litman, R. Efficacy and safety of mij821 in patients with treatment-resistant depression: Results from a randomized, placebo-controlled, proof-of-concept study. Eur. Psychiatry 2021, 64, S334–S335. [Google Scholar] [CrossRef]
- Novartis. Proof of Concept Study Evaluating the Efficacy and Safety of MIJ821 in Patients with Treatment-resistant Depression. ClinicalTrials.gov Identifier: NCT03756129. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03756129?term=MIJ821&cond=Depression&draw=2&rank=2 (accessed on 22 December 2024).
- D’Arrigo, T. Rapastinel Fails Phase 3 Trials. Available online: https://psychnews.psychiatryonline.org/doi/10.1176/appi.pn.2019.pp3a5 (accessed on 22 December 2024).
- Preskorn, S.; Macaluso, M.; Mehra, D.O.; Zammit, G.; Moskal, J.R.; Burch, R.M. Randomized proof of concept trial of GLYX-13, an N-methyl-D-aspartate receptor glycine site partial agonist, in major depressive disorder nonresponsive to a previous antidepressant agent. J. Psychiatr. Pract. 2015, 21, 140–149. [Google Scholar] [CrossRef]
- Gate Neurosciences Inc. AGN-241751 in the Treatment of Major Depressive Disorder. ClinicalTrials.gov Identifier: NCT03586247. Available online: https://clinicaltrials.gov/ct2/show/results/NCT03586427?term=AGN-241751&cond=Depression&draw=2&rank=2&view=results (accessed on 22 December 2024).
- Rajagopal, L.; Huang, M.; He, W.; Ryan, C.; Elzokaky, A.; Banerjee, P.; Meltzer, H.Y. Repeated administration of rapastinel produces exceptionally prolonged rescue of memory deficits in phencyclidine-treated mice. Behav. Brain Res. 2022, 432, 113964. [Google Scholar] [CrossRef]
- VistaGen Therapeutics, Inc. AV-101 as Adjunct Antidepressant Therapy in Patients with Major Depression (ELEVATE). ClinicalTrials.gov identifier: NCT03078322. Available online: https://clinicaltrials.gov/ct2/show/results/NCT03078322 (accessed on 22 December 2024).
- Mataix-Cols, D.; Fernández de la Cruz, L.; Monzani, B.; Rosenfield, D.; Andersson, E.; Pérez-Vigil, A.; Frumento, P.; de Kleine, R.A.; Difede, J.; Dunlop, B.W.; et al. D-Cycloserine Augmentation of Exposure-Based Cognitive Behavior Therapy for Anxiety, Obsessive-Compulsive, and Posttraumatic Stress Disorders: A Systematic Review and Meta-analysis of Individual Participant Data. JAMA Psychiatry 2017, 74, 501–510. [Google Scholar] [CrossRef]
- Schade, S.; Paulus, W. D-Cycloserine in Neuropsychiatric Diseases: A Systematic Review. Int. J. Neuropsychopharmacol. 2016, 19, pyv102. [Google Scholar] [CrossRef] [PubMed]
- Horio, M.; Mori, H.; Hashimoto, K. Is D-cycloserine a prodrug for D-serine in the brain? Biol. Psychiatry 2013, 73, e33–e34. [Google Scholar] [CrossRef] [PubMed]
- Arizanovska, D.; Emodogo, J.A.; Lally, A.P.; Palavicino-Maggio, C.B.; Liebl, D.J.; Folorunso, O.O. Cross species review of the physiological role of D-serine in translationally relevant behaviors. Amino Acids 2023, 55, 1501–1517. [Google Scholar] [CrossRef]
- Guercio, G.D.; Panizzutti, R. Potential and Challenges for the Clinical Use of d-Serine As a Cognitive Enhancer. Front. Psychiatry 2018, 9, 14. [Google Scholar] [CrossRef]
- Meftah, A.; Hasegawa, H.; Kantrowitz, J.T. D-Serine: A Cross Species Review of Safety. Front. Psychiatry 2021, 12, 726365. [Google Scholar] [CrossRef]
- Wierońska, J.M.; Pilc, A. Metabotropic glutamate receptors in the tripartite synapse as a target for new psychotropic drugs. Neurochem. Int. 2009, 55, 85–97. [Google Scholar] [CrossRef]
- Quiroz, J.A.; Tamburri, P.; Deptula, D.; Banken, L.; Beyer, U.; Rabbia, M.; Parkar, N.; Fontoura, P.; Santarelli, L. Efficacy and Safety of Basimglurant as Adjunctive Therapy for Major Depression: A Randomized Clinical Trial. JAMA Psychiatry 2016, 73, 675–684. [Google Scholar] [CrossRef]
- Kent, J.M.; Daly, E.; Kezic, I.; Lane, R.; Lim, P.; De Smedt, H.; De Boer, P.; Van Nueten, L.; Drevets, W.C.; Ceusters, M. Efficacy and safety of an adjunctive mGlu2 receptor positive allosteric modulator to a SSRI/SNRI in anxious depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 67, 66–73. [Google Scholar] [CrossRef]
- Dogra, S.; Conn, P.J. Targeting metabotropic glutamate receptors for the treatment of depression and other stress-related disorders. Neuropharmacology 2021, 196, 108687. [Google Scholar] [CrossRef]
- Wei, Y.; Chang, L.; Hashimoto, K. Molecular mechanisms underlying the antidepressant actions of arketamine: Beyond the NMDA receptor. Mol. Psychiatry 2022, 27, 559–573. [Google Scholar] [CrossRef]
- Leal, G.C.; Souza-Marques, B.; Mello, R.P.; Bandeira, I.D.; Caliman-Fontes, A.T.; Carneiro, B.A.; Faria-Guimarães, D.; Guerreiro-Costa, L.N.F.; Jesus-Nunes, A.P.; Silva, S.S.; et al. Arketamine as adjunctive therapy for treatment-resistant depression: A placebo-controlled pilot study. J. Affect. Disord. 2023, 330, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Atai Life Sciences. Atai Life Sciences Announces Results from Phase 2a Trial of PCN-101 (R-ketamine) for Treatment-Resistant Depression. Available online: https://ir.atai.life/news-releases/news-release-details/atai-life-sciences-announces-results-phase-2a-trial-pcn-101-r (accessed on 22 December 2024).
- Zanos, P.; Highland, J.N.; Stewart, B.W.; Georgiou, P.; Jenne, C.E.; Lovett, J.; Morris, P.J.; Thomas, C.J.; Moaddel, R.; Zarate, C.A., Jr.; et al. (2R,6R)-hydroxynorketamine exerts mGlu(2) receptor-dependent antidepressant actions. Proc. Natl. Acad. Sci. USA 2019, 116, 6441–6450. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Pothula, S.; Liu, R.-J.; Duman, C.H.; Terwilliger, R.; Vlasuk, G.P.; Saiah, E.; Hahm, S.; Duman, R.S. Sestrin modulator NV-5138 produces rapid antidepressant effects via direct mTORC1 activation. J. Clin. Investig. 2019, 129, 2542–2554. [Google Scholar] [CrossRef] [PubMed]
- Fogaça, M.V.; Duman, R.S. Cortical GABAergic Dysfunction in Stress and Depression: New Insights for Therapeutic Interventions. Front. Cell. Neurosci. 2019, 13, 448587. [Google Scholar] [CrossRef]
- Radley, J.J.; Gosselink, K.L.; Sawchenko, P.E. A discrete GABAergic relay mediates medial prefrontal cortical inhibition of the neuroendocrine stress response. J. Neurosci. 2009, 29, 7330–7340. [Google Scholar] [CrossRef]
- Myers, B.; Carvalho-Netto, E.; Wick-Carlson, D.; Wu, C.; Naser, S.; Solomon, M.B.; Ulrich-Lai, Y.M.; Herman, J.P. GABAergic Signaling within a Limbic-Hypothalamic Circuit Integrates Social and Anxiety-Like Behavior with Stress Reactivity. Neuropsychopharmacology 2016, 41, 1530–1539. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef]
- Dudek, K.A.; Dion-Albert, L.; Lebel, M.; LeClair, K.; Labrecque, S.; Tuck, E.; Ferrer Perez, C.; Golden, S.A.; Tamminga, C.; Turecki, G.; et al. Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. Proc. Natl. Acad. Sci. USA 2020, 117, 3326–3336. [Google Scholar] [CrossRef]
- Prévot, T.; Sibille, E. Altered GABA-mediated information processing and cognitive dysfunctions in depression and other brain disorders. Mol. Psychiatry 2021, 26, 151–167. [Google Scholar] [CrossRef]
- Luscher, B.; Shen, Q.; Sahir, N. The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 2011, 16, 383–406. [Google Scholar] [CrossRef]
- Edinoff, A.N.; Odisho, A.S.; Lewis, K.; Kaskas, A.; Hunt, G.; Cornett, E.M.; Kaye, A.D.; Kaye, A.; Morgan, J.; Barrilleaux, P.S.; et al. Brexanolone, a GABA(A) Modulator, in the Treatment of Postpartum Depression in Adults: A Comprehensive Review. Front. Psychiatry 2021, 12, 699740. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Yasuda, K.; Lamba, J.K.; Assem, M.; Davila, J.; Strom, S.; Schuetz, E.G. PXR (NR1I2): Splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators. Toxicol. Appl. Pharmacol. 2004, 199, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Walton, N.; Maguire, J. Allopregnanolone-based treatments for postpartum depression: Why/how do they work? Neurobiol. Stress 2019, 11, 100198. [Google Scholar] [CrossRef] [PubMed]
- Zorumski, C.F.; Paul, S.M.; Covey, D.F.; Mennerick, S. Neurosteroids as novel antidepressants and anxiolytics: GABA-A receptors and beyond. Neurobiol. Stress 2019, 11, 100196. [Google Scholar] [CrossRef]
- Tuem, K.B.; Atey, T.M. Neuroactive Steroids: Receptor Interactions and Responses. Front. Neurol. 2017, 8, 442. [Google Scholar] [CrossRef]
- Rudolph, U.; Knoflach, F. Beyond classical benzodiazepines: Novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 2011, 10, 685–697. [Google Scholar] [CrossRef]
- Althaus, A.L.; Ackley, M.A.; Belfort, G.M.; Gee, S.M.; Dai, J.; Nguyen, D.P.; Kazdoba, T.M.; Modgil, A.; Davies, P.A.; Moss, S.J.; et al. Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator. Neuropharmacology 2020, 181, 108333. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, W.; Liu, Z.; Zhang, T.; Wang, Y.; Li, W. Efficacy and safety of zuranolone in the treatment of major depressive disorder: A meta-analysis. Front. Neurosci. 2023, 17, 1332329. [Google Scholar] [CrossRef]
- Gunduz-Bruce, H.; Silber, C.; Kaul, I.; Rothschild, A.J.; Riesenberg, R.; Sankoh, A.J.; Li, H.; Lasser, R.; Zorumski, C.F.; Rubinow, D.R.; et al. Trial of SAGE-217 in Patients with Major Depressive Disorder. N. Engl. J. Med. 2019, 381, 903–911. [Google Scholar] [CrossRef]
- Parikh, S.V.; Aaronson, S.T.; Mathew, S.J.; Alva, G.; DeBattista, C.; Kanes, S.; Lasser, R.; Bullock, A.; Kotecha, M.; Jung, J.; et al. Efficacy and safety of zuranolone co-initiated with an antidepressant in adults with major depressive disorder: Results from the phase 3 CORAL study. Neuropsychopharmacology 2024, 49, 467–475. [Google Scholar] [CrossRef]
- Clayton, A.H.; Lasser, R.; Parikh, S.V.; Iosifescu, D.V.; Jung, J.; Kotecha, M.; Forrestal, F.; Jonas, J.; Kanes, S.J.; Doherty, J. Zuranolone for the Treatment of Adults With Major Depressive Disorder: A Randomized, Placebo-Controlled Phase 3 Trial. Am. J. Psychiatry 2023, 180, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Dichtel, L.E.; Nyer, M.; Dording, C.; Fisher, L.B.; Cusin, C.; Shapero, B.G.; Pedrelli, P.; Kimball, A.S.; Rao, E.M.; Mischoulon, D.; et al. Effects of Open-Label, Adjunctive Ganaxolone on Persistent Depression Despite Adequate Antidepressant Treatment in Postmenopausal Women: A Pilot Study. J. Clin. Psychiatry 2020, 81, 7602. [Google Scholar] [CrossRef]
- Mickey, B.J.; White, A.T.; Arp, A.M.; Leonardi, K.; Torres, M.M.; Larson, A.L.; Odell, D.H.; Whittingham, S.A.; Beck, M.M.; Jessop, J.E.; et al. Propofol for Treatment-Resistant Depression: A Pilot Study. Int. J. Neuropsychopharmacol. 2018, 21, 1079–1089. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Gepirone NDA 021164 Approval Letter; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2023.
- Subbaiah, M.A.M. Triple Reuptake Inhibitors as Potential Therapeutics for Depression and Other Disorders: Design Paradigm and Developmental Challenges. J. Med. Chem. 2018, 61, 2133–2165. [Google Scholar] [CrossRef]
- Denovo Biopharma FDA Grants Fast Track Designation for Biomarker-Guided DB104 (Liafensine) in Patients with Treatment-Resistant Depression (TRD); U.S. Food and Drug Administration: Silver Spring, MD, USA, 2024.
- Zhu, H.; Wang, W.; Sha, C.; Guo, W.; Li, C.; Zhao, F.; Wang, H.; Jiang, W.; Tian, J. Pharmacological Characterization of Toludesvenlafaxine as a Triple Reuptake Inhibitor. Front. Pharmacol. 2021, 12, 741794. [Google Scholar] [CrossRef] [PubMed]
- Mi, W.; Yang, F.; Li, H.; Xu, X.; Li, L.; Tan, Q.; Wang, G.; Zhang, K.; Tian, F.; Luo, J.; et al. Efficacy, Safety, and Tolerability of Ansofaxine (LY03005) Extended-Release Tablet for Major Depressive Disorder: A Randomized, Double-Blind, Placebo-Controlled, Dose-Finding, Phase 2 Clinical Trial. Int. J. Neuropsychopharmacol. 2022, 25, 252–260. [Google Scholar] [CrossRef]
- Mi, W.; Di, X.; Wang, Y.; Li, H.; Xu, X.; Li, L.; Wang, H.; Wang, G.; Zhang, K.; Tian, F.; et al. A phase 3, multicenter, double-blind, randomized, placebo-controlled clinical trial to verify the efficacy and safety of ansofaxine (LY03005) for major depressive disorder. Transl. Psychiatry 2023, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Intra-Cellular Therapies. Intra-Cellular Therapies Announces Positive Topline Results in Second Phase 3 Trial Evaluating Lumateperone as Adjunctive Therapy in Patients with Major Depressive Disorder; Intra-Cellular Therapies, Inc.: Bedminster, NJ, USA, 2024. [Google Scholar]
- Corponi, F.; Fabbri, C.; Bitter, I.; Montgomery, S.; Vieta, E.; Kasper, S.; Pallanti, S.; Serretti, A. Novel antipsychotics specificity profile: A clinically oriented review of lurasidone, brexpiprazole, cariprazine and lumateperone. Eur. Neuropsychopharmacol. 2019, 29, 971–985. [Google Scholar] [CrossRef]
- Satlin, A.; Durgam, S.; Vanover, K.E.; Davis, R.E.; Huo, J.; Mates, S.; Correll, C. M205. Long-term safety of lumateperone (ITI-007): Metabolic effects in a 1-year study. Schizophr. Bull. 2020, 46, S214. [Google Scholar] [CrossRef]
- Kane, J.M.; Durgam, S.; Satlin, A.; Vanover, K.E.; Chen, R.; Davis, R.; Mates, S. Safety and tolerability of lumateperone for the treatment of schizophrenia: A pooled analysis of late-phase placebo- and active-controlled clinical trials. Int. Clin. Psychopharmacol. 2021, 36, 244–250. [Google Scholar] [CrossRef]
- Melas, P.A.; Wirf, M.; André, H.; Jayaram-Lindström, N.; Mathé, A.A.; Steensland, P. The monoamine stabilizer OSU6162 has anxiolytic-like properties and reduces voluntary alcohol intake in a genetic rat model of depression. Sci. Rep. 2021, 11, 11856. [Google Scholar] [CrossRef] [PubMed]
- Palitsky, R.; Kaplan, D.M.; Peacock, C.; Zarrabi, A.J.; Maples-Keller, J.L.; Grant, G.H.; Dunlop, B.W.; Raison, C.L. Importance of Integrating Spiritual, Existential, Religious, and Theological Components in Psychedelic-Assisted Therapies. JAMA Psychiatry 2023, 80, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Carhart-Harris, R.L.; Erritzoe, D.; Williams, T.; Stone, J.M.; Reed, L.J.; Colasanti, A.; Tyacke, R.J.; Leech, R.; Malizia, A.L.; Murphy, K.; et al. Neural correlates of the psychedelic state as determined by fMRI studies with psilocybin. Proc. Natl. Acad. Sci. USA 2012, 109, 2138–2143. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, F.X.; Kometer, M. The neurobiology of psychedelic drugs: Implications for the treatment of mood disorders. Nat. Rev. Neurosci. 2010, 11, 642–651. [Google Scholar] [CrossRef]
- Olson, D.E. Biochemical Mechanisms Underlying Psychedelic-Induced Neuroplasticity. Biochemistry 2022, 61, 127–136. [Google Scholar] [CrossRef]
- Cavarra, M.; Falzone, A.; Ramaekers, J.G.; Kuypers, K.P.C.; Mento, C. Psychedelic-Assisted Psychotherapy-A Systematic Review of Associated Psychological Interventions. Front. Psychol. 2022, 13, 887255. [Google Scholar] [CrossRef]
- Yaden, D.B.; Nayak, S.M.; Gukasyan, N.; Anderson, B.T.; Griffiths, R.R. The Potential of Psychedelics for End of Life and Palliative Care. Curr. Top. Behav. Neurosci. 2022, 56, 169–184. [Google Scholar] [CrossRef]
- Bogenschutz, M.P.; Ross, S.; Bhatt, S.; Baron, T.; Forcehimes, A.A.; Laska, E.; Mennenga, S.E.; O’Donnell, K.; Owens, L.T.; Podrebarac, S.; et al. Percentage of Heavy Drinking Days Following Psilocybin-Assisted Psychotherapy vs Placebo in the Treatment of Adult Patients With Alcohol Use Disorder: A Randomized Clinical Trial. JAMA Psychiatry 2022, 79, 953–962. [Google Scholar] [CrossRef]
- Goodwin, G.M.; Aaronson, S.T.; Alvarez, O.; Arden, P.C.; Baker, A.; Bennett, J.C.; Bird, C.; Blom, R.E.; Brennan, C.; Burke, L.; et al. Single-dose psilocybin for a treatment resistant episode of depression. N. Engl. J. Med. 2022; in press. [Google Scholar]
- Goodwin, G.M.; Croal, M.; Feifel, D.; Kelly, J.R.; Marwood, L.; Mistry, S.; O’Keane, V.; Peck, S.K.; Simmons, H.; Sisa, C.; et al. Psilocybin for treatment resistant depression in patients taking a concomitant SSRI medication. Neuropsychopharmacology 2023, 48, 1492–1499. [Google Scholar] [CrossRef]
- Haikazian, S.; Chen-Li, D.C.J.; Johnson, D.E.; Fancy, F.; Levinta, A.; Husain, M.I.; Mansur, R.B.; McIntyre, R.S.; Rosenblat, J.D. Psilocybin-assisted therapy for depression: A systematic review and meta-analysis. Psychiatry Res. 2023, 329, 115531. [Google Scholar] [CrossRef]
- Carhart-Harris, R.; Giribaldi, B.; Watts, R.; Baker-Jones, M.; Murphy-Beiner, A.; Murphy, R.; Martell, J.; Blemings, A.; Erritzoe, D.; Nutt, D.J. Trial of Psilocybin versus Escitalopram for Depression. N. Engl. J. Med. 2021, 384, 1402–1411. [Google Scholar] [CrossRef]
- Reckweg, J.T.; Uthaug, M.V.; Szabo, A.; Davis, A.K.; Lancelotta, R.; Mason, N.L.; Ramaekers, J.G. The clinical pharmacology and potential therapeutic applications of 5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT). J. Neurochem. 2022, 162, 128–146. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Sanacora, G.; Woolley, J.; Heinzerling, K.; Dunlop, B.W.; Brown, R.T.; Kakar, R.; Hassman, M.; Trivedi, R.P.; Robison, R.; et al. Single-Dose Psilocybin Treatment for Major Depressive Disorder: A Randomized Clinical Trial. JAMA 2023, 330, 843–853. [Google Scholar] [CrossRef]
- Cao, D.; Yu, J.; Wang, H.; Luo, Z.; Liu, X.; He, L.; Qi, J.; Fan, L.; Tang, L.; Chen, Z.; et al. Structure-based discovery of nonhallucinogenic psychedelic analogs. Science 2022, 375, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Osman, S. A mind-changing approach to the therapeutic use of psychedelics. Nat. Struct. Mol. Biol. 2022, 29, 189. [Google Scholar] [CrossRef] [PubMed]
- Erritzoe, D.; Barba, T.; Greenway, K.T.; Murphy, R.; Martell, J.; Giribaldi, B.; Timmermann, C.; Murphy-Beiner, A.; Jones, M.B.; Nutt, D.; et al. Effect of psilocybin versus escitalopram on depression symptom severity in patients with moderate-to-severe major depressive disorder: Observational 6-month follow-up of a phase 2, double-blind, randomised, controlled trial. eClinicalMedicine 2024, 76, 102799. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, G.; Nowakowska, A.; Atli, M.; Dunlop, B.; Feifel, D.; Hellerstein, D.; Marwood, L.; Shabir, Z.; Mistry, S.; Stansfield, S.; et al. Results from a long-term observational follow-up study of a single dose of psilocybin for a treatment-resistant episode of major depressive disorder. J. Clin. Psychiatry, 2025; in press. [Google Scholar]
- Palitsky, R.; Kaplan, D.M.; Perna, J.; Bosshardt, Z.; Maples-Keller, J.L.; Levin-Aspenson, H.F.; Zarrabi, A.J.; Peacock, C.; Mletzko, T.; Rothbaum, B.O.; et al. A framework for assessment of adverse events occurring in psychedelic-assisted therapies. J. Psychopharmacol. 2024, 38, 690–700. [Google Scholar] [CrossRef]
- Calder, A.E.; Hasler, G. Validation of the Swiss Psychedelic Side Effects Inventory: Standardized assessment of adverse effects in studies of psychedelics and MDMA. J. Affect. Disord. 2024, 365, 258–264. [Google Scholar] [CrossRef]
- Horton, D.M.; Morrison, B.; Schmidt, J. Systematized Review of Psychotherapeutic Components of Psilocybin-Assisted Psychotherapy. Am. J. Psychother. 2021, 74, 140–149. [Google Scholar] [CrossRef]
- Barber, G.S.; Dike, C.C. Ethical and Practical Considerations for the Use of Psychedelics in Psychiatry. Psychiatr. Serv. 2023, 74, 838–846. [Google Scholar] [CrossRef]
- US Center for Drug Evaluation and Research, Food and Drug Administration. Psychedelic Drugs: Considerations for Clinical Investigations. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/psychedelic-drugs-considerations-clinical-investigations (accessed on 27 January 2025).
- Leonard, B.E. The concept of depression as a dysfunction of the immune system. Curr. Immunol. Rev. 2010, 6, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Osimo, E.F.; Baxter, L.J.; Lewis, G.; Jones, P.B.; Khandaker, G.M. Prevalence of low-grade inflammation in depression: A systematic review and meta-analysis of CRP levels. Psychol. Med. 2019, 49, 1958–1970. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.R.; Cavanagh, J.; de Boer, P.; Mondelli, V.; Jones, D.N.C.; Drevets, W.C.; Cowen, P.J.; Harrison, N.A.; Pointon, L.; Pariante, C.M.; et al. Treatment-resistant depression and peripheral C-reactive protein. Br. J. Psychiatry 2019, 214, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.A.; Freitas, T.H.; Maes, M.; De Andrade, N.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N. Peripheral cytokine and chemokine alterations in depression: A meta-analysis of 82 studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef]
- Osimo, E.F.; Pillinger, T.; Rodriguez, I.M.; Khandaker, G.M.; Pariante, C.M.; Howes, O.D. Inflammatory markers in depression: A meta-analysis of mean differences and variability in 5,166 patients and 5,083 controls. Brain Behav. Immun. 2020, 87, 901–909. [Google Scholar] [CrossRef]
- Lucido, M.J.; Bekhbat, M.; Goldsmith, D.R.; Treadway, M.T.; Haroon, E.; Felger, J.C.; Miller, A.H. Aiding and Abetting Anhedonia: Impact of Inflammation on the Brain and Pharmacological Implications. Pharmacol. Rev. 2021, 73, 1084–1117. [Google Scholar] [CrossRef]
- Capuron, L.; Gumnick, J.F.; Musselman, D.L.; Lawson, D.H.; Reemsnyder, A.; Nemeroff, C.B.; Miller, A.H. Neurobehavioral effects of interferon-α in cancer patients: Phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 2002, 26, 643–652. [Google Scholar] [CrossRef]
- Capuron, L.; Raison, C.L.; Musselman, D.L.; Lawson, D.H.; Nemeroff, C.B.; Miller, A.H. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am. J. Psychiatry 2003, 160, 1342–1345. [Google Scholar] [CrossRef]
- Brydon, L.; Harrison, N.A.; Walker, C.; Steptoe, A.; Critchley, H.D. Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol. Psychiatry 2008, 63, 1022–1029. [Google Scholar] [CrossRef]
- Eisenberger, N.I.; Berkman, E.T.; Inagaki, T.K.; Rameson, L.T.; Mashal, N.M.; Irwin, M.R. Inflammation-induced anhedonia: Endotoxin reduces ventral striatum responses to reward. Biol. Psychiatry 2010, 68, 748–754. [Google Scholar] [CrossRef]
- Moieni, M.; Tan, K.M.; Inagaki, T.K.; Muscatell, K.A.; Dutcher, J.M.; Jevtic, I.; Breen, E.C.; Irwin, M.R.; Eisenberger, N.I. Sex Differences in the Relationship Between Inflammation and Reward Sensitivity: A Randomized Controlled Trial of Endotoxin. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2019, 4, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Capuron, L.; Fornwalt, F.B.; Knight, B.T.; Harvey, P.D.; Ninan, P.T.; Miller, A.H. Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J. Affect. Disord. 2009, 119, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Kappelmann, N.; Lewis, G.; Dantzer, R.; Jones, P.B.; Khandaker, G.M. Antidepressant activity of anti-cytokine treatment: A systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol. Psychiatry 2018, 23, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Wittenberg, G.M.; Stylianou, A.; Zhang, Y.; Sun, Y.; Gupta, A.; Jagannatha, P.S.; Wang, D.; Hsu, B.; Curran, M.E.; Khan, S.; et al. Effects of immunomodulatory drugs on depressive symptoms: A mega-analysis of randomized, placebo-controlled clinical trials in inflammatory disorders. Mol. Psychiatry 2020, 25, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.; Arnone, D.; Danese, A.; Papadopoulos, A.; Herane Vives, A.; Cleare, A.J. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur. Neuropsychopharmacol. 2015, 25, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Arteaga-Henríquez, G.; Simon, M.S.; Burger, B.; Weidinger, E.; Wijkhuijs, A.; Arolt, V.; Birkenhager, T.K.; Musil, R.; Müller, N.; Drexhage, H.A. Low-Grade Inflammation as a Predictor of Antidepressant and Anti-Inflammatory Therapy Response in MDD Patients: A Systematic Review of the Literature in Combination with an Analysis of Experimental Data Collected in the EU-MOODINFLAME Consortium. Front. Psychiatry 2019, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Uher, R.; Tansey, K.E.; Dew, T.; Maier, W.; Mors, O.; Hauser, J.; Dernovsek, M.Z.; Henigsberg, N.; Souery, D.; Farmer, A.; et al. An inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline. Am. J. Psychiatry 2014, 171, 1278–1286. [Google Scholar] [CrossRef]
- Cattaneo, A.; Ferrari, C.; Uher, R.; Bocchio-Chiavetto, L.; Riva, M.A.; Pariante, C.M. Absolute Measurements of Macrophage Migration Inhibitory Factor and Interleukin-1-β mRNA Levels Accurately Predict Treatment Response in Depressed Patients. Int. J. Neuropsychopharmacol. 2016, 19, pyw045. [Google Scholar] [CrossRef]
- Jha, M.K.; Minhajuddin, A.; Gadad, B.S.; Greer, T.; Grannemann, B.; Soyombo, A.; Mayes, T.L.; Rush, A.J.; Trivedi, M.H. Can C-reactive protein inform antidepressant medication selection in depressed outpatients? Findings from the CO-MED trial. Psychoneuroendocrinology 2017, 78, 105–113. [Google Scholar] [CrossRef]
- Haroon, E.; Daguanno, A.W.; Woolwine, B.J.; Goldsmith, D.R.; Baer, W.M.; Wommack, E.C.; Felger, J.C.; Miller, A.H. Antidepressant treatment resistance is associated with increased inflammatory markers in patients with major depressive disorder. Psychoneuroendocrinology 2018, 95, 43–49. [Google Scholar] [CrossRef]
- Cattaneo, A.; Ferrari, C.; Turner, L.; Mariani, N.; Enache, D.; Hastings, C.; Kose, M.; Lombardo, G.; McLaughlin, A.P.; Nettis, M.A.; et al. Whole-blood expression of inflammasome- and glucocorticoid-related mRNAs correctly separates treatment-resistant depressed patients from drug-free and responsive patients in the BIODEP study. Transl. Psychiatry 2020, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Wang, N.; Yang, C.; Shi, J.Y.; Yu, H.Y.; Hashimoto, K. Serum interleukin-6 is a predictive biomarker for ketamine’s antidepressant effect in treatment-resistant patients with major depression. Biol. Psychiatry 2015, 77, e19–e20. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wardenaar, K.J.; Bosker, F.J.; Li, J.; Schoevers, R.A. Inflammatory markers and treatment outcome in treatment resistant depression: A systematic review. J. Affect. Disord. 2019, 257, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.L.; Congdon, E.; Olmstead, R.; Njau, S.; Breen, E.C.; Narr, K.L.; Espinoza, R.; Irwin, M.R. Inflammation and Improvement of Depression Following Electroconvulsive Therapy in Treatment-Resistant Depression. J. Clin. Psychiatry 2018, 79, 9042. [Google Scholar] [CrossRef]
- Raison, C.L.; Rutherford, R.E.; Woolwine, B.J.; Shuo, C.; Schettler, P.; Drake, D.F.; Haroon, E.; Miller, A.H. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. JAMA Psychiatry 2013, 70, 31–41. [Google Scholar] [CrossRef]
- Mischoulon, D.; Dunlop, B.W.; Kinkead, B.; Schettler, P.J.; Lamon-Fava, S.; Rakofsky, J.J.; Nierenberg, A.A.; Clain, A.J.; Mletzko Crowe, T.; Wong, A.; et al. Omega-3 Fatty Acids for Major Depressive Disorder With High Inflammation: A Randomized Dose-Finding Clinical Trial. J. Clin. Psychiatry 2022, 83, 42432. [Google Scholar] [CrossRef]
- Pariante, C.M.; Lightman, S.L. The HPA axis in major depression: Classical theories and new developments. Trends Neurosci. 2008, 31, 464–468. [Google Scholar] [CrossRef]
- Belvederi Murri, M.; Pariante, C.; Mondelli, V.; Masotti, M.; Atti, A.R.; Mellacqua, Z.; Antonioli, M.; Ghio, L.; Menchetti, M.; Zanetidou, S.; et al. HPA axis and aging in depression: Systematic review and meta-analysis. Psychoneuroendocrinology 2014, 41, 46–62. [Google Scholar] [CrossRef]
- Iwata, M.; Ota, K.T.; Li, X.Y.; Sakaue, F.; Li, N.; Dutheil, S.; Banasr, M.; Duric, V.; Yamanashi, T.; Kaneko, K.; et al. Psychological Stress Activates the Inflammasome via Release of Adenosine Triphosphate and Stimulation of the Purinergic Type 2X7 Receptor. Biol. Psychiatry 2016, 80, 12–22. [Google Scholar] [CrossRef]
- Iwata, M.; Ota, K.T.; Duman, R.S. The inflammasome: Pathways linking psychological stress, depression, and systemic illnesses. Brain Behav. Immun. 2013, 31, 105–114. [Google Scholar] [CrossRef]
- Bekhbat, M.; Chu, K.; Le, N.A.; Woolwine, B.J.; Haroon, E.; Miller, A.H.; Felger, J.C. Glucose and lipid-related biomarkers and the antidepressant response to infliximab in patients with treatment-resistant depression. Psychoneuroendocrinology 2018, 98, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Bekhbat, M.; Treadway, M.T.; Goldsmith, D.R.; Woolwine, B.J.; Haroon, E.; Miller, A.H.; Felger, J.C. Gene signatures in peripheral blood immune cells related to insulin resistance and low tyrosine metabolism define a sub-type of depression with high CRP and anhedonia. Brain Behav. Immun. 2020, 88, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Welle, J.R.; Woolwine, B.J.; Goldsmith, D.R.; Baer, W.; Patel, T.; Felger, J.C.; Miller, A.H. Associations among peripheral and central kynurenine pathway metabolites and inflammation in depression. Neuropsychopharmacology 2020, 45, 998–1007. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M.; Devinsky, O.; Najjar, A.; Zagzag, D. Neurovascular unit dysfunction with blood-brain barrier hyperpermeability contributes to major depressive disorder: A review of clinical and experimental evidence. J. Neuroinflamm. 2013, 10, 906. [Google Scholar] [CrossRef]
- Köhler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of Anti-inflammatory Treatment on Depression, Depressive Symptoms, and Adverse Effects: A Systematic Review and Meta-analysis of Randomized Clinical Trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhu, X.-M.; Zhang, Q.-E.; Cheng, G.; Cai, D.-B.; He, J.; Ng, C.H.; Ungvari, G.S.; Peng, X.-J.; Ning, Y.-P.; et al. Adjunctive minocycline for major mental disorders: A systematic review. J. Psychopharmacol. 2019, 33, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, A.; Kantsjö, J.B.; Ronchi, F. The Gut-Brain Axis: How Microbiota and Host Inflammasome Influence Brain Physiology and Pathology. Front. Immunol. 2020, 11, 604179. [Google Scholar] [CrossRef]
- Qin, Y.; Havulinna, A.S.; Liu, Y.; Jousilahti, P.; Ritchie, S.C.; Tokolyi, A.; Sanders, J.G.; Valsta, L.; Brożyńska, M.; Zhu, Q.; et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat. Genet. 2022, 54, 134–142. [Google Scholar] [CrossRef]
- Brydges, C.R.; Fiehn, O.; Mayberg, H.S.; Schreiber, H.; Dehkordi, S.M.; Bhattacharyya, S.; Cha, J.; Choi, K.S.; Craighead, W.E.; Krishnan, R.R.; et al. Indoxyl sulfate, a gut microbiome-derived uremic toxin, is associated with psychic anxiety and its functional magnetic resonance imaging-based neurologic signature. Sci. Rep. 2021, 11, 21011. [Google Scholar] [CrossRef]
- Le Merrer, J.; Becker, J.A.J.; Befort, K.; Kieffer, B.L. Reward Processing by the Opioid System in the Brain. Physiol. Rev. 2009, 89, 1379–1412. [Google Scholar] [CrossRef]
- Inagaki, T.K.; Ray, L.A.; Irwin, M.R.; Way, B.M.; Eisenberger, N.I. Opioids and social bonding: Naltrexone reduces feelings of social connection. Soc. Cogn. Affect. Neurosci. 2016, 11, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Drolet, G.; Dumont, É.C.; Gosselin, I.; Kinkead, R.; Laforest, S.; Trottier, J.-F. Role of endogenous opioid system in the regulation of the stress response. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2001, 25, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.M.; Emrich, H.M. Current and historical concepts of opiate treatment in psychiatric disorders. Int. Clin. Psychopharmacol. 1988, 3, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Gerner, R.H.; Catlin, D.H.; Gorelick, D.A.; Hui, K.K.; Li, C.H. β-endorphin: Intravenous infusion causes behavioral change in psychiatric inpatients. Arch. Gen. Psychiatry 1980, 37, 642–647. [Google Scholar] [CrossRef]
- Emrich, H.M.; Vogt, P.; Herz, A. Possible antidepressive effects of opioids: Action of buprenorphine. Ann. N. Y. Acad. Sci. 1982, 398, 108–112. [Google Scholar] [CrossRef]
- Bodkin, J.A.; Zornberg, G.L.; Lukas, S.E.; Cole, J.O. Buprenorphine treatment of refractory depression. J. Clin. Psychopharmacol. 1995, 15, 49–57. [Google Scholar] [CrossRef]
- Karp, J.F.; Butters, M.A.; Begley, A.E.; Miller, M.D.; Lenze, E.J.; Blumberger, D.M.; Mulsant, B.H.; Reynolds, C.F., 3rd. Safety, tolerability, and clinical effect of low-dose buprenorphine for treatment-resistant depression in midlife and older adults. J. Clin. Psychiatry 2014, 75, e785–e793. [Google Scholar] [CrossRef]
- Peciña, M.; Karp, J.F.; Mathew, S.; Todtenkopf, M.S.; Ehrich, E.W.; Zubieta, J.-K. Endogenous opioid system dysregulation in depression: Implications for new therapeutic approaches. Mol. Psychiatry 2019, 24, 576–587. [Google Scholar] [CrossRef]
- Ehrich, E.; Turncliff, R.; Du, Y.; Leigh-Pemberton, R.; Fernandez, E.; Jones, R.; Fava, M. Evaluation of opioid modulation in major depressive disorder. Neuropsychopharmacology 2015, 40, 1448–1455. [Google Scholar] [CrossRef]
- Fava, M.; Memisoglu, A.; Thase, M.E.; Bodkin, J.A.; Trivedi, M.H.; de Somer, M.; Du, Y.; Leigh-Pemberton, R.; DiPetrillo, L.; Silverman, B.; et al. Opioid Modulation With Buprenorphine/Samidorphan as Adjunctive Treatment for Inadequate Response to Antidepressants: A Randomized Double-Blind Placebo-Controlled Trial. Am. J. Psychiatry 2016, 173, 499–508. [Google Scholar] [CrossRef]
- Fava, M.; Thase, M.E.; Trivedi, M.H.; Ehrich, E.; Martin, W.F.; Memisoglu, A.; Nangia, N.; Stanford, A.D.; Yu, M.; Pathak, S. Opioid system modulation with buprenorphine/samidorphan combination for major depressive disorder: Two randomized controlled studies. Mol. Psychiatry 2020, 25, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Fava, M.; Mazzone, E.; Freeman, M.; Flynn, M.; Judge, H.; Hoeppner, B.; Hock, R.S.; Shui, A.; Macaluso, M.; Morrison, M.F.; et al. Double-blind, placebo-controlled, proof-of-concept trial of a kappa-selective opioid receptor antagonist augmentation in treatment-resistant depression. Ann. Clin. Psychiatry 2020, 32, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Savitz, A.; Wajs, E.; Zhang, Y.; Xu, H.; Etropolski, M.; Thase, M.E.; Drevets, W.C. Efficacy and Safety of Seltorexant as Adjunctive Therapy in Major Depressive Disorder: A Phase 2b, Randomized, Placebo-Controlled, Adaptive Dose-Finding Study. Int. J. Neuropsychopharmacol. 2021, 24, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Recourt, K.; de Boer, P.; Zuiker, R.; Luthringer, R.; Kent, J.; van der Ark, P.; Van Hove, I.; van Gerven, J.; Jacobs, G.; van Nueten, L.; et al. The selective orexin-2 antagonist seltorexant (JNJ-42847922/MIN-202) shows antidepressant and sleep-promoting effects in patients with major depressive disorder. Transl. Psychiatry 2019, 9, 216. [Google Scholar] [CrossRef]
- Nollet, M.; Gaillard, P.; Tanti, A.; Girault, V.; Belzung, C.; Leman, S. Neurogenesis-independent antidepressant-like effects on behavior and stress axis response of a dual orexin receptor antagonist in a rodent model of depression. Neuropsychopharmacology 2012, 37, 2210–2221. [Google Scholar] [CrossRef]
- Cuomo, A.; Beccarini Crescenzi, B.; Bolognesi, S.; Goracci, A.; Koukouna, D.; Rossi, R.; Fagiolini, A. S-Adenosylmethionine (SAMe) in major depressive disorder (MDD): A clinician-oriented systematic review. Ann. Gen. Psychiatry 2020, 19, 50. [Google Scholar] [CrossRef]
- Mayberg, H.S.; Dunlop, B.W. Balancing the beautiful and the good in pursuit of biomarkers for depression. World Psychiatry 2023, 22, 265–267. [Google Scholar] [CrossRef]
- Mancuso, E.; Sampogna, G.; Boiano, A.; Della Rocca, B.; Di Vincenzo, M.; Lapadula, M.V.; Martinelli, F.; Lucci, F.; Luciano, M. Biological correlates of treatment resistant depression: A review of peripheral biomarkers. Front. Psychiatry 2023, 14, 1291176. [Google Scholar] [CrossRef]
- Gkesoglou, T.; Bargiota, S.I.; Iordanidou, E.; Vasiliadis, M.; Bozikas, V.P.; Agorastos, A. Prognostic Significance of Blood-Based Baseline Biomarkers in Treatment-Resistant Depression: A Literature Review of Available Studies on Treatment Response. Brain Sci. 2022, 12, 940. [Google Scholar] [CrossRef]
- Meshkat, S.; Alnefeesi, Y.; Jawad, M.Y.; Di Vincenzo, J.D.; Rodrigues, N.B.; Ceban, F.; Lui, L.M.; McIntyre, R.S.; Rosenblat, J.D. Brain-Derived Neurotrophic Factor (BDNF) as a biomarker of treatment response in patients with Treatment Resistant Depression (TRD): A systematic review & meta-analysis. Psychiatry Res. 2022, 317, 114857. [Google Scholar] [CrossRef]
- Brown, L.C.; Stanton, J.D.; Bharthi, K.; Maruf, A.A.; Müller, D.J.; Bousman, C.A. Pharmacogenomic Testing and Depressive Symptom Remission: A Systematic Review and Meta-Analysis of Prospective, Controlled Clinical Trials. Clin. Pharmacol. Ther. 2022, 112, 1303–1317. [Google Scholar] [CrossRef]
- Oslin, D.W.; Lynch, K.G.; Shih, M.-C.; Ingram, E.P.; Wray, L.O.; Chapman, S.R.; Kranzler, H.R.; Gelernter, J.; Pyne, J.M.; Stone, A.; et al. Effect of Pharmacogenomic Testing for Drug-Gene Interactions on Medication Selection and Remission of Symptoms in Major Depressive Disorder: The PRIME Care Randomized Clinical Trial. JAMA 2022, 328, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, Y.; Jiang, J.; Lucas, M.V.; Fonzo, G.A.; Rolle, C.E.; Cooper, C.; Chin-Fatt, C.; Krepel, N.; Cornelssen, C.A.; et al. An electroencephalographic signature predicts antidepressant response in major depression. Nat. Biotechnol. 2020, 38, 439–447. [Google Scholar] [CrossRef]
- Kelley, M.E.; Choi, K.S.; Rajendra, J.K.; Craighead, W.E.; Rakofsky, J.J.; Dunlop, B.W.; Mayberg, H.S. Establishing Evidence for Clinical Utility of a Neuroimaging Biomarker in Major Depressive Disorder: Prospective Testing and Implementation Challenges. Biol. Psychiatry 2021, 90, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Fyer, A.J.; Schneier, F.R.; Simpson, H.B.; Choo, T.H.; Tacopina, S.; Kimeldorf, M.B.; Steinglass, J.E.; Wall, M.; Walsh, B.T. Heterogeneity in Fear Processing across and within Anxiety, Eating, and Compulsive Disorders. J. Affect. Disord. 2020, 275, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Abramovitch, A.; Short, T.; Schweiger, A. The C Factor: Cognitive dysfunction as a transdiagnostic dimension in psychopathology. Clin. Psychol. Rev. 2021, 86, 102007. [Google Scholar] [CrossRef] [PubMed]
- Guineau, M.G.; Ikani, N.; Rinck, M.; Collard, R.M.; van Eijndhoven, P.; Tendolkar, I.; Schene, A.H.; Becker, E.S.; Vrijsen, J.N. Anhedonia as a transdiagnostic symptom across psychological disorders: A network approach. Psychol. Med. 2023, 53, 3908–3919. [Google Scholar] [CrossRef]
- Mandakh, B.; Zhihao, L.; Dunlop, B.W.; Treadway, M.T.; Mehta, N.D.; Revill, K.P.; Lucido, M.J.; Changdo, H.; Andrea, A.; Wommack, E.C.; et al. Sustained effects of repeated levodopa (L-DOPA) administration on reward circuitry, effort-based motivation, and anhedonia in depressed patients with higher inflammation. Brain Behav. Immun. 2024, 125, 240–248. [Google Scholar] [CrossRef]
- Huhn, A.S.; Meyer, R.E.; Harris, J.D.; Ayaz, H.; Deneke, E.; Stankoski, D.M.; Bunce, S.C. Evidence of anhedonia and differential reward processing in prefrontal cortex among post-withdrawal patients with prescription opiate dependence. Brain Res. Bull. 2016, 123, 102–109. [Google Scholar] [CrossRef]
- Felger, J.C.; Li, Z.; Haroon, E.; Woolwine, B.J.; Jung, M.Y.; Hu, X.; Miller, A.H. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol. Psychiatry 2016, 21, 1358–1365. [Google Scholar] [CrossRef]
- Yin, L.; Xu, X.; Chen, G.; Mehta, N.D.; Haroon, E.; Miller, A.H.; Luo, Y.; Li, Z.; Felger, J.C. Inflammation and decreased functional connectivity in a widely-distributed network in depression: Centralized effects in the ventral medial prefrontal cortex. Brain Behav. Immun. 2019, 80, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Zhu, J.; Zuckerman, H.; Rosenblat, J.D.; Brietzke, E.; Pan, Z.; Subramanieapillai, M.; Park, C.; Lee, Y.; McIntyre, R.S. Pharmacological interventions targeting anhedonia in patients with major depressive disorder: A systematic review. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 92, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Krystal, A.D.; Pizzagalli, D.A.; Smoski, M.; Mathew, S.J.; Nurnberger, J.; Lisanby, S.H.; Iosifescu, D.; Murrough, J.W.; Yang, H.; Weiner, R.D.; et al. A randomized proof-of-mechanism trial applying the ‘fast-fail’ approach to evaluating κ-opioid antagonism as a treatment for anhedonia. Nat. Med. 2020, 26, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.K.; Juarez, B.; Ku, S.M.; Zhang, H.; Calizo, R.C.; Walsh, J.J.; Chaudhury, D.; Zhang, S.; Hawkins, A.; Dietz, D.M.; et al. KCNQ channel openers reverse depressive symptoms via an active resilience mechanism. Nat. Commun. 2016, 7, 11671. [Google Scholar] [CrossRef]
- Costi, S.; Morris, L.S.; Kirkwood, K.A.; Hoch, M.; Corniquel, M.; Vo-Le, B.; Iqbal, T.; Chadha, N.; Pizzagalli, D.A.; Whitton, A.; et al. Impact of the KCNQ2/3 Channel Opener Ezogabine on Reward Circuit Activity and Clinical Symptoms in Depression: Results From a Randomized Controlled Trial. Am. J. Psychiatry 2021, 178, 437–446. [Google Scholar] [CrossRef]
- Bekhbat, M.; Li, Z.; Mehta, N.D.; Treadway, M.T.; Lucido, M.J.; Woolwine, B.J.; Haroon, E.; Miller, A.H.; Felger, J.C. Functional connectivity in reward circuitry and symptoms of anhedonia as therapeutic targets in depression with high inflammation: Evidence from a dopamine challenge study. Mol. Psychiatry 2022, 27, 4113–4121. [Google Scholar] [CrossRef]
- Ventorp, F.; Lindahl, J.; van Westen, D.; Jensen, J.; Björkstrand, J.; Lindqvist, D. Preliminary Evidence of Efficacy and Target Engagement of Pramipexole in Anhedonic Depression. Psychiatr. Res. Clin. Pract. 2022, 4, 42–47. [Google Scholar] [CrossRef]
- Grunebaum, M.F.; Galfalvy, H.C.; Choo, T.H.; Keilp, J.G.; Moitra, V.K.; Parris, M.S.; Marver, J.E.; Burke, A.K.; Milak, M.S.; Sublette, M.E.; et al. Ketamine for Rapid Reduction of Suicidal Thoughts in Major Depression: A Midazolam-Controlled Randomized Clinical Trial. Am. J. Psychiatry 2018, 175, 327–335. [Google Scholar] [CrossRef]
- Fu, D.J.; Ionescu, D.F.; Li, X.; Lane, R.; Lim, P.; Sanacora, G.; Hough, D.; Manji, H.; Drevets, W.C.; Canuso, C.M. Esketamine Nasal Spray for Rapid Reduction of Major Depressive Disorder Symptoms in Patients Who Have Active Suicidal Ideation With Intent: Double-Blind, Randomized Study (ASPIRE I). J. Clin. Psychiatry 2020, 81, 6605. [Google Scholar] [CrossRef]
- Ionescu, D.F.; Fu, D.J.; Qiu, X.; Lane, R.; Lim, P.; Kasper, S.; Hough, D.; Drevets, W.C.; Manji, H.; Canuso, C.M. Esketamine Nasal Spray for Rapid Reduction of Depressive Symptoms in Patients with Major Depressive Disorder Who Have Active Suicide Ideation with Intent: Results of a Phase 3, Double-Blind, Randomized Study (ASPIRE II). Int. J. Neuropsychopharmacol. 2020, 24, 22–31. [Google Scholar] [CrossRef]
- Hochschild, A.; Keilp, J.G.; Madden, S.P.; Burke, A.K.; Mann, J.J.; Grunebaum, M.F. Ketamine vs midazolam: Mood improvement reduces suicidal ideation in depression. J. Affect. Disord. 2022, 300, 10–16. [Google Scholar] [CrossRef]
- Canuso, C.M.; Singh, J.B.; Fedgchin, M.; Alphs, L.; Lane, R.; Lim, P.; Pinter, C.; Hough, D.; Sanacora, G.; Manji, H.; et al. Efficacy and Safety of Intranasal Esketamine for the Rapid Reduction of Symptoms of Depression and Suicidality in Patients at Imminent Risk for Suicide: Results of a Double-Blind, Randomized, Placebo-Controlled Study. Am. J. Psychiatry 2018, 175, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Perlis, R.H. Hard Outcomes: Clinical Trials to Reduce Suicide. Am. J. Psychiatry 2011, 168, 1009–1011. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Y.; Edmiston, E.K.; Womer, F.Y.; Zhang, R.; Zhao, P.; Jiang, X.; Wu, F.; Kong, L.; Zhou, Y.; et al. Structural and Functional Abnormities of Amygdala and Prefrontal Cortex in Major Depressive Disorder With Suicide Attempts. Front. Psychiatry 2020, 10, 923. [Google Scholar] [CrossRef]
- Du, L.; Zeng, J.; Liu, H.; Tang, D.; Meng, H.; Li, Y.; Fu, Y. Fronto-limbic disconnection in depressed patients with suicidal ideation: A resting-state functional connectivity study. J. Affect. Disord. 2017, 215, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, S.-W.; Myung, W.; Han, C.E.; Fava, M.; Mischoulon, D.; Papakostas, G.I.; Seo, S.W.; Cho, H.; Seong, J.-K.; et al. Reduced orbitofrontal-thalamic functional connectivity related to suicidal ideation in patients with major depressive disorder. Sci. Rep. 2017, 7, 15772. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.M.; Nieto, S.; Bosler, L.; Wong, M.; Bishop, I.; Mooney, L.; Cahill, C.M. Mechanisms Underlying the Anti-Suicidal Treatment Potential of Buprenorphine. Adv. Drug Alcohol Res. 2021, 1, 10009. [Google Scholar] [CrossRef]
- Yovell, Y.; Bar, G.; Mashiah, M.; Baruch, Y.; Briskman, I.; Asherov, J.; Lotan, A.; Rigbi, A.; Panksepp, J. Ultra-Low-Dose Buprenorphine as a Time-Limited Treatment for Severe Suicidal Ideation: A Randomized Controlled Trial. Am. J. Psychiatry 2016, 173, 491–498. [Google Scholar] [CrossRef]
- Masson, G. Novartis Cuts Depression Study, Nearly Erasing $210M Cadent Acquisition from Pipeline. Available online: https://www.fiercebiotech.com/biotech/novartis-discontinues-depression-study-after-benefit-risk-analysis (accessed on 22 December 2024).
- Rush, A.J. Challenges of research on treatment-resistant depression: A clinician’s perspective. World Psychiatry 2023, 22, 415–417. [Google Scholar] [CrossRef]
- Rush, A.J.; Sackeim, H.A.; Conway, C.R.; Bunker, M.T.; Hollon, S.D.; Demyttenaere, K.; Young, A.H.; Aaronson, S.T.; Dibué, M.; Thase, M.E.; et al. Clinical research challenges posed by difficult-to-treat depression. Psychol. Med. 2022, 52, 419–432. [Google Scholar] [CrossRef]
- Amasi-Hartoonian, N.; Pariante, C.M.; Cattaneo, A.; Sforzini, L. Understanding treatment-resistant depression using “omics” techniques: A systematic review. J. Affect. Disord. 2022, 318, 423–455. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, C.; Corponi, F.; Souery, D.; Kasper, S.; Montgomery, S.; Zohar, J.; Rujescu, D.; Mendlewicz, J.; Serretti, A. The Genetics of Treatment-Resistant Depression: A Critical Review and Future Perspectives. Int. J. Neuropsychopharmacol. 2019, 22, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Polo, A.J.; Makol, B.A.; Castro, A.S.; Colón-Quintana, N.; Wagstaff, A.E.; Guo, S. Diversity in randomized clinical trials of depression: A 36-year review. Clin. Psychol. Rev. 2019, 67, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Monahan, K.; Weyandt, L.; Shepard, E. Diversity inclusion in clinical trials investigating esketamine for depression: A systematic review. Exp. Clin. Psychopharmacol. 2023, 31, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Nierenberg, A.A.; Stewart, J.W.; Warden, D.; Niederehe, G.; Thase, M.E.; Lavori, P.W.; Lebowitz, B.D. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR* D report. Am. J. Psychiatry 2006, 163, 1905–1917. [Google Scholar] [CrossRef]
- Keller, M.B.; Coryell, W.H.; Endicott, J.; Maser, J.D.; Schettler, P.J. Clinical Guide to Depression and Bipolar Disorder: Findings from the Collaborative Depression Study; American Psychiatric Pub: Washington, DC, USA, 2013. [Google Scholar]
- Chen, M.H.; Cheng, C.M.; Gueorguieva, R.; Lin, W.C.; Li, C.T.; Hong, C.J.; Tu, P.C.; Bai, Y.M.; Tsai, S.J.; Krystal, J.H.; et al. Maintenance of antidepressant and antisuicidal effects by D-cycloserine among patients with treatment-resistant depression who responded to low-dose ketamine infusion: A double-blind randomized placebo-control study. Neuropsychopharmacology 2019, 44, 2112–2118. [Google Scholar] [CrossRef]
- Patten, D.K.; Schultz, B.G.; Berlau, D.J. The safety and efficacy of low-dose naltrexone in the management of chronic pain and inflammation in multiple sclerosis, fibromyalgia, Crohn’s disease, and other chronic pain disorders. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2018, 38, 382–389. [Google Scholar] [CrossRef]
- Dunlop, B.W.; Binder, E.B.; Iosifescu, D.; Mathew, S.J.; Neylan, T.C.; Pape, J.C.; Carrillo-Roa, T.; Green, C.; Kinkead, B.; Grigoriadis, D.; et al. Corticotropin-Releasing Factor Receptor 1 Antagonism Is Ineffective for Women With Posttraumatic Stress Disorder. Biol. Psychiatry 2017, 82, 866–874. [Google Scholar] [CrossRef]
- Borges, S.; Chen, Y.-F.; Laughren, T.P.; Temple, R.; Patel, H.D.; David, P.A.; Mathis, M.; Unger, E.; Yang, P.; Khin, N.A. Review of maintenance trials for major depressive disorder: A 25-year perspective from the US Food and Drug Administration. J. Clin. Psychiatry 2014, 75, 18305. [Google Scholar] [CrossRef]
- Keller, M.B.; Trivedi, M.H.; Thase, M.E.; Shelton, R.C.; Kornstein, S.G.; Nemeroff, C.B.; Friedman, E.S.; Gelenberg, A.J.; Kocsis, J.H.; Dunner, D.L. The Prevention of Recurrent Episodes of Depression with Venlafaxine for Two Years (PREVENT) study: Outcomes from the acute and continuation phases. Biol. Psychiatry 2007, 62, 1371–1379. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Compound | Proposed Mechanism | Clinical Trial Identifier(s) | Phase | Notes |
|---|---|---|---|---|
| Arketamine (PCN-101) | AMPAR agonist, sigma-1 agonist | NCT05414422 | 2 | Adjunctive therapy;Failed to separate from placebo |
| AXS-05 (bupropion-dextromethorphan) | Uncompetitive NMDAR antagonist, sigma-1 agonist | NCT04634669, NCT04971291, NCT04039022, NCT02741791 | 3 | Monotherapy; Approved as monotherapy for MDD in August 2022 |
| Esketamine (CLE-100) | NMDAR antagonist | NCT04103892 | 2 | Oral formulation; adjunctive therapy |
| Esmethadone (REL-1017) | NMDAR antagonist | NCT04855747, NCT03051256 | 3 | RELIANCE studies; adjunctive therapy; unlikely to reach primary endpoint based on interim analysis Also evaluated as monotherapy or adjunctive for MDD (NCT05081167, NCT04855760) |
| (2R,6R)-hydroxynorketamine | Weak NMDAR antagonist, opioid receptor positive allosteric modulator | NCT06511908 | 2 | Augmentation |
| MIJ821 | NMDAR GluN2B negative allosteric modulator | NCT03756129, NCT05454410, NCT04722666 | 2 | Adjunctive therapy |
| Nitrous oxide | NMDAR antagonist, iNOS inducer, cerebral vasodilator | NCT03283670, NCT05007028, NCT04957368 | 2 | Monotherapy/adjunctive therapy; Also evaluated as monotherapy (NCT02139540, NCT03869736, NCT05357040) |
| NV-5138 | Sestrin2 modulator, mTORC1 pathway activator | NCT05066672 | 2 | Monotherapy |
| TS-161 | mGluR2/3 antagonist | NCT04821271 | 2 | Monotherapy |
| Drug/Compound | Proposed Mechanism | Clinical Trial Identifier(s) | Phase | Notes |
|---|---|---|---|---|
| GABAergic Modulators | ||||
| Propofol | GABAA receptor positive allosteric modulator | NCT03684447 | 2/3 | Not clearly defined as monotherapy vs. adjunctive therapy |
| Monoaminergic Modulators | ||||
| Ayahuasca | 5-HT2 agonist, MAOI | NCT02914769 | 1/2 | Response and remission rates significant at 7 days |
| DMT | 5-HT2 agonist | NCT06524830 | 2 | Monotherapy; administered as buccal film |
| 5-MeO-DMT | 5-HT2 agonist | NCT04698603, NCT05800860, NCT05660642, NCT05870540 | 2 | Monotherapy; primarily open label studies |
| Liafensine | DAT, NET, SERT antagonist | NCT05113771 | 2 | Monotherapy; FDA fast-track designation granted based on success of Phase 2 trial; currently in trial using a proprietary pharmacogenomic biomarker, DGM4 |
| Lumateperone | 5-HT2 antagonist, D2 antagonist | NCT05850689, NCT05061706, NCT04985942 | 3 | Adjunctive therapy |
| OSU-6162 | D2 and 5-HT2 partial agonist | NCT05641623 | 2 | Adjunctive therapy |
| Psilocybin | 5-HT2 agonist | NCT05220410, NCT05029466, NCT04519957, NCT06512220, NCT04670081, NCT05624268, NCT05711940, NCT06518720, NCT06132178, NCT06341426, NCT05383313, NCT05710237 | 2/3 | Monotherapy or in conjunction with psychotherapy or neuromodulation; also evaluated as for monotherapy for non-TRD MDD (NCT03429075, NCT03866174) |
| Anti-inflammatory and Immune Modulators | ||||
| Eicosapentaenoic acid (EPA) | Anti-inflammatory omega-3 fatty acid | NCT05774665 | 2 | Adjunctive therapy; 4 g of EPA-enriched omega-3 fatty acid supplement |
| Ixekizumab | IL-17A neutralizing antibody | NCT04979910 | 2 | Adjunctive therapy; CRP > 2 mg/L as part of inclusion criteria |
| Tofacitinib | JAK1/3 inhibitor | NCT04141904 | 2 | Adjunctive therapy; CRP > 1 mg/L as part of inclusion criteria. Trial terminated due to COVID with no new studies registered |
| Fecal microbiota transplant | Microbiome seeding | NCT04805879 | 3 | Adjunctive therapy |
| Additional Mechanisms | ||||
| Seltorexant | Orexin type 2 receptor antagonist | NCT04951609, NCT04532749, NCT04513912, NCT04533529, NCT06559306 | 3 | Adjunctive therapy |
| Drug/Compound | Proposed Mechanism | Clinical Trial Identifier(s) | Phase | Notes |
|---|---|---|---|---|
| Transdiagnostic Approach—Anhedonia | ||||
| Aticaprant | Κ-opioid receptor antagonist | NCT05455684, NCT05455684, NCT05550532, NCT06514742 | 3 | MDD + moderate to severe anhedonia with inadequate response to SSRI or SNRI |
| ALTO-203 | H3 inverse agonist | NCT06391593 | 2 | MDD + anhedonia; monotherapy |
| Ansofaxine | DAT, NET, SERT antagonist | NCT06270433 | NR | MDD + anhedonia; using desvenlafaxine as an active comparator |
| Baricitinib | JAK1/2 inhibitor | NCT05849038 | 2 | HIV with MDD + anhedonia |
| Buspirone | 5-HT1A agonist | NCT05357547 | 2 | Reward processing in healthy controls; trial completed in 2023 with no results posted to date |
| Estradiol | Estrogen receptor agonist | NCT05282277, NCT06610305 | 4 | First study investigating anhedonia and/or psychosis in perimenopausal women; second study investigating effect of estradiol vs. progesterone vs. placebo on MDD with perimenstrual symptom worsening + anhedonia |
| Insulin | Glucose regulation | NCT03915613 | 1/2 | Proof of concept study establishing a role for insulin/insulin resistance in anhedonia and behavioral deficits in MDD |
| NBI-1065846 (TAK-041) | GPR139 agonist | NCT05165394 | 2 | MDD + anhedonia; no separation from placebo on measures of anhedonia or overall depression. Also evaluated in schizophrenia + anhedonia (negative results; NCT03319953); no further trials listed |
| Pitolisant | H3 agonist, sigma1 agonist | NCT05849675 | NR | Healthy volunteers; assessing effect on functional connectivity. |
| Pramipexole | D3 receptor agonist | NCT05355337, NCT05825235 | 4 | MDD + anhedonia |
| Psilocybin | 5-HT2 agonist | NCT06230757 | 2 | TRD + anhedonia |
| Sinemet (carbidopa/levodopa) | Dopamine agonist | NCT06075771, NCT03243552, NCT05909267 | 2/4 | MDD + anhedonia; CRP > 2 mg/L as inclusion criteria; second study investigating ASD with social anhedonia and the impact of levodopa vs. placebo and social skills training third study investigating impact in MDD + anhedonia, no CRP cutoff listed |
| XEN1101 | KCNQ2/3 agonist | NCT04827901 | 2 | MDD + anhedonia completed in 2024 with no results posted to date |
| Transdiagnostic Approach—Suicidal Ideation | ||||
| Buprenorphine | Partial mu agonist | NCT04116528, NCT03646058 | 3 | First study investigating ketamine followed by buprenorphine for sustained anti-suicidal effect; second study investigating analgesic buprenorphine dosing as augmentation to antidepressant treatment in acute suicidality |
| Estradiol | Estrogen receptor agonist | NCT04112368, NCT03498313, NCT06191289 | 2/4 | Treatment of peri-menstrual suicidal ideation |
| D-cycloserine | NMDAR glycine site partial agonist | NCT06121284 | 2 | Augmentation of intermittent theta burst rTMS |
| Ketamine | NMDAR antagonist | NCT04669665, NCT06366334, NCT05468840 | 2/3/4 | MDD + current suicidal ideation; active suicidal ideation in the emergency room setting; suicidality in opiate-use disorder; Ten additional active studies |
| Nitrous oxide | NMDAR antagonist, iNOS inducer, cerebral vasodilator | NCT05710887, NCT06430489, NCT06636357 | 2 | Non-psychotic MDD + acute suicidality in ED setting |
| NRX-101 (D-cycloserine + lurasidone) | NMDAR glycine site partial agonist + antipsychotic | NCT03395392, NCT03396068 | 2 | Bipolar depression + sub-acute suicidal ideation/behavior |
| Psilocybin | 5-HT2 agonist | NCT05220410 | 2 | TRD + chronic suicidal ideation; open label |
| Tetrahydrocannabinol | CB receptor agonist | NCT06381180 | 1/2 | PTSD with chronic risk for suicidality |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucido, M.J.; Dunlop, B.W. Emerging Medications for Treatment-Resistant Depression: A Review with Perspective on Mechanisms and Challenges. Brain Sci. 2025, 15, 161. https://doi.org/10.3390/brainsci15020161
Lucido MJ, Dunlop BW. Emerging Medications for Treatment-Resistant Depression: A Review with Perspective on Mechanisms and Challenges. Brain Sciences. 2025; 15(2):161. https://doi.org/10.3390/brainsci15020161
Chicago/Turabian StyleLucido, Michael J., and Boadie W. Dunlop. 2025. "Emerging Medications for Treatment-Resistant Depression: A Review with Perspective on Mechanisms and Challenges" Brain Sciences 15, no. 2: 161. https://doi.org/10.3390/brainsci15020161
APA StyleLucido, M. J., & Dunlop, B. W. (2025). Emerging Medications for Treatment-Resistant Depression: A Review with Perspective on Mechanisms and Challenges. Brain Sciences, 15(2), 161. https://doi.org/10.3390/brainsci15020161