
Label-Free Quantitative Proteomic Analysis of Nitrogen Starvation in Arabidopsis Root Reveals New Aspects of H2S Signaling by Protein Persulfidation




Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Ma Terial and Growth Conditions
2.2. Immunoblot Analysis
2.3. Amino Acid Determination by UPLC-MS/MS
2.4. Protein Persulfidation Enrichment by Tag-Switch Method
2.5. LC-MS/MS
2.6. Raw Data Processing and Analysis
3. Results
3.1. Identification and Quantitative Comparison of the Persulfidation Patterns between Nitrogen-Sufficient and Nitrogen-Deprivation Conditions
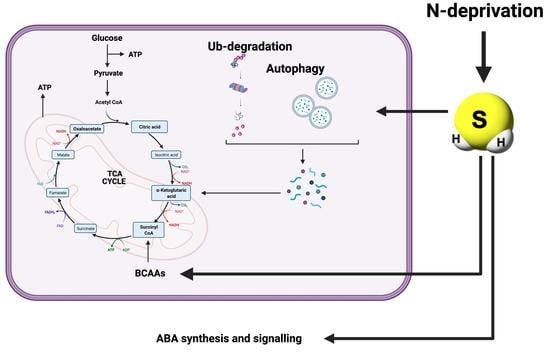
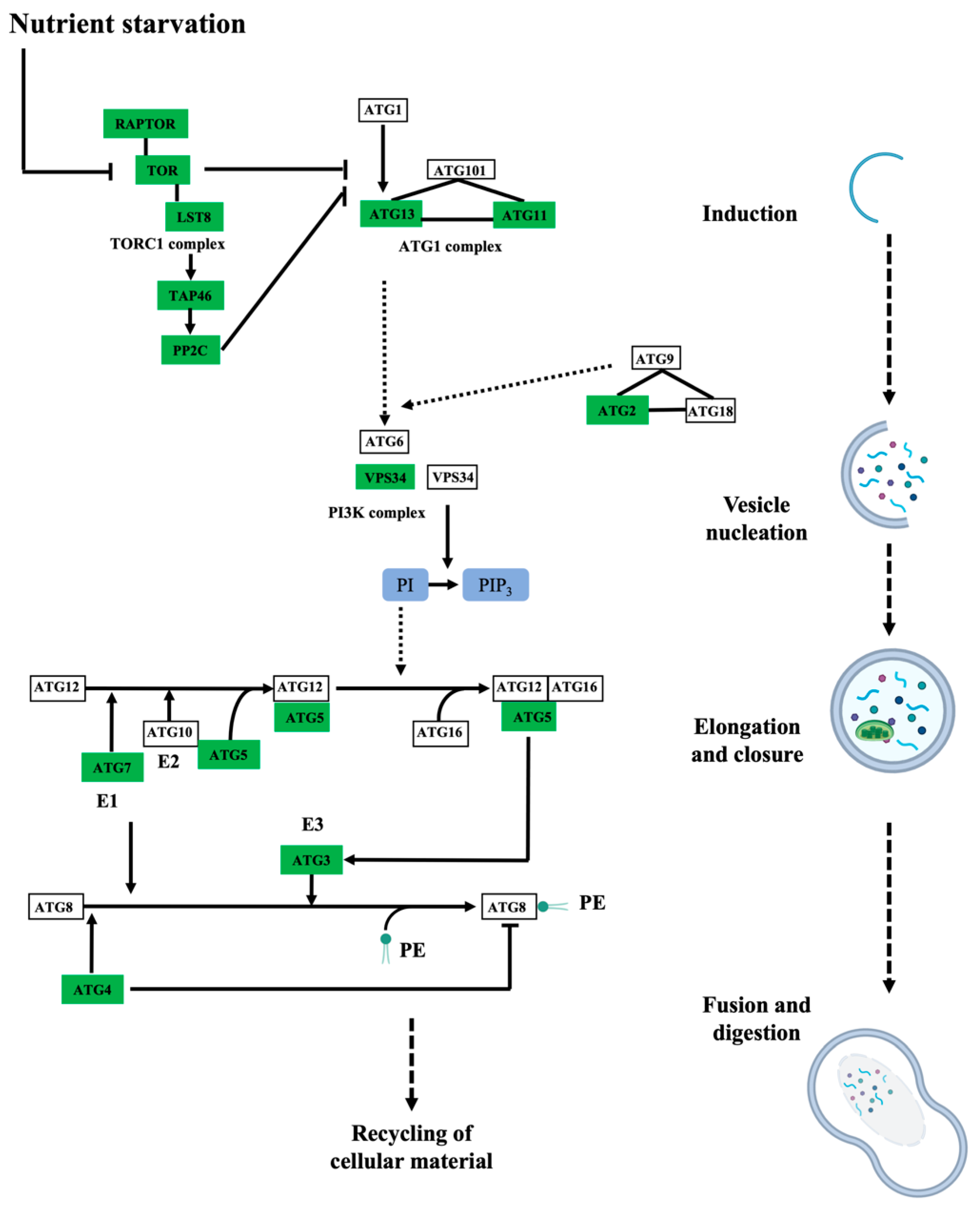
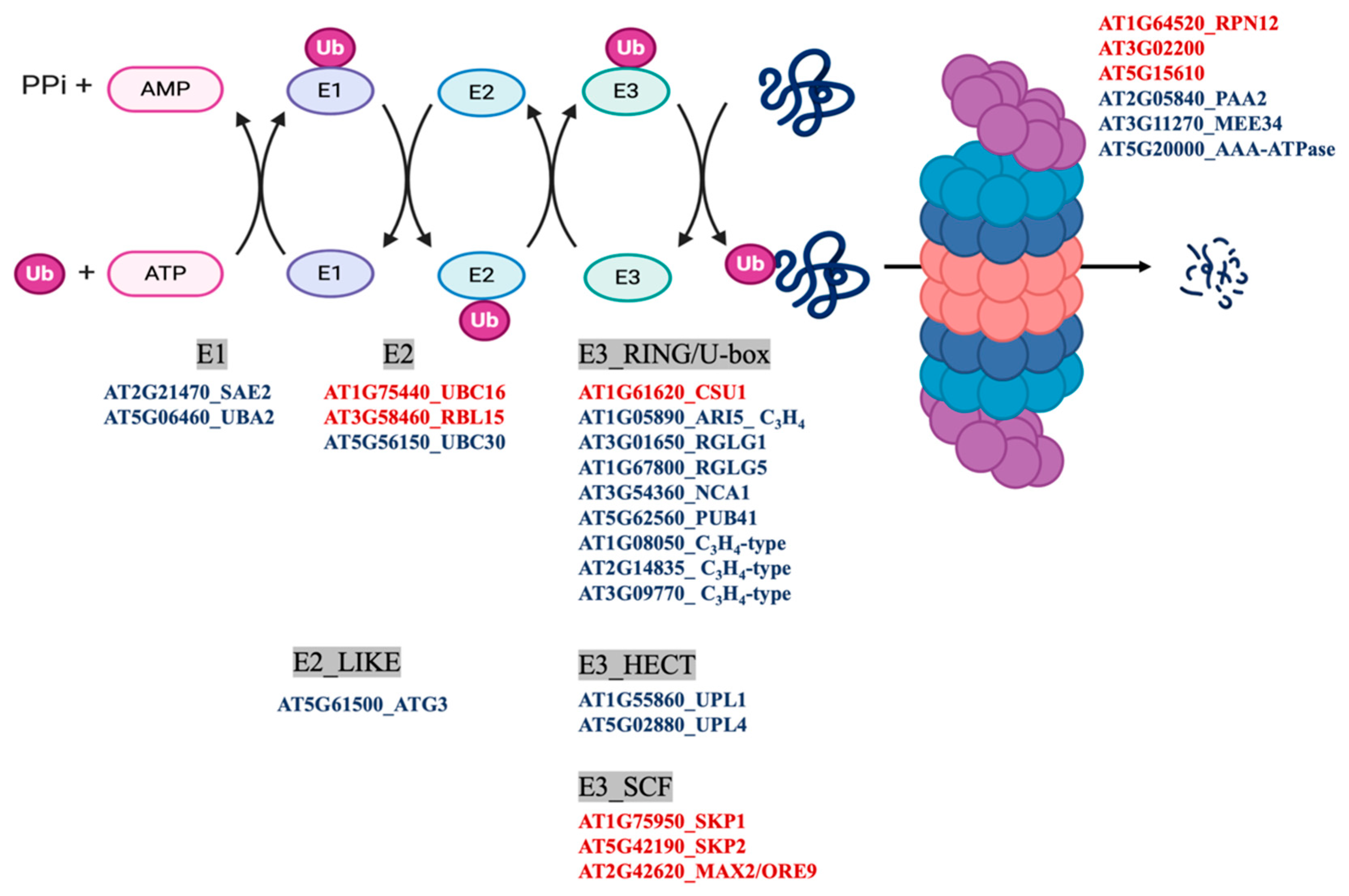
3.2. Protein Persulfidation Have Impact on the Regulation of Protein Degradation and Autophagy Process
3.3. Plant Hormone Signaling Components Are Targets of Persulfidation
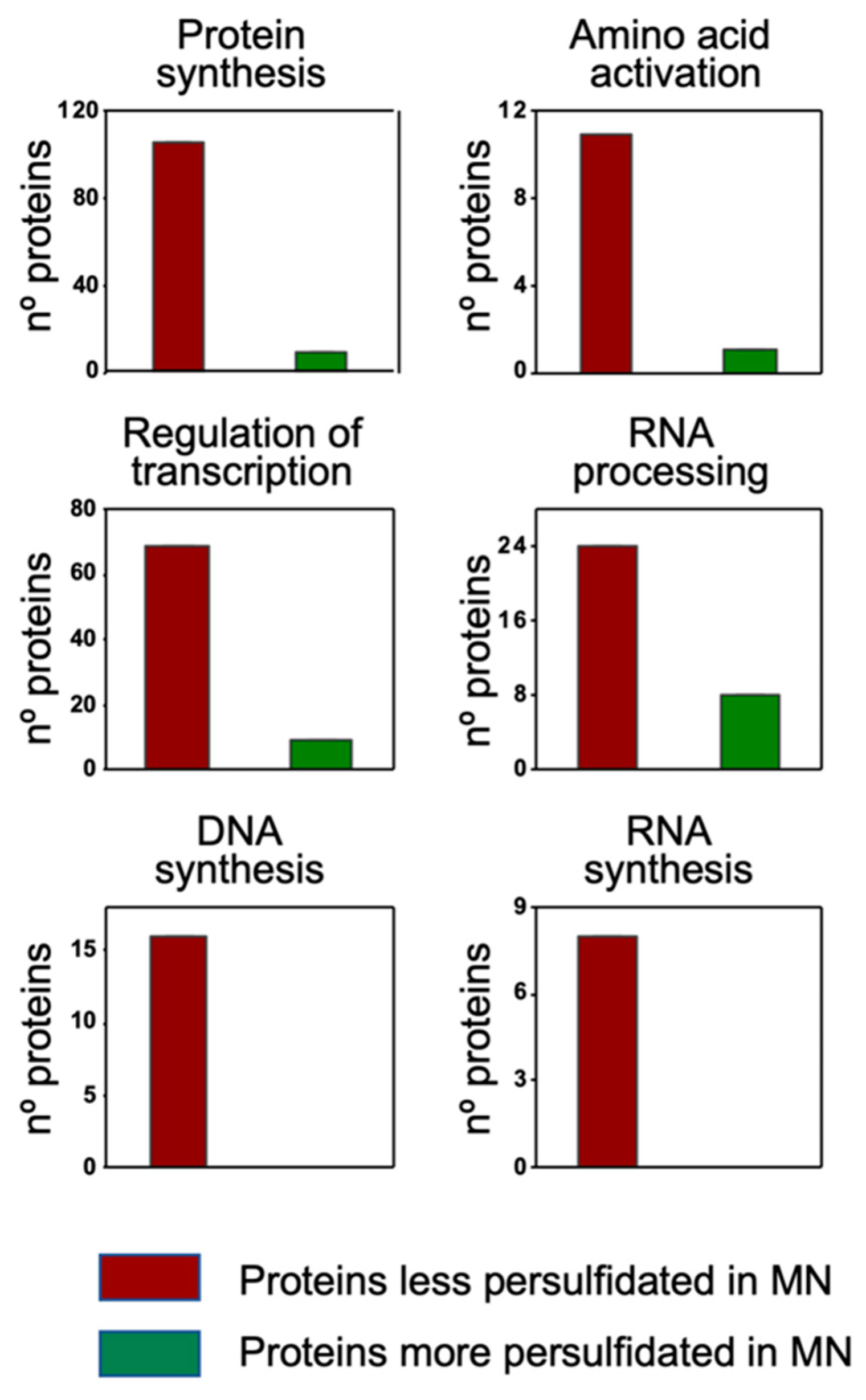
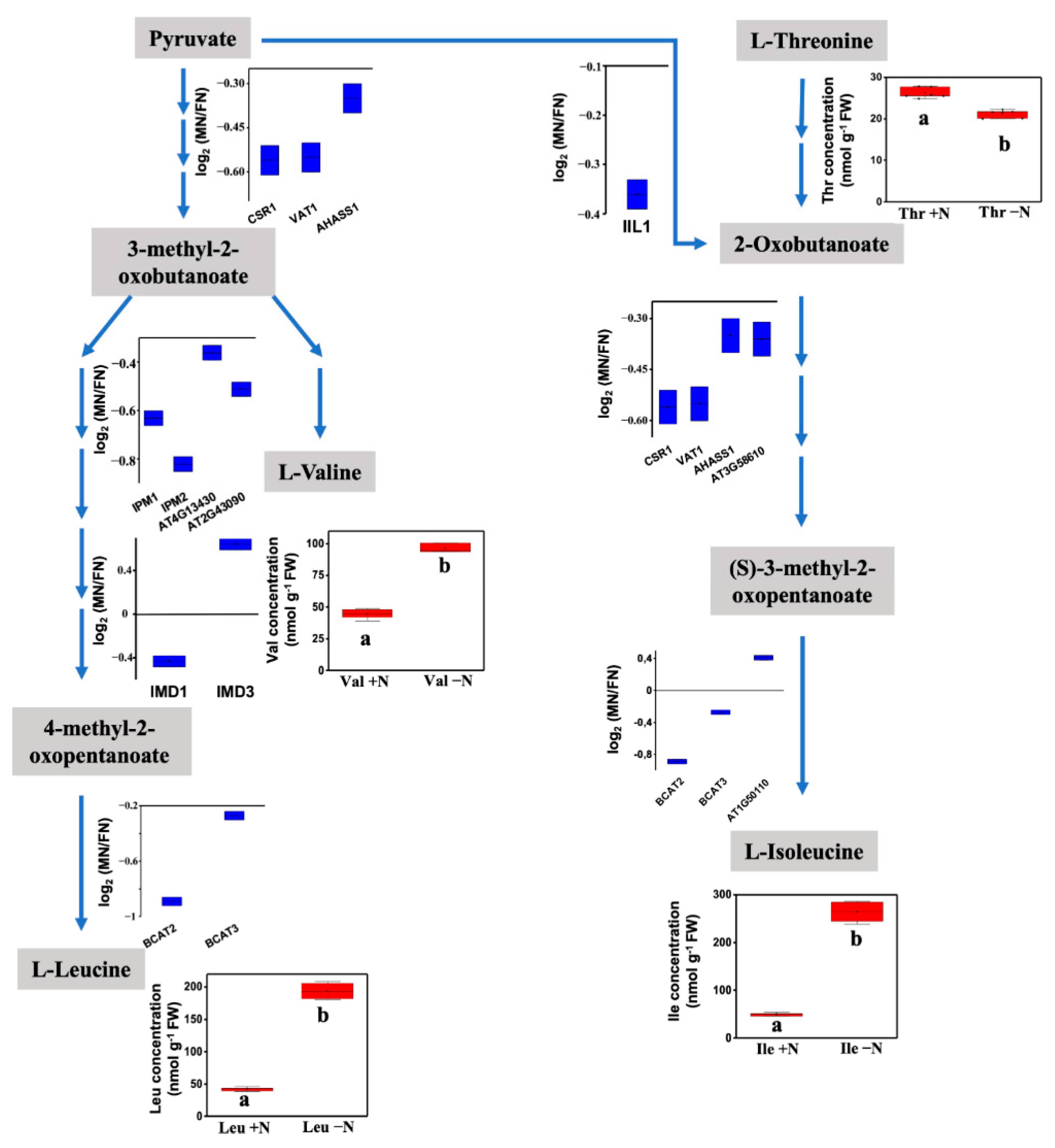
3.4. Cellular Processes and Branched-Chain Amino Acid Biosynthesis Are Regulated by Persulfidation under N Deprivation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, R. Gasotransmitters: Growing pains and joys. Trends Biochem. Sci. 2014, 39, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen Sulfide and Polysulfide Signaling. Antioxid. Redox Signal. 2017, 27, 619–621. [Google Scholar] [CrossRef]
- Aroca, A.; Gotor, C.; Bassham, D.C.; Romero, L.C. Hydrogen Sulfide: From a Toxic Molecule to a Key Molecule of Cell Life. Antioxidants 2020, 9, 621. [Google Scholar] [CrossRef] [PubMed]
- Gotor, C.; Garcia, I.; Aroca, A.; Laureano-Marin, A.M.; Arenas-Alfonseca, L.; Jurado-Flores, A.; Moreno, I.; Romero, L.C. Signaling by hydrogen sulfide and cyanide through post-translational modification. J. Exp. Bot. 2019, 70, 4251–4265. [Google Scholar] [CrossRef]
- Walsh, B.J.C.; Giedroc, D.P. H(2)S and reactive sulfur signaling at the host-bacterial pathogen interface. J. Biol. Chem. 2020, 295, 13150–13168. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Zhang, Y.; Palmer, L.D.; Kehl-Fie, T.E.; Skaar, E.P.; Trinidad, J.C.; Giedroc, D.P. Hydrogen Sulfide and Reactive Sulfur Species Impact Proteome S-Sulfhydration and Global Virulence Regulation in Staphylococcus aureus. ACS Infect. Dis. 2017, 3, 744–755. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018, 149, 101–109. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, M.; Zhou, H.; Zhao, D.; Gotor, C.; Romero, L.C.; Shen, J.; Ge, Z.; Zhang, Z.; Shen, W.; et al. Hydrogen sulfide, a signaling molecule in plant stress responses. J. Integr. Plant Biol. 2021, 63, 146–160. [Google Scholar] [CrossRef]
- Gotor, C.; Laureano-Marín, A.M.; Arenas-Alfonseca, L.; Moreno, I.; Aroca, Á.; García, I.; Romero, L.C. Advances in Plant Sulfur Metabolism and Signaling. In Progress in Botany; Cánovas, F.M., Lüttge, U., Matyssek, R., Eds.; Springer: Cham, Swizerland, 2017; Volume 78, pp. 45–66. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]
- Aroca, A.; Gotor, C.; Romero, L.C. Hydrogen Sulfide Signaling in Plants: Emerging Roles of Protein Persulfidation. Front. Plant Sci. 2018, 9, 1369. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef]
- Aroca, A.; Schneider, M.; Scheibe, R.; Gotor, C.; Romero, L.C. Hydrogen Sulfide Regulates the Cytosolic/Nuclear Partitioning of Glyceraldehyde-3-Phosphate Dehydrogenase by Enhancing its Nuclear Localization. Plant Cell Physiol. 2017, 58, 983–992. [Google Scholar] [CrossRef]
- Doka, E.; Ida, T.; Dagnell, M.; Abiko, Y.; Luong, N.C.; Balog, N.; Takata, T.; Espinosa, B.; Nishimura, A.; Cheng, Q.; et al. Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 2020, 6, eaax8358. [Google Scholar] [CrossRef] [PubMed]
- Aroca, A.; Benito, J.M.; Gotor, C.; Romero, L.C. Persulfidation proteome reveals the regulation of protein function by hydrogen sulfide in diverse biological processes in Arabidopsis. J. Exp. Bot. 2017, 68, 4915–4927. [Google Scholar] [CrossRef] [PubMed]
- Comas, F.; Latorre, J.; Ortega, F.; Rodriguez, M.A.; Kern, M.; Lluch, A.; Ricart, W.; Bluher, M.; Gotor, C.; Romero, L.C.; et al. Activation of endogenous H2S biosynthesis or supplementation with exogenous H2S enhances adipose tissue adipogenesis and preserves adipocyte physiology in humans. Antioxid. Redox Signal. 2021. [Google Scholar] [CrossRef]
- Zhang, D.; Macinkovic, I.; Devarie-Baez, N.O.; Pan, J.; Park, C.M.; Carroll, K.S.; Filipovic, M.R.; Xian, M. Detection of protein S-sulfhydration by a tag-switch technique. Angew. Chem. Int. Ed. Engl. 2014, 53, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zhang, J.; Zhou, M.; Zhou, H.; Gotor, C.; Romero, L.C.; Shen, J.; Yuan, X.; Xie, Y. Current approaches for detection of hydrogen sulfide and persulfidation in biological systems. Plant Physiol. Biochem. 2020, 155, 367–373. [Google Scholar] [CrossRef]
- Aroca, Á.; Serna, A.; Gotor, C.; Romero, L.C. S-Sulfhydration: A Cysteine Posttranslational Modification in Plant Systems. Plant Physiol. 2015, 168, 334–342. [Google Scholar] [CrossRef]
- Scuffi, D.; Álvarez, C.; Laspina, N.; Gotor, C.; Lamattina, L.; García-Mata, C. Hydrogen Sulfide Generated by l-Cysteine Desulfhydrase Acts Upstream of Nitric Oxide to Modulate Abscisic Acid-Dependent Stomatal Closure. Plant Physiol. 2014, 166, 2065–2076. [Google Scholar] [CrossRef]
- Scuffi, D.; Nietzel, T.; Di Fino, L.M.; Meyer, A.J.; Lamattina, L.; Schwarzländer, M.; Laxalt, A.M.; García-Mata, C. Hydrogen Sulfide Increases Production of NADPH Oxidase-Dependent Hydrogen Peroxide and Phospholipase D-Derived Phosphatidic Acid in Guard Cell Signaling. Plant Physiol. 2018, 176, 2532. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, M.; Ge, Z.; Shen, J.; Zhou, C.; Gotor, C.; Romero, L.C.; Duan, X.; Liu, X.; Wu, D.; et al. Abscisic acid-triggered guard cell l-cysteine desulfhydrase function and in situ hydrogen sulfide production contributes to heme oxygenase-modulated stomatal closure. Plant Cell Environ. 2020, 43, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Jia, H.; Wang, X.; Shi, C.; Wang, X.; Ma, P.; Wang, J.; Ren, M.; Li, J. Hydrogen Sulfide Positively Regulates Abscisic Acid Signaling through Persulfidation of SnRK2.6 in Guard Cells. Mol. Plant 2020, 13, 732–744. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, J.; Zhou, M.; Zhou, H.; Cui, B.; Gotor, C.; Romero, L.C.; Fu, L.; Yang, J.; Foyer, C.H.; et al. Persulfidation-based Modification of Cysteine Desulfhydrase and the NADPH Oxidase RBOHD Controls Guard Cell Abscisic Acid Signaling. Plant Cell 2020, 32, 1000–1017. [Google Scholar] [CrossRef]
- Gotor, C.; Garcia, I.; Crespo, J.L.; Romero, L.C. Sulfide as a signaling molecule in autophagy. Autophagy 2013, 9, 609–611. [Google Scholar] [CrossRef]
- Gotor, C.; Laureano-Marin, A.M.; Moreno, I.; Aroca, A.; Garcia, I.; Romero, L.C. Signaling in the plant cytosol: Cysteine or sulfide? Amino Acids 2015, 47, 2155–2164. [Google Scholar] [CrossRef]
- Alvarez, C.; Calo, L.; Romero, L.C.; Garcia, I.; Gotor, C. An O-acetylserine(thiol)lyase homolog with L-cysteine desulfhydrase activity regulates cysteine homeostasis in Arabidopsis. Plant Physiol. 2010, 152, 656–669. [Google Scholar] [CrossRef]
- Alvarez, C.; Garcia, I.; Moreno, I.; Perez-Perez, M.E.; Crespo, J.L.; Romero, L.C.; Gotor, C. Cysteine-generated sulfide in the cytosol negatively regulates autophagy and modulates the transcriptional profile in Arabidopsis. Plant Cell 2012, 24, 4621–4634. [Google Scholar] [CrossRef] [PubMed]
- Laureano-Marin, A.M.; Moreno, I.; Romero, L.C.; Gotor, C. Negative Regulation of Autophagy by Sulfide Is Independent of Reactive Oxygen Species. Plant Physiol. 2016, 171, 1378–1391. [Google Scholar] [CrossRef] [PubMed]
- Laureano-Marin, A.M.; Aroca, A.; Perez-Perez, M.E.; Yruela, I.; Jurado-Flores, A.; Moreno, I.; Crespo, J.L.; Romero, L.C.; Gotor, C. Abscisic Acid-Triggered Persulfidation of the Cys Protease ATG4 Mediates Regulation of Autophagy by Sulfide. Plant Cell 2020, 32, 3902–3920. [Google Scholar] [CrossRef]
- Bermudez, M.A.; Galmes, J.; Moreno, I.; Mullineaux, P.M.; Gotor, C.; Romero, L.C. Photosynthetic adaptation to length of day is dependent on S-sulfocysteine synthase activity in the thylakoid lumen. Plant Physiol. 2012, 160, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Krstic, J.; Reinisch, I.; Schindlmaier, K.; Galhuber, M.; Berger, N.; Kupper, N.; Moyschewitz, E.; Auer, M.; Michenthaler, H.; Nössing, C.; et al. Fasting reverses drug-resistance in hepatocellular carcinoma through p53-dependent metabolic synergism. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Masclaux-Daubresse, C.; Chen, Q.; Havé, M. Regulation of nutrient recycling via autophagy. Curr. Opin. Plant Biol. 2017, 39, 8–17. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 1–382. [Google Scholar] [CrossRef]
- Lam, H.M.; Coschigano, K.T.; Oliveira, I.C.; Melo-Oliveira, R.; Coruzzi, G.M. The Molecular-Genetics of Nitrogen Assimilation into Amino Acids in Higher Plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 569–593. [Google Scholar] [CrossRef]
- Huang, J.; Willems, P.; Wei, B.; Tian, C.; Ferreira, R.B.; Bodra, N.; Martínez Gache, S.A.; Wahni, K.; Liu, K.; Vertommen, D.; et al. Mining for protein S-sulfenylation in Arabidopsis uncovers redox-sensitive sites. Proc. Natl. Acad. Sci. USA 2019, 116, 21256–21261. [Google Scholar] [CrossRef]
- Klie, S.; Nikoloski, Z. The Choice between MapMan and Gene Ontology for Automated Gene Function Prediction in Plant Science. Front. Genet. 2012, 3, 115. [Google Scholar] [CrossRef]
- Belda-Palazon, B.; Julian, J.; Coego, A.; Wu, Q.; Zhang, X.; Batistic, O.; Alquraishi, S.A.; Kudla, J.; An, C.; Rodriguez, P.L. ABA inhibits myristoylation and induces shuttling of the RGLG1 E3 ligase to promote nuclear degradation of PP2CA. Plant J. 2019, 98, 813–825. [Google Scholar] [CrossRef]
- Yu, J.; Kang, L.; Li, Y.; Wu, C.; Zheng, C.; Liu, P.; Huang, J. RING finger protein RGLG1 and RGLG2 negatively modulate MAPKKK18 mediated drought stress tolerance in Arabidopsis. J. Integr. Plant Biol. 2020, 63, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Botterweg-Paredes, E.; Doll, J.; Eguen, T.; Blaakmeer, A.; Matton, S.; Xie, Y.; Lunding, B.S.; Zentgraf, U.; Guan, C.; et al. Multi-level analysis of the interactions between REVOLUTA and MORE AXILLARY BRANCHES 2 in controlling plant development reveals parallel, independent and antagonistic functions. Development 2020, 147, dev183681. [Google Scholar] [CrossRef]
- Wang, L.; Wang, B.; Yu, H.; Guo, H.; Lin, T.; Kou, L.; Wang, A.; Shao, N.; Ma, H.; Xiong, G.; et al. Transcriptional regulation of strigolactone signalling in Arabidopsis. Nature 2020, 583, 277–281. [Google Scholar] [CrossRef]
- Bunsick, M.; Toh, S.; Wong, C.; Xu, Z.; Ly, G.; McErlean, C.S.P.; Pescetto, G.; Nemrish, K.E.; Sung, P.; Li, J.D.; et al. SMAX1-dependent seed germination bypasses GA signalling in Arabidopsis and Striga. Nat. Plants 2020, 6, 646–652. [Google Scholar] [CrossRef]
- Ruan, J.; Zhou, Y.; Zhou, M.; Yan, J.; Khurshid, M.; Weng, W.; Cheng, J.; Zhang, K. Jasmonic Acid Signaling Pathway in Plants. Int. J. Mol. Sci. 2019, 20, 2479. [Google Scholar] [CrossRef] [PubMed]
- Planas-Riverola, A.; Gupta, A.; Betegón-Putze, I.; Bosch, N.; Ibañes, M.; Caño-Delgado, A.I. Brassinosteroid signaling in plant development and adaptation to stress. Development 2019, 146, dev.151894. [Google Scholar] [CrossRef] [PubMed]
- Soto-Burgos, J.; Zhuang, X.; Jiang, L.; Bassham, D.C. Dynamics of Autophagosome Formation. Plant Physiol. 2018, 176, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Mugume, Y.; Bassham, D.C. New advances in autophagy in plants: Regulation, selectivity and function. Semin. Cell Dev. Biol. 2018, 80, 113–122. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017, 89, 789–804. [Google Scholar] [CrossRef]
- Mergner, J.; Frejno, M.; List, M.; Papacek, M.; Chen, X.; Chaudhary, A.; Samaras, P.; Richter, S.; Shikata, H.; Messerer, M.; et al. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 2020, 579, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Weiss, M.; Simonovic, M.; Haertinger, G.; Schrimpf, S.P.; Hengartner, M.O.; von Mering, C. PaxDb, a database of protein abundance averages across all three domains of life. Mol. Cell. Proteom. 2012, 11, 492–500. [Google Scholar] [CrossRef]
- Marshall, R.S.; Vierstra, R.D. Autophagy: The Master of Bulk and Selective Recycling. Annu. Rev. Plant Biol. 2018, 69, 173–208. [Google Scholar] [CrossRef]
- Taherbhoy, A.M.; Tait, S.W.; Kaiser, S.E.; Williams, A.H.; Deng, A.; Nourse, A.; Hammel, M.; Kurinov, I.; Rock, C.O.; Green, D.R.; et al. Atg8 transfer from Atg7 to Atg3: A distinctive E1-E2 architecture and mechanism in the autophagy pathway. Mol. Cell 2011, 44, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Benchoam, D.; Cuevasanta, E.; Moller, M.N.; Alvarez, B. Hydrogen Sulfide and Persulfides Oxidation by Biologically Relevant Oxidizing Species. Antioxidants 2019, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, X.; Peirats-Llobet, M.; Belda-Palazon, B.; Wang, X.; Cui, S.; Yu, X.; Rodriguez, P.L.; An, C. Ubiquitin Ligases RGLG1 and RGLG5 Regulate Abscisic Acid Signaling by Controlling the Turnover of Phosphatase PP2CA. Plant Cell 2016, 28, 2178–2196. [Google Scholar] [CrossRef]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Ko, H.S.; Lee, Y.I.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef]
- Nelson, D.C.; Scaffidi, A.; Dun, E.A.; Waters, M.T.; Flematti, G.R.; Dixon, K.W.; Beveridge, C.A.; Ghisalberti, E.L.; Smith, S.M. F-box protein MAX2 has dual roles in karrikin and strigolactone signaling in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2011, 108, 8897–8902. [Google Scholar] [CrossRef]
- Makino, A.; Osmond, B. Effects of nitrogen nutrition on nitrogen partitioning between chloroplasts and mitochondria in pea and wheat. Plant Physiol. 1991, 96, 355–362. [Google Scholar] [CrossRef]
- Chen, Y.; Murchie, E.H.; Hubbart, S.; Horton, P.; Peng, S. Effects of season-dependent irradiance levels and nitrogen-deficiency on photosynthesis and photoinhibition in field-grown rice (Oryza sativa). Physiol. Plant. 2003, 117, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.Y.; Chen, M.X.; Chan, W.L.; Yang, F.; Tian, Y.; Song, T.; Xie, L.J.; Zhou, Y.; Xiao, S.; Zhang, J.; et al. SWATH-MS quantitative proteomic investigation of nitrogen starvation in Arabidopsis reveals new aspects of plant nitrogen stress responses. J. Proteom. 2018, 187, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Brunold, C.; Suter, M. Regulation of Sulfate Assimilation by Nitrogen Nutrition in the Duckweed Lemna minor L. Plant Physiol. 1984, 76, 579–583. [Google Scholar] [CrossRef]
- Takahashi, H.; Saito, K. Subcellular localization of spinach cysteine synthase isoforms and regulation of their gene expression by nitrogen and sulfur. Plant Physiol. 1996, 112, 273–280. [Google Scholar] [CrossRef]
- Takahashi, H.; Kopriva, S.; Giordano, M.; Saito, K.; Hell, R. Sulfur assimilation in photosynthetic organisms: Molecular functions and regulations of transporters and assimilatory enzymes. Annu. Rev. Plant Biol. 2011, 62, 157–184. [Google Scholar] [CrossRef] [PubMed]
- Romero, L.C.; Aroca, M.A.; Laureano-Marin, A.M.; Moreno, I.; Garcia, I.; Gotor, C. Cysteine and cysteine-related signaling pathways in Arabidopsis thaliana. Mol. Plant 2014, 7, 264–276. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster 1. Enrichment Score: 5.8 | |||||
| Term | Count | p Value | Fold Enrichment | Benjamini | FDR |
| GO:0009082~branched-chain amino acid biosynthetic process | 13 | 2.44 × 10−9 | 8.57 | 3.63 × 10−7 | 3.52 × 10−7 |
| GO:0009098~leucine biosynthetic process | 13 | 1.08 × 10−8 | 7.79 | 1.32 × 10−6 | 1.28 × 10−6 |
| GO:0009099~valine biosynthetic process | 10 | 1.12 × 10−6 | 7.75 | 7.89 × 10−5 | 7.64 × 10−5 |
| GO:0009097~isoleucine biosynthetic process | 7 | 3.31 × 10−4 | 6.59 | 1.19 × 10−2 | 1.16 × 10−2 |
| GO:0006532~aspartate biosynthetic process | 6 | 4.53 × 10−4 | 7.91 | 1.49 × 10−2 | 1.44 × 10−2 |
| Cluster 2. Enrichment Score: 1.7 | |||||
| Term | Count | p Value | Fold Enrichment | Benjamini | FDR |
| GO:0006544~glycine metabolic process | 4 | 0.012055 | 7.53 | 2.31 × 10−1 | 2.24 × 10−1 |
| GO:0035999~tetrahydrofolate interconversion | 5 | 0.022806 | 4.39 | 3.73 × 10−1 | 3.61 × 10−1 |
| GO:0006563~L-serine metabolic process | 4 | 0.025800 | 5.86 | 4.02 × 10−1 | 3.89 × 10−1 |
| Cluster 3. Enrichment Score: 1.2 | |||||
| Term | Count | p Value | Fold Enrichment | Benjamini | FDR |
| GO:0006032~chitin catabolic process | 6 | 0.03689 | 3.16 | 4.75 × 10−1 | 4.61 × 10−1 |
| GO:0016998~cell wall macromolecule catabolic process | 6 | 0.04293 | 3.04 | 5.38 × 10−1 | 5.21 × 10−1 |
| GO:0000272~polysaccharide catabolic process | 5 | 0.05997 | 3.296 | 6.64 × 10−1 | 6.43 × 10−1 |
| GO:0006040~amino sugar metabolic process | 4 | 0.09970 | 3.51 | 9.50 × 10−1 | 9.21 × 10−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurado-Flores, A.; Romero, L.C.; Gotor, C. Label-Free Quantitative Proteomic Analysis of Nitrogen Starvation in Arabidopsis Root Reveals New Aspects of H2S Signaling by Protein Persulfidation. Antioxidants 2021, 10, 508. https://doi.org/10.3390/antiox10040508
Jurado-Flores A, Romero LC, Gotor C. Label-Free Quantitative Proteomic Analysis of Nitrogen Starvation in Arabidopsis Root Reveals New Aspects of H2S Signaling by Protein Persulfidation. Antioxidants. 2021; 10(4):508. https://doi.org/10.3390/antiox10040508
Chicago/Turabian StyleJurado-Flores, Ana, Luis C. Romero, and Cecilia Gotor. 2021. "Label-Free Quantitative Proteomic Analysis of Nitrogen Starvation in Arabidopsis Root Reveals New Aspects of H2S Signaling by Protein Persulfidation" Antioxidants 10, no. 4: 508. https://doi.org/10.3390/antiox10040508
APA StyleJurado-Flores, A., Romero, L. C., & Gotor, C. (2021). Label-Free Quantitative Proteomic Analysis of Nitrogen Starvation in Arabidopsis Root Reveals New Aspects of H2S Signaling by Protein Persulfidation. Antioxidants, 10(4), 508. https://doi.org/10.3390/antiox10040508