
Beneficial Role of Exercise in the Modulation of mdx Muscle Plastic Remodeling and Oxidative Stress

 , and
, and
Abstract
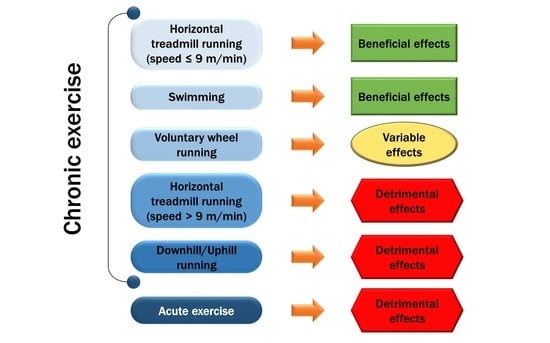
:
1. Introduction
2. Duchenne Muscular Dystrophy
3. The mdx Model
4. Effects of Exercise on Plastic Remodeling of mdx Muscle
4.1. Effects of Forced Running on Plastic Remodeling of mdx Muscle
4.2. Effects of Swimming Exercise on Plastic Remodeling of mdx Muscle
4.3. Effects of Voluntary Running on Plastic Remodeling of mdx Muscle
5. Mitochondria Impairment and Redox Equilibrium in DMD Muscles
6. Exercise Modulation of mdx Muscle Oxidative Stress and Inflammation
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| A. Chronic Exercise | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Sex/Age | Protocol | Muscle | Hindlimb Muscle (Morphological and Functional Changes) | Respiratory Function | Cardiac Function | Degeneration/ Regeneration | Fibrosis | Metabolic Adaptive Changes | Inflammatory Markers | Oxidant/ Antioxidant Markers | Exercise Muscle Effects |
| Faist, V., et al. (2001) [281] | 4 weeks (young mice) 16 weeks (adult mice) | Pre-training + LIT Pre-training: 4 to 8.3 m/min, 2 × 5 to 2 × 30 min/day, 8 days Training: 8.3 m/min, 2 × 30 min/day, 6 weeks | Femoris GAST QUAD | Young mice: = body weight Adult mice: = body weight | Young mice: ↑ CK Adult mice: = CK | Young mice: ↓ oxygen consumption ↓ RCI = pADP/OC Adult mice: ↑ oxygen consumption = RCI = pADP/OC | Young mice: ↑ TBARS ↑ Lipofuscin = peroxyl radicals ↓ GSH = SOD activity = α-tocopherol Adult mice: = TBARS = Lipofuscin = peroxyl radicals = GSH = SOD activity = α-tocopherol | (−) Young (±) Adult | ||||
| Fernandes, D.C., et al. (2019) [73] | 11 weeks | LIT 9 m/min, 30 min/ day, 3 days/week, 60 days | GAST TA | ↓ collagen fiber area | ↑ TBARS ↑ carbonylated protein in GAST and TA ↑ SOD activity ↑ FRAP activity in TA | (±) | ||||||
| Fontana, S., et al. (2015) [110] | Male 8 weeks | Pre-training + LIT Pre-training: 3.2 m/min, 15 to 30 min/day, 5 days/week, 2 weeks. Training: workload progressive increase, 4 to 4.8 m/min, 30 to 60 min/day, 5 days/week, 4 weeks | QUAD | ↑ SOD1 ↓ CA3 | (+) | |||||||
| Frinchi, M., et al. (2013) [77] | Male 8 weeks | Pre-training + LIT Pre-training: 3.2 m/min, 15 to 30 min/day, 5 days/week, 2 weeks. Training: workload progressive increase, 4 to 4.8 m/min, 30 to 60 min/day, 5 days/week, 4 weeks | GAST QUAD | ↓ degenerative myofibers ↓ Cx39 | (+) | |||||||
| Gaiad, T.P., et al. (2017) [74] | Male 8 weeks | Pre-training + LIT Pre-training: Speed progressive increase. Training: 9 m/min, 30 min/day, 3 days/ week, 60 days | TA | = CLN | ↓ fibrosis deposition (↑ Col 3 and = Col 1) | (+) | ||||||
| Hermes, T.A., et al. (2018) [119] | Male and female 2 months | LIT 6 m/min, 15 to 30 min/day, 2 days/week, 4 weeks | DIA | ↑ CK (in males) = CNF | ↑ inflammatory area (in males) | (−) males (±) females | ||||||
| Hoepers, A., et al. (2020) [108] | Male 28 days | Pre-training + LIT Pre-training: 7 days, 4 m/min. Training: 6 and 9 m/min, 30 min/day, 2 days/week, 8 weeks | GAST | ↓ activity of complex II, II-III and IV | ↓ lipid peroxidation ↓ carbonylated proteins ↑ free thiols | (+) | ||||||
| Kaczor, J.J., et al. (2007) [109] | 28 days | LIT 9 m/min, 30 min/day, 2 days/week, 8 weeks | DIA, EDL, GAST SOL | = CLN in DIA | ↓ MDA in white GAST ↓ carbonylated protein = OGDH activity in GAST = COX activity in GAST = SOD1 and SOD2 activity in GAST = CAT activity in GAST = GPX in EDL and SOL | (+) GAST (±) EDL, SOL, DIA | ||||||
| Morici, G., et al. (2017) [76] | Male 8 weeks | Pre-training + LIT Pre-training: 3.2 m/min, 15 to 30 min/day, 5 days/week, 2 weeks. Training: workload progressive increase, 4 to 4.8 m/min, 30 to 60 min/day, 5 days/week, 4 weeks | DIA, GAST QUAD | ↑ regeneration area ↓ necrotic area in DIA | ↓ NF-kB in QUAD | (+) | ||||||
| Pinto, P.A.F., et al. (2018) [75] | Male 11 weeks | LIT 9 m/min, 30 min/day, 3 days/week, 8 weeks | Lateral GAST | = CLN | ↓ Col fibers type I = Col fibers type III | (+) | ||||||
| Zelikovich, A.S., et al. (2019) [79] | 16–20 weeks | LIT 4 m/min or 8 m/min, 30 min/day, 3 days/week, 6 months (Each session consisted of 7’30’’ warm up followed by 22’30’’ training at target speed) | DIA, GAST Heart TA | ↑ grip strength ↑ tetanic force in TA ↑ specific force in TA ↑ resistance to fatigue in TA ↑ type IIa fibers in TA ↓ Adipocyte CSA in GAST | ↑ MV ↓ Ti ↓ Te | ↓ LVIDD and LVPWT thickness | = CNF in GAST, DIA and Heart = serum CK | = fibrosis in GAST, DIA and Heart | ↑ PGC-1α in Heart Trend ↑ PGC-1α in GAST ↑ Adipoq serum and in GAST | (+) | ||
| Zeman, J.R., et al. (2000) [78] | 10 weeks | LIT 9 m/min, 1 h/day, 5 days/week, 10 weeks | GAST Plantaris SOL | ↓ necrotic area in GAST and SOL ↑ necrotic area in Plantaris | (+) GAST, SOL (−) Plantaris | |||||||
| Burdi, R., et al. (2009) [92] | Male 4–5 weeks | MIT 12 m/min, 30 min/day, 2 days/week, 4–8 weeks | DIA, EDL GAST TA | ↓ forelimb strength | = isometric tetanic tension in DIA | ↑ CK | = antioxidant activity ↑ plasma ROS level ↑ O-2 in TA | (−) EDL, GAST, TA (±) DIA | ||||
| Burdi, R., et al. (2006) [99] | Male 4–5 weeks | MIT 12 m/min, 30 min/day, 2 days/week, 4–8 weeks | DIA, EDL GAST TA | ↓ forelimb strength | = serum CK ↓ gCl in EDL = gCl in DIA = gK | ↓ TGFβ1 in DIA Trend ↑ TGFβ1 in TA and GAST | (−) EDL, GAST, TA (±) DIA | |||||
| Camerino, G.M., et al. (2014) [88] | Male 4–5 weeks | MIT A: 12 m/min, 30 min/day, 2 days/week, 4 weeks. B: 12 m/min, 30 min/day, 2 days/week, 12 weeks | EDL GAST SOL | A: ↓ forelimbs strength ↑ fatigue ↓ MHC2b = SERCA1 = SERCA2 ↓ HDAC5 = MHC isoforms = Utrn = Cn = MEF2 isoforms ↓ HDAC5 B: ↓ forelimbs strength = MHC isoforms = SERCA1 ↓ SERCA2 ↑ Cn = MEF2 isoforms ↓ HDAC5 | A: = Fst = Myog = IGF1 = Svil ↓ BNIP3 ↓ LC3 B: ↓ Fst ↓ Myog = IGF1 = Svil ↓ BNIP3 ↓ LC3 | A: = TGFβ = Mstn = Atrogin1 B: = TGFβ = Mstn = Atrogin1 | A: = PGC-1α = Sirt1 = Pparγ = COX4 = CS = Utrn = VEGF-a and –b B: ↓ PGC-1α ↓ Sirt1 ↓ Pparγ = COX4 = CS = Utrn = VEGF-a and -b | A: = TNFα = Adipoq = Adipor1 B: = TNFα = Adipoq = Adipor1 | A: = Tubα = NOX2 = c-Src B: = Tubα = NOX2 = c-Src | A: (−) B: (−) | ||
| Capogrosso, R.F., et al. (2017) [86] | Male 4–5 weeks | MIT A: 12 m/min, 30 min/day, 2 days/week, 4 weeks B: 12 m/min, 30 min/day, 2 days/week, 12 to 20 weeks | DIA EDL GAST TA | A: ↓ forelimbs strength = twitch and tetanic tension = torque = resistance to eccentric contractions = fatigue in EDL B: ↓ forelimbs strength ↓ twitch and tetanic tension = torque = resistance to eccentric contractions = fatigue ↑ AChR1 ↓ MHC1 in EDL | A: = twitch and tetanic tension = fatigue B: = twitch and tetanic tension = fatigue | B: ↓ Fst in EDL = pAMPK/ AMPK in TA = CNF in GAST and DIA = TGFβ1 in GAST = MMP9 serum | B: = Col 1 = ITGA7 = Eln in EDL | B: ↑ PGC-1α ↓ Sirt1 = FIH1 = CD31 in EDL | B: = Tubα = NOX2 in EDL | A: (−) EDL, TA (±) DIA B: (−) EDL, TA (±) DIA, GAST | ||
| De Luca, A., et al. (2003) [90] | Male 3–4 weeks | MIT 12 m/min, 30 min/day, 2 days/week, 4–8 weeks | DIA EDL TA | ↓ forelimb strength ↑ RM in EDL = RM in DIA ↓ gCl in EDL = gCl in DIA | ↑ necrotic cell in TA | ↑ inflammatory infiltrated cells in TA | (−) EDL, TA (±) DIA | |||||
| De Luca, A., et al. (2005) [96] | Male 4–5 weeks | MIT 12 m/min, 30 min/day, 2 days/week, 4–8 weeks | DIA EDL, GAST | = body weight ↓ forelimb strength ↓ % fiber expressing slow MHC in EDL | ↑ serum CK = Utrn level in DIA | Trend ↑ TGFβ1 in GAST Trend ↑ % area of connective tissue in GAST | (−) EDL, GAST (±) DIA | |||||
| Fraysse, B., et al. (2004) [85] | 3–4 weeks | MIT 12 m/min, 30 min/day, 2 days/week, 4–8 weeks | EDL | ↓ forelimb strength Any fast-to-slow fiber transition | ↑ resting [Ca2+]i ↑ Sarcolemmal permeability = CK | (−) | ||||||
| Gamberi, T., et al. (2018) [95] | Male 4–5 weeks | MIT 12 m/min, 30 min/day, 2 days/week, 4 weeks | TA | ↑ Tnnt3 ↑ Tnnt2 ↑ MYOZ1 ↑ Actin ↑ LDB3 ↓ MYLPF ↓ TPM1 ↓ MYL1 | ↑ CK | ↑ ALDOA ↑ TPI1 ↑ Eno3 ↑ UGPR ↑ FH ↑ MDH2 ↑ ALDH4A1 ↑ complex II ↑ AK1 ↓ Complex I, III ↓ NDUFB7 ↓ ATP5a1 = Complex IV, V = PGC1-α = Sirt1 | (−) | |||||
| Hall, J.E., et al. (2007) [126] | Male 4 weeks | Pre-training + MIT Pre-training: workload progressive increase 8 to 12 m/min, 4 weeks. Training: 12 m/min, 30 min/day, 6 weeks | EDL GAST QUAD | ↓ serum CK | (+) | |||||||
| Morales, M.G., et al. (2013) [82] | Male 8 weeks | MIT 12 m/min, 30 min/day, 3 days/week, 6 months | GAST | ↓ Net Force | ↑ degeneration | ↑ Col 3 ↑ Fn ↑ CTGF ↑ TGFβ ↑ SMAD3 phosphorylation | (−) | |||||
| Pessina, P., et al. (2014) [81] | 3–4–5 months | MIT 12 m/min, 30 min/day (with a rest of 5 minutes every 10 minutes of exercise), 3 days/week, 1, 2 or 3 months | GAST | ↓ tetanic force | ↑ fibrosis (↑ Col 1, ↑ Fn deposition, ↑ TGFβ1 and CTGF mRNA, ↑ SMAD2/3 protein level) | (−) | ||||||
| Radley-Crabb, H., et al. (2012) [87] | Male 8–12 weeks | MIT 12 m/min, 30 min/day, 2 days/week, 4 weeks | DIA EDL GAST QUAD TA Triceps | ↓ forelimb grip strenght | Necrosis: DIA (↑ (0 min, ↑ 24 h, = 96 h) GAST (trend ↑ 0 min, trend ↑ 24 h, = 96 h) QUAD (trend ↑ 0 min, ↑ 24 h, = 96 h) TA (= 0 min, ↑ 24 h, ↓ 96 h) Triceps (= 0 min, ↑ 24 h, = 96 h) ↑ CK (24 h) = CK (96 h) | ↓ IL-1β (96 h) ↓ IL-6 (96 h) ↓ TNF (0 min) = TNF (24 h) in QUAD | ↑ thiol oxidation (0 min) = thiol oxidation (24 h) = MDA in QUAD | (−) 0 min, 24 h (±) 96 h | ||||
| Rocco, A.B., et al. (2014) [80] | Male 8 weeks | Pre-training +MIT Pre-training: 5 m/min, 5 min/day, 3 days. Training: (5 min at 0 m/min + 2 min at 5 m/min + 8 min at 8 m/min + 30 min at 12 m/min)/day, 2 days/week, 4 weeks | DIA, GAST Pectoralis QUAD TA TB | = muscle mass | ↑ Col in all muscles except DIA | = VO2max = time to exhaustion, = speed at exhaustion = total energy = VO2cv = VO2min | (−) GAST, TA, QUAD, TB, Pectoralis (±) DIA | |||||
| Schill, K.E., et al. (2016) [15] | Male and female 4 weeks | MIT 12 m/min, 30 min/day, 2 days/week, 4 weeks | Abdominal Heart QUAD | ↓ exhaustion times | ↑ intracellular IgG in Heart = intracellular IgG in QUAD | ↑ hydroxyproline in QUAD and Heart = hydroxyproline in Abdominal ↑ Col 1 in Heart | ↓ basal oxygen consumption = maximal oxygen consumption | ↑ GSH oxidized by ROS in QUAD and Abdominal Trend ↑ GSH oxidized by ROS in Heart | (−) | |||
| Van Putten, M., et al. (2012) [16] | Female 4 weeks | MIT 12 m/min, 30 min/day, 3 days/week, 12 weeks | QUAD | ↑ CK | ↑ fibrosis in QUAD | (−) | ||||||
| Vita, G.L., et al. (2020) [106] | 4–5 weeks | MIT 12 m/min, 30 min/day, 2 days/week, 8–10 weeks | DIA, GAST TA | = Minimum telomere length | ↑ TRF1 in TA ↓ TRF1 in DIA = TRF1 in GAST = PARP1 in GAST, TA and DIA ↑ MTERT protein level and activity in DIA = MTERT in GAST and TA | (±) | ||||||
| B. Acute exercise | ||||||||||||
| Reference | Sex/Age | Protocol | Muscle | Hindlimb Muscle (Morphological and Functional Changes) | Respiratory Function | Cardiac Function | Degeneration /Regeneration | Fibrosis | Metabolic Adaptive Changes | Inflammatory Markers | Oxidant /Antioxidant Markers | Exercise Muscle Effects |
| Camerino, G.M., et al. (2014) [88] | Male 16 weeks | MIT 12 m/min, 30 min | GAST | = Mhc1 = Cn = Serca2 | = Fst | = PGC-1α = Sirt1 = PPARγ | (±) | |||||
| Capogrosso, R.F., et al. (2017) [86] | Male 16 weeks | MIT 12 m/min, 30 min | DIA EDL | ↓ twitch and tetanic tension ↓ TTP ↓ HRT ↑ resistance to eccentric contractions = fatigue | ↓ twitch and tetanic tension = fatigue | (−) | ||||||
| Radley-Crabb, H., et al. (2012) [87] | Male 8–12 weeks | MIT 12 m/min, 30 min | EDL GAST QUAD TA Triceps | ↑ necrosis in QUAD (24 h, 48 h) = necrosis in TA, EDL, Triceps and GAST ↑ CK (0 h) = CK (24 h) | ↑ IL-1β (2 h) ↑ IL-6 (0 min, 2 h) = IL1β and IL-6 (24 h), ↓ TNF (0 min, 2 h, 24 h) in QUAD | ↑ thiol oxidation (0 min, 2 h) = thiol oxidation (24 h) in QUAD | (−) QUAD (±) EDL, GAST, TA, Triceps | |||||
| Terrill, J.R., et al. (2012) [111] | Male 12 weeks | MIT 12 m/min, 30 min | GAST QUAD | ↑ necrosis ↑ CK | ↑ thiol oxidation | (−) | ||||||
| A. Chronic Exercise | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Sex/Age | Protocol | Muscle | Hindlimb Muscle (Morphological and Functional Changes) | Respiratory Function | Cardiac Function | Degeneration/ Regeneration | Fibrosis | Metabolic Adaptive Changes | Inflammatory Markers | Oxidant/ Antioxidant Markers | Exercise Muscle Effects |
| Anderson, C.L., et al. (2006) [93] | Male 7 weeks | −15°, 10 m/min, 10 min/day, 3 days | BB DIA EDL GAST SOL TA | ↑ damaged fibers in TA, BB and DIA | (−) TA, BB, DIA | |||||||
| Brussee, V., et al. (1997) [25] | Male 8 weeks | −15°, 10 m/min, 10 min/day, 3 days | BB DIA EDL, GAST, SOL TA TB | ↑ degenerating muscle fibers | (−) | |||||||
| Cerri, D.G., et al. (2009) [94] | Male 4 weeks | Downhill, 12 m/min, 20 min/day, 3 days/week, 5 weeks | DIA, GAST | ↑ Gal-1 | (−) | |||||||
| Fowler, W.M., et al. (1990) [100] | 3 weeks | +18°, 4 m/min, 3 weeks | EDL, SOL | ↑ twitch tension ↑ rate of twitch tension development ↑ rate of twitch tension relaxation ↑ twitch/tetanus in SOL | ↓ CLN ↓ necrosis ↓ fibers splitting ↓ moth-eaten fibers | (+) | ||||||
| Kobayashi, Y.M., et al. (2012) [98] | Male 10 weeks | −15°, 5 min at 3 m/min + 10 min at 15 m/min, 2 weeks | EDL GAST QUAD | ↓ strength in EDL | ↑ myoglobinuria = CK | ↑ oedema and inflammation in QUAD and GAST | (−) | |||||
| Mokhtarian, A., et al. (1995) [91] | 14 days | +15°, 4 m/min, 30 min/days, 7 days | EDL, SOL | = ratio of type I and type II myofibers in SOL = ratio of type IIa and type IIb myofibers in EDL | ↑ necrosis | (−) | ||||||
| Nakamura, A., et al. (2002) [118] | Male 6 weeks | +7°, 15 m/min, 60 min/day, 2 days/week, 5 weeks + 23 m/min, 60 min/day, 2 days/week, 5 weeks | Heart | ↑ heart/body weight | ↑ % area of dystrophic lesion ↑ pERK1/2 ↑ p38 MAPK ↑ calcineurin = pJNK1 | ↑ fibrotic area | ↑ inflammatory cells | (−) | ||||
| Nakamura, A., et al. (2005) [84] | Male 6 weeks | +7°, 15 m/min, 60 min/day, 2 days/week, 5 weeks + 23 m/min, 60 min/day, 2 days/week, 5 weeks | GAST | ↑ pERK1/2 ↑ p38 MAPK ↑ JNK2 = JNK1 | ↑ MMP9 | (−) | ||||||
| Okano, T., et al. (2005) [89] | Male 6 weeks | +7°, 15 m/min, 60 min/day, 2 days/week, 5 weeks + 23 m/min, 60 min/day, 2 days/week, 5 weeks | GAST QUAD SOL TA | = body weight = Serum albumin levels ↓ large-sized DRGs in SOL, GAST and QUAD = small-sized DRGs Trend ↓ medium-sized DRGs | ↓ IGF1 mRNA in SOL, GAST and TA ↓ MyoD mRNA in SOL, GAST and TA | = Col 3 in SOL and GAST | (−) | |||||
| Taniguti, A.P., et al. (2011) [83] | Male and female 6 months | −17°, 17 m/min, 60 min/day, 7 weeks | BB DIA Heart, TA | ↓ grip strength and normalized forelimb muscle strength = body mass | ↑ serum CK | ↑ TGFβ1 in BB and Heart ↑ fibrosis in TA and BB = fibrosis in DIA and Heart | (−) TA, BB, Heart (±) DIA | |||||
| B. Acute exercise | ||||||||||||
| Reference | Sex/Age | Protocol | Muscle | Hindlimb Muscle (Morphological and Functional Changes) | Respiratory Function | Cardiac Function | Degeneration/ Regeneration | Fibrosis | Metabolic Adaptive Changes | Inflammatory Markers | Oxidant/ Antioxidant Markers | Exercise Muscle Effects |
| Clarke, M.S.F., et al. (1993) [116] | 12 weeks | −16°, 0.6 m/min, 5 min | Triceps | ↑ damaged myofibers | ↓ bFGF | (−) | ||||||
| Lindsay, A., et al. (2018) [215] | Male 3 months | −10°, workload progressive increase 10 to 15 m/min, 5 min + 15 m/min, 30 min | ↑ Isoxan thopterin | (−) | ||||||||
| Mathur, S., et al. (2011) [115] | Male 5–12 months | −14°, workload progressive increase 8 to 10 m/min, 45 min | Lower hindlimbs | ↑ membrane breakdown ↑ T2 | (−) | |||||||
| Quinlan, J.G., et al. (2006) [112] | 5–7 weeks | −10°, 8 m/min, 10 min + workload progressive increase 8 to 16 m/min, up to 90 min or fatigue | QUAD SOL TB | ↑ membrane breakdown in QUAD | (−) | |||||||
| Vilquin, J.T., et al. (1998) [113] | Male and female 9–14 months | −16°, 10 m/min, 5 min | ↑ serum CK (1 h) = serum CK (3 days) | (−) 1 h (±) 3 days | ||||||||
| Whitehead, N.P., et al. (2006) [114] | Male 7–10 weeks | −17°, 10 m/min, 45 min | EDL | ↓ isometric force | ↑ membrane breakdown | (−) | ||||||
| A. Chronic Exercise | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Sex/Age | Protocol | Muscle | Hindlimb Muscle (Morphological and Functional Changes) | Respiratory Function | Cardiac Function | Degeneration/ Regeneration | Fibrosis | Metabolic Adaptive Changes | Inflammatory Markers | Oxidant/ Antioxidant Markers | Exercise Muscle Effects |
| Hayes, A., et al. (1998) [129] | Male 24 months | LIT 25 min/day, 4 or 5 days/week, 10 weeks | EDL SOL | ↑ relative tetanic tension = absolute force ↓ RT1/2 in SOL = % of type I, type IIa and type IIb fibers | (+) | |||||||
| Hyzewicz, J., et al. (2015) [128] | Male 4 weeks | LIT 30 min/day, 4 days/week, 4 weeks | GAST | ↑ grip strength ↑ fast isoform of Tnnt | ↑ MyBP-C | ↑ CK (1 h) = CK(2 h) | ↑ respiratory chain protein ↑ UDPGP ↑ CA3 | ↓ carbonylated proteins (VDAC1, fast isoform of Tnnt, MyBP-C and PGM1) | (+) | |||
| Hyzewicz, J., et al. (2017) [127] | Male 4 weeks | LIT 30 min/day, 4 days/week, 4 weeks | GAST | ↑ grip strength | = CK = CNF = IgG+ cells | ↓ CCL2 ↓ TIMP-1 = C5α ↑ CD68+/ CD11b+ cells ↑ F4/80+/ CD11+cells Trend ↓ Ly6C+ / CD11b+ cells Trend ↑ Ly6C+/CD11b-cells ↓ iNOS ↑ CD68 = CD206 = CD163 | (+) | |||||
| Matsakas, A., et al. (2013) [132] | Male 6–8 weeks | LIT 30 min/day, 4 days | GAST TA | ↑ CK | ↑ Hypoxia in GAST and TA | (−) | ||||||
| Barbin, I.C.C., et al. (2016) [133] | Male 11 months | MIT 60 min/day, 6 days/week, 2 months | DIA Heart | ↑ wall thickness/lumen diameter of the pulmonary trunk | ↑ degenerating cardio- myocytes | ↑ fibrosis in DIA and Heart ↑ pro-MMP2, pre-MMP2 and active MMP2 in DIA and Heart ↑ TGFβ in Heart | (−) | |||||
| Hayes, A., et al. (1993) [130] | 5 weeks | Pre-training + HIT Pre-training: workload progressive increase 5 min to 2 h/day Training: 2 h/day, 5 days/week, 5 weeks | EDL SOL | ↓ muscle mass in EDL (↓ type IIa fiber area) and SOL (↓ type I and type IIa fiber area) ↑ RT1/2 in EDL ↑ twitch tension in SOL ↑ resistance to fatigue in EDL and SOL | (+) | |||||||
| Lynch, G.S., et al. (1993) [131] | 5 weeks | Pre training + HIT Pre-training: workload progressive increase 5 min to 2 h/day Training: 2 h/day, 5 days/week, 5 weeks | EDL SOL | ↑ intermediate type fiber in SOL ↑ sensitivity to Sr2* in type IIa fiber in EDL ↓ sensitivity to Ca2+ and Sr2+ in EDL type IIB fibers ↓ sensitivity to Ca2+ and Sr2+ in SOL type IIA fibers | (+) | |||||||
| B. Acute exercise | ||||||||||||
| Reference | Sex/Age | Protocol | Muscle | Hindlimb muscle (Morphological and Functional Changes) | Respiratory Function | Cardiac Function | Degeneration/ Regeneration | Fibrosis | Metabolic Adaptive Changes | Inflammatory Markers | Oxidant/ Antioxidant Markers | Exercise Muscle Effects |
| Bouchentouf, M., et al. (2006) [134] | Male 2 months | LIT 20 min | TA | ↑ fiber damage | (−) | |||||||
| A. Chronic Exercise | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Sex/Age | Protocol | Muscle | Hindlimb Muscle (Morphological and Functional Changes) | Respiratory Function | Cardiac Function | Degeneration/ Regeneration | Fibrosis | Metabolic Adaptive Changes | Inflammatory Markers | Oxidant/ Antioxidant Markers | Exercise Muscle Effects |
| Baltgalvis, K.A., et al. (2012) [141] | Male and female 4 weeks | 12 weeks low-resistance wheel running | DIA GAST Heart, TA | = muscle mass ↑ maximal isometric torque in GAST = concentric and passive torque in GAST ↑ fatigue resistance ↑ type IIx fiber in GAST | ↑ Heart mass | = CK = CNF in TA and DIA ↓ CNF in GAST | ↑ CS, β-HAD in GAST = CS, β-HAD in TA ↑ COXIV in GAST | (+) GAST, Heart (±) TA, DIA | ||||
| Brereton, D., et al. (2012) [153] | 4 months | 4 weeks | erector spinae muscle | ↑ Fibrosis ↑ Kyphosis | (−) | |||||||
| Bueno, Jr. C.R., et al. (2012) [137] | Male 2 months | 1 month | TB | ↑ aerobic capacity ↑ CSA = Myoglobin | ↓ ubiquitinated proteins ↑ CK | ↑ p-AMPKα (Thr172) = AMPKα ↑ p-ACC/ACC = Pparδ = SCD | (+) | |||||
| Call, J.A., et al. (2008) [136] | Male 21 days | 3 weeks | EDL GAST Heart SOL | ↑ muscle mass in GAST ↑ weekly distance ↑ tetanic stress output in EDL ↑ active stifness in EDL ↑ total contractile protein in EDL ↑ actin in EDL ↑ myosin in EDL shift to fatigue-resistant fibers (↑ Type I fibers and ↓ Type IIb fibers) in EDL | ↑ muscle mass in Heart | ↑ CS in Heart = CS in GAST and SOL ↑ β-HAD in GAST and Heart = β-HAD in SOL | (+) EDL, GAST, Heart (±) SOL | |||||
| Call, J.A., et al. (2010) [144] | Male 4–5 weeks | 12 weeks of voluntary free wheel or resistance wheel running | EDL GAST Heart QUAD SOL TA Triceps | ↑ grip strength = dorsiflexion torque and whole body tension ↑ specific tetanic force in SOL ↑ specific eccentric force (only free wheel) in SOL = absolute twitch, tetanic or eccentric force in SOL ↑ relative mass in SOL and TA (only free wheel) ↑ relative mass in Triceps (only in Resist Wheel) = relative mass in GAST and EDL ↑ β-dystroglycan in GAST ↑ Vinculin in SOL = α-7 integrin in SOL, GAST and Triceps = Talin in SOL, GAST and Triceps | = relative mass in Heart | = CK | (+) (±) Heart | |||||
| Coles, C.A., et al. (2020) [149] | Male 9 weeks | 3 weeks | QUAD | ↑ necrosis ↑ CNF | ↑ Extracelluar matrix genes (i.e. Col 1a2, Col 3a1, Fn1, PCOLCE, etc.) ↓ EGLN1 | ↑ F4/80+ cell ↑ M1 macrophages (F4/80+/CD206-) Trend ↑ M2a macrophages (CD206+/CD163-) = M2c (CD206+/CD163+) ↑ inflammation related genes (i.e. CD14, IFi30, IL10rα, etc.) ↓ Pde4d ↑ Receptors recognising extracellular DAMPs (TLR2, TLR4, TLR6, P2XR4, etc.) | (+) | |||||
| Costas, J.M., et al. (2010) [160] | Female 7 weeks | 4 weeks | Heart | = body mass ↓ LVLWT ↑ LVSLD | ↑ fibrosis | (−) | ||||||
| Delacroix, C., et al. (2018) [135] | 2–4 months | 1 week | EDL GAST Plantaris TA | ↑muscle excitability in TA and EDL (↑ CMAP ↑ SLC8A1 ↓ SCN4A ↓ Cacna1s) ↑ calcineurine pathway in TA and EDL (↑Rcan3, ↓ Cmya5, ↓ Myoz1 = Rcan1) ↑ fast to slow transition in TA (↑ MHC2, MHC7, Tnni1) = MHC3 ↓ MHC4, Tnni2 ↓ muscle fragility in TA and EDL No effect in GAST and Plantaris | = Myog in TA | = TGFβ = Col 1a1 in TA | ↑ TFAM ↑ Nrf1 ↑ PGC-1β = PGC-1α = SDHB = NDUF5 in TA | = TNF in TA | (+) TA, EDL (±) GAST, Plantaris | |||
| Dupont-Versteegden, E.E., et al. (1994) [158] | 3 weeks | 37–49 weeks | DIA SOL | ↑ muscle/body weight in SOL = fatigue in SOL | ↑ active tension in DIA ↑ CT in DIA = fatigue in DIA | (+) | ||||||
| Dupont-Versteegden, E.E., et al. (1996) [139] | Male 3 weeks | 9 (A) or 49 (B) weeks | DIA SOL | ↑ muscle/body weight in SOL (A and B) ↑ Myosin in SOL (B) | ↑ active tension of DIA (B) ↑ contraction time of DIA (A) ↑ Fatigability of DIA (A) | = CK | A: (+) B: (+) | |||||
| Ferry, A., et al. (2015) [146] | Male and female 7 months | 3 months | Heart TA | = force drop = specific maximal force ↑ absolute maximal force (only in female) ↑ muscle weight (only in female) | = ejection and shortening fraction = BNP = MHC7 ↑ ANF | = fibrosis in Heart = Col 1a1 = Col 3a1 = CTFG = TGFβ1 | (+) TA in female (±) Heart | |||||
| Gordon, B.S., et al. (2014) [140] | Male and female 4 weeks | 12 weeks | QUAD SOL | = CSA in QUAD | ↑ CLN in QUAD ↑ Utrn in QUAD Trend ↑ Utrn in SOL | Trend ↓ CD45+ | (+) | |||||
| Hayes, A., et al. (1996) [142] | Male 4 weeks | 16 weeks | EDL SOL | ↑ absolute and relative muscle mass of SOL ↑ absolute twitch of SOL ↑ tetanic tension of SOL ↑ Fatigue resistance of EDL and SOL ↑ Type IIa fibers in EDL ↓ Type IIb fibers in EDL ↑ Type I fibers in SOL ↓ Type IIa fibers in SOL | (+) SOL, EDL | |||||||
| Hourdé, C., et al. (2013) [143] | Female 4–5 weeks | 4.5 months | Heart TA | ↑ Absolute maximal force ↑ Muscle weight ↓ susceptibility to contraction induced-injury = MHC1, MHC2x, MHC2b ↑ MHC2a mRNA but not protein ↓ ACTG1 | ↓ ejection and shortening fraction ↓ LVPWT ↑ LVEDD | = regeneration (= MHC, eMHC, IL6, Myog, MyoD, CyclinD1) | = fibrosis (= Col 1α1, TGFβ1, PKC) | = BNIP3, LC3, Atrogin = Mstn, Fst, REDD1, REDD2 = PGC1-α = Sirt1 = SERCA2 | = NFAT | (+) TA (−) Heart | ||
| Hulmi, J.J., et al. (2013) [282] | Male 6–7 weeks | 7 weeks | EDL, GAST MQF SOL TA | = weight mass | = CK in GAST | ↑ PGC-1α ↑ LC3 ↑ CS ↑ Cyt-C ↑ SDH in GAST | = TNFα in GAST | (+) GAST | ||||
| Hulmi, J.J., et al. (2016) [284] | Male 6–7 weeks | 7 weeks | GAST | = grip strenght, Shift to smaller fibers | ↑ p-Sirt1 = pAMPKα at Thr172 = AMPK = p-AMPK/AMPK = PDI = IRE1α = pERK = eIF2α = p-eIF2α = GRP78 = TxNIP = TRX = Hsp70 = Hsp60 = Hsp90 = Hsp47 = Hsp25 | ↑ protein carbonyl levels ↑ GSSG ↑ GSSG/GSH ↑ TPOR = GSH, GRD, GST = GPX | (−) | |||||
| Hunt, L.C., et al. (2011) [150] | Male 10 weeks | 2 weeks | QUAD | ↑ necrosis = LIFR = LIF = IL-6 | (−) | |||||||
| Kogelman, B., et al. (2018) [159] | Female 15 months | 3–4 days/week, 8 weeks | Anterior and posterior hindlimb Heart QUAD | Left ventricular functions: = Heart/body mass = ejection fraction = end systolic volume = stroke volume = cardiac output Right ventricular functions: ↑ end systolic volume Trend ↓ ejection fraction = stroke volume = cardiac output | = fibrosis = Col 1a1 = Lox | = Lgals3 = CD68 | (±) | |||||
| Landisch, M.R., et al. (2008) [138] | Male and female 4 weeks | 8 weeks | EDL SOL TA | ↑ muscle mass in SOL ↑ type IIa fiber and ↓ type IIb fibers in EDL ↓ large fibers in SOL and EDL | = CNF in EDL and SOL = eMHC in SOL | ↑ CS activity in EDL = CS activity in SOL and TA = CCO activity in EDL, SOL and TA = β-HAD activity in TA | (+) EDL, SOL (±) TA | |||||
| Lim, J.H., et al. (2004) [151] | 28 days | 12 h/day, 8 weeks | SOL | ↑ degeneration (↑ apoptotic myonuclei, ↑ Bax, ↓ Bcl2) | (−) | |||||||
| Selsby, J.T., et al. (2013) [147] | Male 4 weeks | 1 year | DIA EDL GAST Heart QUAD SOL TA | ↑ absolute mass of GAST and SOL = absolute mass of EDL, TA, QUAD = specific tension in EDL and soleus ↑ CSA and tetanic force in SOL | ↓ specific tension | ↑ Heart mass ↑ LVDd and LVDs ↑ LVEDV ↑ LVESV ↑stroke volume | (+) SOL, GAST, Heart (±) EDL, TA, QUAD (−) DIA | |||||
| Smythe, G.M., et al. (2012) [148] | Male 10–12 weeks | 2 weeks | GAST QUAD | = CSA | ↑ necrosis in QUAD ↑ CNF in QUAD | (−) | ||||||
| Wineinger, M.A., et al. (1998) [145] | 6 months | 11 months | EDL, SOL | ↑ muscle mass in SOL ↑ fatigue resistance in EDL | (+) | |||||||
| B. Acute exercise | ||||||||||||
| Reference | Sex/Age | Protocol | Muscle | Hindlimb Muscle (Morphological and Functional Changes) | Respiratory Function | Cardiac Function | Degeneration/ Regeneration | Fibrosis | Metabolic Adaptive Changes | Inflammatory Markers | Oxidant/ Antioxidant Markers | Exercise Muscle Effects |
| Archer, J.D., et al. (2006) [157] | 4 weeks | 24 h | DIA GAST QUAD TA | ↑ fiber damage in DIA, QUAD, GAST, TA ↑ Myf5 in QUAD | (−) | |||||||
| Sandri, M., et al. (1995) [155] | 4 weeks | 16 h | EDL SOL TA | ↑ DNA Fragmentation ↑ apoptotic myonuclei ↑ ubiquitinated protein | (−) | |||||||
| Sandri, M., et al. (1997) [156] | 4 weeks | 16 h | TA | ↑ DNA Fragmentation ↑ apoptotic myonuclei ↑ ubiquitinated protein ↓ Bcl2 = BAG1 = FASL | (−) | |||||||
| Reference | Sex/Age | Protocol | Muscle | Low Intensity Treadmill Outcomes |
|---|---|---|---|---|
| Fernandes, D.C., et al. (2019) [73] | 11 weeks | 9 m/min, 30 min/day, 3 days/week, 60 days | GAST TA | Reduces collagen deposition and induces a modulation of the redox status. |
| Fontana, S., et al. (2015) [110] | Male 8 weeks | Pre-training: 3.2 m/min, 15 to 30 min/day, 5 days/week, 2 weeks Training: workload progressive increase, 4 to 4.8 m/min, 30 to 60 min/day, 5 days/week, 4 weeks | QUAD | Contributes to reduce cell degeneration process, by counteracting oxidative stress. |
| Frinchi, M., et al. (2013) [77] | Male 8 weeks | Pre-training: 3.2 m/min, 15 to 30 min/day, 5 days/week, 2 weeks Training: workload progressive increase, 4 to 4.8 m/min, 30 to 60 min/day, 5 days/week, 4 weeks | GAST QUAD | Induces a strong beneficial effect on the degeneration-regeneration process. |
| Gaiad, T.P., et al (2017) [74] | Male 8 weeks | 9 m/min, 30 min/day, 3 days/week, 60 days | TA | Induces adaptations in extracellular matrix, increasing elasticity of dystrophic muscle tissue and delaying fibrosis deposition. |
| Hoepers, A., et al. (2020) [108] | Male 28 days | Pre training: 7 days at 4 m/min Training: 6 or 9 m/min, 30 min/day, 2 days/week, 8 weeks | GAST | Reverses lipid and protein damage and increases the antioxidant activity. |
| Kaczor, J.J., et al. (2007) [109] | 28 days | 9 m/min, 30 min/day, 2 days/week, 8 weeks | GAST EDL SOL | Decreases oxidative stress markers in white muscle. |
| Morici, G., et al. (2017) [76] | Male 8 weeks | Pre-training: 3.2 m/min, 15 to 30 min/day, 5 days/week, 2 weeks Training: workload progressive increase, 4 to 4.8 m/min, 30 to 60 min/day, 5 days/week, 4 weeks | DIA GAST QUAD | Induces a trend for regeneration areas to be larger than necrosis areas in diaphragm and modulates the inflammatory status of hindlimb muscle. |
| Pinto, P.A.F., et al. (2018) [75] | Male 11 weeks | 9 m/min, 30 min/day, 3 days/week, 8 weeks | Lateral GAST | Reduces intramuscular fibrosis deposition and does not exacerbates markers of muscle injury. |
| Zelikovich, A.S., et al. (2019) [79] | 4–5 months | 4 m/min or 8 m/min, 30 min/day, 3 days/week, 6 months (Each exercise session consisted of 7’30’’ warm up followed by a 22’30’’ training at target speed) | DIA GAST Heart QUAD TA | Improves tetanic and specific force in TA muscle, increases respiratory capacity, attenuates cardiac decline associated with disease progression, increases adiponectin and reduces adipocyte cross sectional area, and induces a modest increase in expression of the PGC-1α gene in the gastrocnemius muscle. |
References
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Motohashi, Y.; Miyatake, S.; Komaki, H.; Takeda, S.; Aoki, Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: From discovery to clinical trials. Am. J. Transl. Res. 2016, 8, 2471–2489. [Google Scholar] [PubMed]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-induced skeletal muscle remodeling and metabolic adaptation: Redox signaling and role of autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef] [PubMed]
- Grange, R.W.; Call, J.A. Recommendations to define exercise prescription for Duchenne muscular dystrophy. Exerc. Sport Sci. Rev. 2007, 35, 12–17. [Google Scholar] [CrossRef]
- Markert, C.D.; Case, L.E.; Carter, G.T.; Furlong, P.A.; Grange, R.W. Exercise and Duchenne muscular dystrophy: Where we have been and where we need to go. Muscle Nerve 2012, 45, 746–751. [Google Scholar] [CrossRef]
- Franchi, M.V.; Reeves, N.D.; Narici, M.V. Skeletal Muscle Remodeling in Response to Eccentric vs. Concentric Loading: Morphological, Molecular, and Metabolic Adaptations. Front. Physiol. 2017, 8, 447. [Google Scholar] [CrossRef] [Green Version]
- McGlory, C.; Devries, M.C.; Phillips, S.M. Skeletal muscle and resistance exercise training; the role of protein synthesis in recovery and remodeling. J. Appl. Physiol. 2017, 122, 541–548. [Google Scholar] [CrossRef]
- Hyzewicz, J.; Ruegg, U.T.; Takeda, S. Comparison of Experimental Protocols of Physical Exercise for mdx Mice and Duchenne Muscular Dystrophy Patients. J. Neuromuscul. Dis. 2015, 2, 325–342. [Google Scholar] [CrossRef] [Green Version]
- Spaulding, H.R.; Selsby, J.T. Is Exercise the Right Medicine for Dystrophic Muscle? Med. Sci. Sports Exerc. 2018, 50, 1723–1732. [Google Scholar] [CrossRef]
- Gianola, S.; Pecoraro, V.; Lambiase, S.; Gatti, R.; Banfi, G.; Moja, L. Efficacy of muscle exercise in patients with muscular dystrophy: A systematic review showing a missed opportunity to improve outcomes. PLoS ONE 2013, 8, e65414. [Google Scholar] [CrossRef]
- Kostek, M. Precision Medicine and Exercise Therapy in Duchenne Muscular Dystrophy. Sports 2019, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- Kostek, M.C.; Gordon, B. Exercise Is an Adjuvant to Contemporary Dystrophy Treatments. Exerc. Sport Sci. Rev. 2018, 46, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Louboutin, J.P.; Fichter-Gagnepain, V.; Thaon, E.; Fardeau, M. Morphometric analysis of mdx diaphragm muscle fibres. Comparison with hindlimb muscles. Neuromuscul. Disord. 1993, 3, 463–469. [Google Scholar] [CrossRef]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifiro, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Schill, K.E.; Altenberger, A.R.; Lowe, J.; Periasamy, M.; Villamena, F.A.; Rafael-Fortney, J.A.; Devor, S.T. Muscle damage, metabolism, and oxidative stress in mdx mice: Impact of aerobic running. Muscle Nerve 2016, 54, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Van Putten, M.; Hulsker, M.; Nadarajah, V.D.; van Heiningen, S.H.; van Huizen, E.; van Iterson, M.; Admiraal, P.; Messemaker, T.; den Dunnen, J.T.; ’t Hoen, P.A.; et al. The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS ONE 2012, 7, e31937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrillo, S.; Pelosi, L.; Piemonte, F.; Travaglini, L.; Forcina, L.; Catteruccia, M.; Petrini, S.; Verardo, M.; D’Amico, A.; Musaro, A.; et al. Oxidative stress in Duchenne muscular dystrophy: Focus on the NRF2 redox pathway. Hum. Mol. Genet. 2017, 26, 2781–2790. [Google Scholar] [CrossRef]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef]
- Dhawan, J.; Rando, T.A. Stem cells in postnatal myogenesis: Molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 2005, 15, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Snijders, T.; Nederveen, J.P.; McKay, B.R.; Joanisse, S.; Verdijk, L.B.; van Loon, L.J.; Parise, G. Satellite cells in human skeletal muscle plasticity. Front. Physiol. 2015, 6, 283. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kwak, H.B.; Thompson, L.V.; Lawler, J.M. Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar] [PubMed]
- Smith, L.R.; Hammers, D.W.; Sweeney, H.L.; Barton, E.R. Increased collagen cross-linking is a signature of dystrophin-deficient muscle. Muscle Nerve 2016, 54, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, B.; Karpati, G.; Carpenter, S. Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J. Neurol. Sci. 1990, 100, 9–13. [Google Scholar] [CrossRef]
- Brussee, V.; Tardif, F.; Tremblay, J.P. Muscle fibers of mdx mice are more vulnerable to exercise than those of normal mice. Neuromuscul. Disord. 1997, 7, 487–492. [Google Scholar] [CrossRef]
- Ricotti, V.; Ridout, D.A.; Muntoni, F. Steroids in Duchenne muscular dystrophy. Neuromuscul. Disord. 2013, 23, 696–697. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules 2015, 20, 18168–18184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Westering, T.L.; Betts, C.A.; Wood, M.J. Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy. Molecules 2015, 20, 8823–8855. [Google Scholar] [CrossRef]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes 2020, 11, 837. [Google Scholar] [CrossRef]
- Fairclough, R.J.; Wood, M.J.; Davies, K.E. Therapy for Duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat. Rev. Genet. 2013, 14, 373–378. [Google Scholar] [CrossRef]
- Iftikhar, M.; Frey, J.; Shohan, M.J.; Malek, S.; Mousa, S.A. Current and emerging therapies for Duchenne muscular dystrophy and spinal muscular atrophy. Pharmacol. Ther. 2020, 107719. [Google Scholar] [CrossRef]
- Voet, N.B.; van der Kooi, E.L.; van Engelen, B.G.; Geurts, A.C. Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst. Rev. 2019, 12, CD003907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yucel, N.; Chang, A.C.; Day, J.W.; Rosenthal, N.; Blau, H.M. Humanizing the mdx mouse model of DMD: The long and the short of it. NPJ Regen. Med. 2018, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Putten, M.; Lloyd, E.M.; de Greef, J.C.; Raz, V.; Willmann, R.; Grounds, M.D. Mouse models for muscular dystrophies: An overview. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [Green Version]
- Bulfield, G.; Siller, W.G.; Wight, P.A.; Moore, K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA 1984, 81, 1189–1192. [Google Scholar] [CrossRef] [Green Version]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kelly, A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef]
- Chamberlain, J.S.; Metzger, J.; Reyes, M.; Townsend, D.; Faulkner, J.A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007, 21, 2195–2204. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Long, C.; Yue, Y.; Duan, D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum. Mol. Genet. 2009, 18, 1209–1220. [Google Scholar] [CrossRef] [Green Version]
- Collins, C.A.; Morgan, J.E. Duchenne’s muscular dystrophy: Animal models used to investigate pathogenesis and develop therapeutic strategies. Int. J. Exp. Pathol. 2003, 84, 165–172. [Google Scholar] [CrossRef]
- Duddy, W.; Duguez, S.; Johnston, H.; Cohen, T.V.; Phadke, A.; Gordish-Dressman, H.; Nagaraju, K.; Gnocchi, V.; Low, S.; Partridge, T. Muscular dystrophy in the mdx mouse is a severe myopathy compounded by hypotrophy, hypertrophy and hyperplasia. Skelet. Muscle 2015, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Torres, L.F.; Duchen, L.W. The mutant mdx: Inherited myopathy in the mouse: Morphological studies of nerves, muscles and end-plates. Brain 1987, 110(Pt. 2), 269–299. [Google Scholar] [CrossRef]
- Spencer, M.J.; Montecino-Rodriguez, E.; Dorshkind, K.; Tidball, J.G. Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin. Immunol. 2001, 98, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Sussman, M. Duchenne muscular dystrophy. J. Am. Acad. Orthop. Surg. 2002, 10, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Hartel, J.V.; Granchelli, J.A.; Hudecki, M.S.; Pollina, C.M.; Gosselin, L.E. Impact of prednisone on TGF-beta1 and collagen in diaphragm muscle from mdx mice. Muscle Nerve 2001, 24, 428–432. [Google Scholar] [CrossRef]
- Dangain, J.; Vrbova, G. Muscle development in mdx mutant mice. Muscle Nerve 1984, 7, 700–704. [Google Scholar] [CrossRef]
- Coulton, G.R.; Curtin, N.A.; Morgan, J.E.; Partridge, T.A. The mdx mouse skeletal muscle myopathy: II. Contractile properties. Neuropathol. Appl. Neurobiol. 1988, 14, 299–314. [Google Scholar] [CrossRef]
- Coulton, G.R.; Morgan, J.E.; Partridge, T.A.; Sloper, J.C. The mdx mouse skeletal muscle myopathy: I. A histological, morphometric and biochemical investigation. Neuropathol. Appl. Neurobiol. 1988, 14, 53–70. [Google Scholar] [CrossRef]
- Bostick, B.; Yue, Y.; Long, C.; Duan, D. Prevention of dystrophin-deficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ. Res. 2008, 102, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Bostick, B.; Yue, Y.; Long, C.; Marschalk, N.; Fine, D.M.; Chen, J.; Duan, D. Cardiac expression of a mini-dystrophin that normalizes skeletal muscle force only partially restores heart function in aged Mdx mice. Mol. Ther. 2009, 17, 253–261. [Google Scholar] [CrossRef]
- Hakim, C.H.; Grange, R.W.; Duan, D. The passive mechanical properties of the extensor digitorum longus muscle are compromised in 2- to 20-mo-old mdx mice. J. Appl. Physiol. 2011, 110, 1656–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefaucheur, J.P.; Pastoret, C.; Sebille, A. Phenotype of dystrophinopathy in old mdx mice. Anat. Rec. 1995, 242, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Lynch, G.S.; Hinkle, R.T.; Chamberlain, J.S.; Brooks, S.V.; Faulkner, J.A. Force and power output of fast and slow skeletal muscles from mdx mice 6-28 months old. J. Physiol. 2001, 535, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Pastoret, C.; Sebille, A. mdx mice show progressive weakness and muscle deterioration with age. J. Neurol. Sci. 1995, 129, 97–105. [Google Scholar] [CrossRef]
- Fluck, M. Functional, structural and molecular plasticity of mammalian skeletal muscle in response to exercise stimuli. J. Exp. Biol. 2006, 209, 2239–2248. [Google Scholar] [CrossRef] [Green Version]
- Liguori, G. ACSM’s Guidelines for Exercise Testing and Prescription; ACSM: Indianapolis, IN, USA, 2021. [Google Scholar]
- Jetté, M.; Sidney, K.; Blümchen, G. Metabolic equivalents (METS) in exercise testing, exercise prescription, and evaluation of functional capacity. Clin. Cardiol. 1990, 13, 555–565. [Google Scholar] [CrossRef]
- Ainsworth, B.E.; Haskell, W.L.; Herrmann, S.D.; Meckes, N.; Bassett, D.R., Jr.; Tudor-Locke, C.; Greer, J.L.; Vezina, J.; Whitt-Glover, M.C.; Leon, A.S. 2011 Compendium of Physical Activities: A second update of codes and MET values. Med. Sci. Sports Exerc. 2011, 43, 1575–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pette, D. The adaptive potential of skeletal muscle fibers. Can. J. Appl. Physiol. 2002, 27, 423–448. [Google Scholar] [CrossRef]
- Fluck, M.; Hoppeler, H. Molecular basis of skeletal muscle plasticity—From gene to form and function. Rev. Physiol. Biochem. Pharmacol. 2003, 146, 159–216. [Google Scholar]
- Qaisar, R.; Bhaskaran, S.; Van Remmen, H. Muscle fiber type diversification during exercise and regeneration. Free Radic. Biol. Med. 2016, 98, 56–67. [Google Scholar] [CrossRef]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Kim, H.J.; Lee, W.J.; Seong, J.K. A comparison of the metabolic effects of treadmill and wheel running exercise in mouse model. Lab. Anim. Res. 2020, 36, 3. [Google Scholar] [CrossRef] [Green Version]
- Dehghani, M.; Kargarfard, M.; Rabiee, F.; Nasr-Esfahani, M.H.; Ghaedi, K. A comparative study on the effects of acute and chronic downhill running vs uphill running exercise on the RNA levels of the skeletal muscles PGC1-α, FNDC5 and the adipose UCP1 in BALB/c mice. Gene 2018, 679, 369–376. [Google Scholar] [CrossRef]
- Vernillo, G.; Giandolini, M.; Edwards, W.B.; Morin, J.B.; Samozino, P.; Horvais, N.; Millet, G.Y. Biomechanics and Physiology of Uphill and Downhill Running. Sports Med. 2017, 47, 615–629. [Google Scholar] [CrossRef]
- Kemi, O.J.; Loennechen, J.P.; Wisloff, U.; Ellingsen, O. Intensity-controlled treadmill running in mice: Cardiac and skeletal muscle hypertrophy. J. Appl. Physiol. 2002, 93, 1301–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Huang, Y.; Zhang, Y.; Huang, H.; Hong, S.; Liu, T. Impacts of exercise interventions on different diseases and organ functions in mice. J. Sport Health Sci. 2020, 9, 53–73. [Google Scholar] [CrossRef]
- Yin, X.; Zhao, Y.; Zheng, Y.L.; Wang, J.Z.; Li, W.; Lu, Q.J.; Huang, Q.N.; Zhang, C.Y.; Chen, X.; Ma, J.Z. Time-Course Responses of Muscle-Specific MicroRNAs Following Acute Uphill or Downhill Exercise in Sprague-Dawley Rats. Front. Physiol. 2019, 10, 1275. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.-Y.; Hsu, C.-C.; Su, C.-P.; Lin, C.-F.; Lin, Y.-A.; Lin, C.-L.; Hsu, M.-C. Comparison of lower limb muscle activation during downhill, level and uphill running. Isokinet. Exerc. Sci. 2010, 18, 163–168. [Google Scholar] [CrossRef]
- Zhou, L.; Lu, H. Targeting fibrosis in Duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 2010, 69, 771–776. [Google Scholar] [CrossRef]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Munoz-Canoves, P. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed. Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef]
- Mahdy, M.A.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef]
- Fernandes, D.C.; Cardoso-Nascimento, J.J.A.; Garcia, B.C.C.; Costa, K.B.; Rocha-Vieira, E.; Oliveira, M.X.; Machado, A.S.D.; Santos, A.P.; Gaiad, T.P. Low intensity training improves redox status and reduces collagen fibers on dystrophic muscle. J. Exerc. Rehabil. 2019, 15, 213–223. [Google Scholar] [CrossRef]
- Gaiad, T.P.; Oliveira, M.X.; Lobo, A.R., Jr.; Liborio, L.R.; Pinto, P.A.F.; Fernandes, D.C.; Santos, A.P.; Ambrosio, C.E.; Machado, A.S.D. Low-intensity training provokes adaptive extracellular matrix turnover of a muscular dystrophy model. J. Exerc. Rehabil. 2017, 13, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Pinto, P.A.F.; Machado, A.S.D.; Libório, L.R.; Santos, A.P.; Oliveira, M.X.; Gaiad, T.P. Low Intensity Training Provokes Adaptations on Muscle Fibrosis of a Muscular Dystrophy Model. Int. J. Morphol. 2018, 36, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Morici, G.; Frinchi, M.; Pitruzzella, A.; Di Liberto, V.; Barone, R.; Pace, A.; Di Felice, V.; Belluardo, N.; Cappello, F.; Mudo, G.; et al. Mild Aerobic Exercise Training Hardly Affects the Diaphragm of mdx Mice. J. Cell. Physiol. 2017, 232, 2044–2052. [Google Scholar] [CrossRef]
- Frinchi, M.; Macaluso, F.; Licciardi, A.; Perciavalle, V.; Coco, M.; Belluardo, N.; Morici, G.; Mudo, G. Recovery of damaged skeletal muscle in mdx mice through low-intensity endurance exercise. Int. J. Sports Med. 2014, 35, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zeman, R.J.; Peng, H.; Danon, M.J.; Etlinger, J.D. Clenbuterol reduces degeneration of exercised or aged dystrophic (mdx) muscle. Muscle Nerve 2000, 23, 521–528. [Google Scholar] [CrossRef]
- Zelikovich, A.S.; Quattrocelli, M.; Salamone, I.M.; Kuntz, N.L.; McNally, E.M. Moderate exercise improves function and increases adiponectin in the mdx mouse model of muscular dystrophy. Sci. Rep. 2019, 9, 5770. [Google Scholar] [CrossRef]
- Rocco, A.B.; Levalley, J.C.; Eldridge, J.A.; Marsh, S.A.; Rodgers, B.D. A novel protocol for assessing exercise performance and dystropathophysiology in the mdx mouse. Muscle Nerve 2014, 50, 541–548. [Google Scholar] [CrossRef]
- Pessina, P.; Cabrera, D.; Morales, M.G.; Riquelme, C.A.; Gutierrez, J.; Serrano, A.L.; Brandan, E.; Munoz-Canoves, P. Novel and optimized strategies for inducing fibrosis in vivo: Focus on Duchenne Muscular Dystrophy. Skelet. Muscle 2014, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Morales, M.G.; Cabrera, D.; Cespedes, C.; Vio, C.P.; Vazquez, Y.; Brandan, E.; Cabello-Verrugio, C. Inhibition of the angiotensin-converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN-2). Cell Tissue Res. 2013, 353, 173–187. [Google Scholar] [CrossRef]
- Taniguti, A.P.; Pertille, A.; Matsumura, C.Y.; Santo Neto, H.; Marques, M.J. Prevention of muscle fibrosis and myonecrosis in mdx mice by suramin, a TGF-beta1 blocker. Muscle Nerve 2011, 43, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Yoshida, K.; Ueda, H.; Takeda, S.; Ikeda, S. Up-regulation of mitogen activated protein kinases in mdx skeletal muscle following chronic treadmill exercise. Biochim. Biophys. Acta 2005, 1740, 326–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraysse, B.; Liantonio, A.; Cetrone, M.; Burdi, R.; Pierno, S.; Frigeri, A.; Pisoni, M.; Camerino, C.; De Luca, A. The alteration of calcium homeostasis in adult dystrophic mdx muscle fibers is worsened by a chronic exercise in vivo. Neurobiol. Dis. 2004, 17, 144–154. [Google Scholar] [CrossRef]
- Capogrosso, R.F.; Mantuano, P.; Cozzoli, A.; Sanarica, F.; Massari, A.M.; Conte, E.; Fonzino, A.; Giustino, A.; Rolland, J.F.; Quaranta, A.; et al. Contractile efficiency of dystrophic mdx mouse muscle: In vivo and ex vivo assessment of adaptation to exercise of functional end points. J. Appl. Physiol. 2017, 122, 828–843. [Google Scholar] [CrossRef]
- Radley-Crabb, H.; Terrill, J.; Shavlakadze, T.; Tonkin, J.; Arthur, P.; Grounds, M. A single 30 min treadmill exercise session is suitable for ‘proof-of concept studies’ in adult mdx mice: A comparison of the early consequences of two different treadmill protocols. Neuromuscul. Disord. 2012, 22, 170–182. [Google Scholar] [CrossRef]
- Camerino, G.M.; Cannone, M.; Giustino, A.; Massari, A.M.; Capogrosso, R.F.; Cozzoli, A.; De Luca, A. Gene expression in mdx mouse muscle in relation to age and exercise: Aberrant mechanical-metabolic coupling and implications for pre-clinical studies in Duchenne muscular dystrophy. Hum. Mol. Genet. 2014, 23, 5720–5732. [Google Scholar] [CrossRef] [Green Version]
- Okano, T.; Yoshida, K.; Nakamura, A.; Sasazawa, F.; Oide, T.; Takeda, S.; Ikeda, S. Chronic exercise accelerates the degeneration-regeneration cycle and downregulates insulin-like growth factor-1 in muscle of mdx mice. Muscle Nerve 2005, 32, 191–199. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Pierno, S.; Liantonio, A.; Cetrone, M.; Camerino, C.; Fraysse, B.; Mirabella, M.; Servidei, S.; Ruegg, U.T.; Conte Camerino, D. Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J. Pharmacol. Exp. Ther. 2003, 304, 453–463. [Google Scholar] [CrossRef]
- Mokhtarian, A.; Lefaucheur, J.P.; Even, P.C.; Sebille, A. Effects of treadmill exercise and high-fat feeding on muscle degeneration in mdx mice at the time of weaning. Clin. Sci. 1995, 89, 447–452. [Google Scholar] [CrossRef]
- Burdi, R.; Rolland, J.F.; Fraysse, B.; Litvinova, K.; Cozzoli, A.; Giannuzzi, V.; Liantonio, A.; Camerino, G.M.; Sblendorio, V.; Capogrosso, R.F.; et al. Multiple pathological events in exercised dystrophic mdx mice are targeted by pentoxifylline: Outcome of a large array of in vivo and ex vivo tests. J. Appl. Physiol. 2009, 106, 1311–1324. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.L.; De Repentigny, Y.; Cifelli, C.; Marshall, P.; Renaud, J.M.; Worton, R.G.; Kothary, R. The mouse dystrophin muscle promoter/enhancer drives expression of mini-dystrophin in transgenic mdx mice and rescues the dystrophy in these mice. Mol. Ther. 2006, 14, 724–734. [Google Scholar] [CrossRef]
- Cerri, D.G.; Rodrigues, L.C.; Stowell, S.R.; Araujo, D.D.; Coelho, M.C.; Oliveira, S.R.; Bizario, J.C.; Cummings, R.D.; Dias-Baruffi, M.; Costa, M.C. Degeneration of dystrophic or injured skeletal muscles induces high expression of Galectin-1. Glycobiology 2008, 18, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamberi, T.; Fiaschi, T.; Valocchia, E.; Modesti, A.; Mantuano, P.; Rolland, J.F.; Sanarica, F.; De Luca, A.; Magherini, F. Proteome analysis in dystrophic mdx mouse muscle reveals a drastic alteration of key metabolic and contractile proteins after chronic exercise and the potential modulation by anti-oxidant compounds. J. Proteom. 2018, 170, 43–58. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Nico, B.; Liantonio, A.; Didonna, M.P.; Fraysse, B.; Pierno, S.; Burdi, R.; Mangieri, D.; Rolland, J.F.; Camerino, C.; et al. A multidisciplinary evaluation of the effectiveness of cyclosporine a in dystrophic mdx mice. Am. J. Pathol. 2005, 166, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Bizario, J.C.; Cerri, D.G.; Rodrigues, L.C.; Oliveira, G.L.; Nomizo, A.; de Araujo, D.D.; Fukuhara, P.S.; Ribeiro, J.C.; de Castro, F.A.; Costa, M.C. Imatinib mesylate ameliorates the dystrophic phenotype in exercised mdx mice. J. Neuroimmunol. 2009, 212, 93–101. [Google Scholar] [CrossRef]
- Kobayashi, Y.M.; Rader, E.P.; Crawford, R.W.; Campbell, K.P. Endpoint measures in the mdx mouse relevant for muscular dystrophy pre-clinical studies. Neuromuscul. Disord. 2012, 22, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdi, R.; Didonna, M.P.; Pignol, B.; Nico, B.; Mangieri, D.; Rolland, J.F.; Camerino, C.; Zallone, A.; Ferro, P.; Andreetta, F.; et al. First evaluation of the potential effectiveness in muscular dystrophy of a novel chimeric compound, BN 82270, acting as calpain-inhibitor and anti-oxidant. Neuromuscul. Disord. 2006, 16, 237–248. [Google Scholar] [CrossRef]
- Fowler, W.M., Jr.; Abresch, R.T.; Larson, D.B.; Sharman, R.B.; Entrikin, R.K. High-repetitive submaximal treadmill exercise training: Effect on normal and dystrophic mice. Arch. Phys. Med. Rehabil. 1990, 71, 552–557. [Google Scholar]
- Shadrach, J.L.; Wagers, A.J. Stem cells for skeletal muscle repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2297–2306. [Google Scholar] [CrossRef]
- Schmidt, M.; Schuler, S.C.; Huttner, S.S.; von Eyss, B.; von Maltzahn, J. Adult stem cells at work: Regenerating skeletal muscle. Cell. Mol. Life Sci. 2019, 76, 2559–2570. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Serra, C.; Lee, G.; Wagner, K.R. Stem cell-based therapies for Duchenne muscular dystrophy. Exp. Neurol. 2020, 323, 113086. [Google Scholar] [CrossRef]
- Judson, R.N.; Rossi, F.M.V. Towards stem cell therapies for skeletal muscle repair. NPJ Regen. Med. 2020, 5, 10. [Google Scholar] [CrossRef]
- Lindsay, A.; Larson, A.A.; Verma, M.; Ervasti, J.M.; Lowe, D.A. Isometric resistance training increases strength and alters histopathology of dystrophin-deficient mouse skeletal muscle. J. Appl. Physiol. 2019, 126, 363–375. [Google Scholar] [CrossRef]
- Vita, G.L.; Aguennouz, M.; Sframeli, M.; Sanarica, F.; Mantuano, P.; Oteri, R.; Polito, F.; Licata, N.; Romeo, S.; Distefano, M.G.; et al. Effect of exercise on telomere length and telomere proteins expression in mdx mice. Mol. Cell. Biochem. 2020, 470, 189–197. [Google Scholar] [CrossRef]
- Lund, T.C.; Grange, R.W.; Lowe, D.A. Telomere shortening in diaphragm and tibialis anterior muscles of aged mdx mice. Muscle Nerve 2007, 36, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Hoepers, A.; Alberti, A.; Freiberger, V.; Ventura, L.; Grigollo, L.R.; Andreu, C.S.; da Silva, B.B.; Martins, D.F.; Junior, R.J.N.; Streck, E.L.; et al. Effect of Aerobic Physical Exercise in an Animal Model of Duchenne Muscular Dystrophy. J. Mol. Neurosci. 2020, 70, 1552–1564. [Google Scholar] [CrossRef]
- Kaczor, J.J.; Hall, J.E.; Payne, E.; Tarnopolsky, M.A. Low intensity training decreases markers of oxidative stress in skeletal muscle of mdx mice. Free Radic. Biol. Med. 2007, 43, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Fontana, S.; Schillaci, O.; Frinchi, M.; Giallombardo, M.; Morici, G.; Di Liberto, V.; Alessandro, R.; De Leo, G.; Perciavalle, V.; Belluardo, N.; et al. Reduction in mdx mouse muscle degeneration by low-intensity endurance exercise: A proteomic analysis in quadriceps muscle of exercised compared with sedentary mdx mice. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrill, J.R.; Radley-Crabb, H.G.; Grounds, M.D.; Arthur, P.G. N-Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscul. Disord. 2012, 22, 427–434. [Google Scholar] [CrossRef]
- Quinlan, J.G.; Wong, B.L.; Niemeier, R.T.; McCullough, A.S.; Levin, L.; Emanuele, M. Poloxamer 188 failed to prevent exercise-induced membrane breakdown in mdx skeletal muscle fibers. Neuromuscul. Disord. 2006, 16, 855–864. [Google Scholar] [CrossRef]
- Vilquin, J.T.; Brussee, V.; Asselin, I.; Kinoshita, I.; Gingras, M.; Tremblay, J.P. Evidence of mdx mouse skeletal muscle fragility in vivo by eccentric running exercise. Muscle Nerve 1998, 21, 567–576. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Streamer, M.; Lusambili, L.I.; Sachs, F.; Allen, D.G. Streptomycin reduces stretch-induced membrane permeability in muscles from mdx mice. Neuromuscul. Disord. 2006, 16, 845–854. [Google Scholar] [CrossRef]
- Mathur, S.; Vohra, R.S.; Germain, S.A.; Forbes, S.; Bryant, N.D.; Vandenborne, K.; Walter, G.A. Changes in muscle T2 and tissue damage after downhill running in mdx mice. Muscle Nerve 2011, 43, 878–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.S.; Khakee, R.; McNeil, P.L. Loss of cytoplasmic basic fibroblast growth factor from physiologically wounded myofibers of normal and dystrophic muscle. J. Cell Sci. 1993, 106(Pt. 1), 121–133. [Google Scholar]
- Poche, H.; Hopfenmuller, W.; Hoffmann, M. Detection and identification of myoglobin in serum by immunoblotting. Effect of exercise on patients with Duchenne muscular dystrophy. Clin. Physiol. Biochem. 1987, 5, 103–111. [Google Scholar]
- Nakamura, A.; Yoshida, K.; Takeda, S.; Dohi, N.; Ikeda, S. Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Lett. 2002, 520, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Hermes, T.A.; Kido, L.A.; Macedo, A.B.; Mizobuti, D.S.; Moraes, L.H.R.; Somazz, M.C.; Cagnon, V.H.A.; Minatel, E. Sex influences diaphragm muscle response in exercised mdx mice. Cell Biol. Int. 2018, 42, 1611–1621. [Google Scholar] [CrossRef]
- Grounds, M.D.; Radley, H.G.; Lynch, G.S.; Nagaraju, K.; De Luca, A. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol. Dis. 2008, 31, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Tiidus, P.M. Estrogen and gender effects on muscle damage, inflammation, and oxidative stress. Can. J. Appl. Physiol. 2000, 25, 274–287. [Google Scholar] [CrossRef]
- Tiidus, P.M. Oestrogen and sex influence on muscle damage and inflammation: Evidence from animal models. Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 509–513. [Google Scholar] [CrossRef]
- Tiidus, P.M. Influence of estrogen on skeletal muscle damage, inflammation, and repair. Exerc. Sport Sci. Rev. 2003, 31, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Enns, D.L.; Tiidus, P.M. Estrogen influences satellite cell activation and proliferation following downhill running in rats. J. Appl. Physiol. 2008, 104, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Velders, M.; Schleipen, B.; Fritzemeier, K.H.; Zierau, O.; Diel, P. Selective estrogen receptor-beta activation stimulates skeletal muscle growth and regeneration. FASEB J. 2012, 26, 1909–1920. [Google Scholar] [CrossRef]
- Hall, J.E.; Kaczor, J.J.; Hettinga, B.P.; Isfort, R.J.; Tarnopolsky, M.A. Effects of a CRF2R agonist and exercise on mdx and wildtype skeletal muscle. Muscle Nerve 2007, 36, 336–341. [Google Scholar] [CrossRef]
- Hyzewicz, J.; Tanihata, J.; Kuraoka, M.; Nitahara-Kasahara, Y.; Beylier, T.; Ruegg, U.T.; Vater, A.; Takeda, S. Low-Intensity Training and the C5a Complement Antagonist NOX-D21 Rescue the mdx Phenotype through Modulation of Inflammation. Am. J. Pathol. 2017, 187, 1147–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyzewicz, J.; Tanihata, J.; Kuraoka, M.; Ito, N.; Miyagoe-Suzuki, Y.; Takeda, S. Low intensity training of mdx mice reduces carbonylation and increases expression levels of proteins involved in energy metabolism and muscle contraction. Free Radic. Biol. Med. 2015, 82, 122–136. [Google Scholar] [CrossRef] [Green Version]
- Hayes, A.; Williams, D.A. Contractile function and low-intensity exercise effects of old dystrophic (mdx) mice. Am. J. Physiol. 1998, 274, C1138–C1144. [Google Scholar] [CrossRef]
- Hayes, A.; Lynch, G.S.; Williams, D.A. The effects of endurance exercise on dystrophic mdx mice: I. Contractile and histochemical properties of intact muscles. Proc. Biol. Sci. 1993, 253, 19–25. [Google Scholar]
- Lynch, G.S.; Hayes, A.; Lam, M.H.; Williams, D.A. The effects of endurance exercise on dystrophic mdx mice: II. Contractile properties of skinned muscle fibres. Proc. Biol. Sci. 1993, 253, 27–33. [Google Scholar]
- Matsakas, A.; Yadav, V.; Lorca, S.; Narkar, V. Muscle ERRgamma mitigates Duchenne muscular dystrophy via metabolic and angiogenic reprogramming. FASEB J. 2013, 27, 4004–4016. [Google Scholar] [CrossRef] [PubMed]
- Barbin, I.C.; Pereira, J.A.; Bersan Rovere, M.; de Oliveira Moreira, D.; Marques, M.J.; Santo Neto, H. Diaphragm degeneration and cardiac structure in mdx mouse: Potential clinical implications for Duchenne muscular dystrophy. J. Anat. 2016, 228, 784–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchentouf, M.; Benabdallah, B.F.; Mills, P.; Tremblay, J.P. Exercise improves the success of myoblast transplantation in mdx mice. Neuromuscul. Disord. 2006, 16, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Delacroix, C.; Hyzewicz, J.; Lemaitre, M.; Friguet, B.; Li, Z.; Klein, A.; Furling, D.; Agbulut, O.; Ferry, A. Improvement of Dystrophic Muscle Fragility by Short-Term Voluntary Exercise through Activation of Calcineurin Pathway in mdx Mice. Am. J. Pathol. 2018, 188, 2662–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Call, J.A.; Voelker, K.A.; Wolff, A.V.; McMillan, R.P.; Evans, N.P.; Hulver, M.W.; Talmadge, R.J.; Grange, R.W. Endurance capacity in maturing mdx mice is markedly enhanced by combined voluntary wheel running and green tea extract. J. Appl. Physiol. 2008, 105, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Bueno Jr, C.R.; Pantaleao, L.C.; Voltarelli, V.A.; Bozi, L.H.; Brum, P.C.; Zatz, M. Combined effect of AMPK/PPAR agonists and exercise training in mdx mice functional performance. PLoS ONE 2012, 7, e45699. [Google Scholar] [CrossRef] [Green Version]
- Landisch, R.M.; Kosir, A.M.; Nelson, S.A.; Baltgalvis, K.A.; Lowe, D.A. Adaptive and nonadaptive responses to voluntary wheel running by mdx mice. Muscle Nerve 2008, 38, 1290–1303. [Google Scholar] [CrossRef] [Green Version]
- Dupont-Versteegden, E.E. Exercise and clenbuterol as strategies to decrease the progression of muscular dystrophy in mdx mice. J. Appl. Physiol. 1996, 80, 734–741. [Google Scholar] [CrossRef]
- Gordon, B.S.; Lowe, D.A.; Kostek, M.C. Exercise increases utrophin protein expression in the mdx mouse model of Duchenne muscular dystrophy. Muscle Nerve 2014, 49, 915–918. [Google Scholar] [CrossRef]
- Baltgalvis, K.A.; Call, J.A.; Cochrane, G.D.; Laker, R.C.; Yan, Z.; Lowe, D.A. Exercise training improves plantar flexor muscle function in mdx mice. Med. Sci. Sports Exerc. 2012, 44, 1671–1679. [Google Scholar] [CrossRef] [Green Version]
- Hayes, A.; Williams, D.A. Beneficial effects of voluntary wheel running on the properties of dystrophic mouse muscle. J. Appl. Physiol. 1996, 80, 670–679. [Google Scholar] [CrossRef]
- Hourde, C.; Joanne, P.; Medja, F.; Mougenot, N.; Jacquet, A.; Mouisel, E.; Pannerec, A.; Hatem, S.; Butler-Browne, G.; Agbulut, O.; et al. Voluntary physical activity protects from susceptibility to skeletal muscle contraction-induced injury but worsens heart function in mdx mice. Am. J. Pathol. 2013, 182, 1509–1518. [Google Scholar] [CrossRef]
- Call, J.A.; McKeehen, J.N.; Novotny, S.A.; Lowe, D.A. Progressive resistance voluntary wheel running in the mdx mouse. Muscle Nerve 2010, 42, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Wineinger, M.A.; Abresch, R.T.; Walsh, S.A.; Carter, G.T. Effects of aging and voluntary exercise on the function of dystrophic muscle from mdx mice. Am. J. Phys. Med. Rehabil. 1998, 77, 20–27. [Google Scholar] [CrossRef]
- Ferry, A.; Benchaouir, R.; Joanne, P.; Peat, R.A.; Mougenot, N.; Agbulut, O.; Butler-Browne, G. Effect of voluntary physical activity initiated at age 7 months on skeletal hindlimb and cardiac muscle function in mdx mice of both genders. Muscle Nerve 2015, 52, 788–794. [Google Scholar] [CrossRef]
- Selsby, J.T.; Acosta, P.; Sleeper, M.M.; Barton, E.R.; Sweeney, H.L. Long-term wheel running compromises diaphragm function but improves cardiac and plantarflexor function in the mdx mouse. J. Appl. Physiol. 2013, 115, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Smythe, G.M.; White, J.D. Voluntary wheel running in dystrophin-deficient (mdx) mice: Relationships between exercise parameters and exacerbation of the dystrophic phenotype. PLoS Curr. 2011, 3, RRN1295. [Google Scholar] [CrossRef]
- Coles, C.A.; Gordon, L.; Hunt, L.C.; Webster, T.; Piers, A.T.; Kintakas, C.; Woodman, K.; Touslon, S.L.; Smythe, G.M.; White, J.D.; et al. Expression profiling in exercised mdx suggests a role for extracellular proteins in the dystrophic muscle immune response. Hum. Mol. Genet. 2020, 29, 353–368. [Google Scholar] [CrossRef]
- Hunt, L.C.; Coles, C.A.; Gorman, C.M.; Tudor, E.M.; Smythe, G.M.; White, J.D. Alterations in the expression of leukemia inhibitory factor following exercise: Comparisons between wild-type and mdx muscles. PLoS Curr. 2011, 3, RRN1277. [Google Scholar] [CrossRef]
- Lim, J.H.; Kim, D.Y.; Bang, M.S. Effects of exercise and steroid on skeletal muscle apoptosis in the mdx mouse. Muscle Nerve 2004, 30, 456–462. [Google Scholar] [CrossRef]
- Podhorska-Okolow, M.; Sandri, M.; Zampieri, S.; Brun, B.; Rossini, K.; Carraro, U. Apoptosis of myofibres and satellite cells: Exercise-induced damage in skeletal muscle of the mouse. Neuropathol. Appl. Neurobiol. 1998, 24, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Brereton, D.; Plochocki, J.; An, D.; Costas, J.; Simons, E. The effects of glucocorticoid and voluntary exercise treatment on the development of thoracolumbar kyphosis in dystrophin-deficient mice. PLoS Curr. 2012, 4, e4ffdff160de168b. [Google Scholar] [CrossRef] [PubMed]
- DiMario, J.X.; Uzman, A.; Strohman, R.C. Fiber regeneration is not persistent in dystrophic (MDX) mouse skeletal muscle. Dev. Biol. 1991, 148, 314–321. [Google Scholar] [CrossRef]
- Sandri, M.; Carraro, U.; Podhorska-Okolov, M.; Rizzi, C.; Arslan, P.; Monti, D.; Franceschi, C. Apoptosis, DNA damage and ubiquitin expression in normal and mdx muscle fibers after exercise. FEBS Lett. 1995, 373, 291–295. [Google Scholar] [CrossRef]
- Sandri, M.; Podhorska-Okolow, M.; Geromel, V.; Rizzi, C.; Arslan, P.; Franceschi, C.; Carraro, U. Exercise induces myonuclear ubiquitination and apoptosis in dystrophin-deficient muscle of mice. J. Neuropathol. Exp. Neurol. 1997, 56, 45–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, J.D.; Vargas, C.C.; Anderson, J.E. Persistent and improved functional gain in mdx dystrophic mice after treatment with L-arginine and deflazacort. FASEB J. 2006, 20, 738–740. [Google Scholar] [CrossRef]
- Dupont-Versteegden, E.E.; McCarter, R.J.; Katz, M.S. Voluntary exercise decreases progression of muscular dystrophy in diaphragm of mdx mice. J. Appl. Physiol. 1994, 77, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Kogelman, B.; Putker, K.; Hulsker, M.; Tanganyika-de Winter, C.; van der Weerd, L.; Aartsma-Rus, A.; van Putten, M. Voluntary exercise improves muscle function and does not exacerbate muscle and heart pathology in aged Duchenne muscular dystrophy mice. J. Mol. Cell. Cardiol. 2018, 125, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Costas, J.M.; Nye, D.J.; Henley, J.B.; Plochocki, J.H. Voluntary exercise induces structural remodeling in the hearts of dystrophin-deficient mice. Muscle Nerve 2010, 42, 881–885. [Google Scholar] [CrossRef]
- Chinet, A.E.; Even, P.C.; Decrouy, A. Dystrophin-dependent efficiency of metabolic pathways in mouse skeletal muscles. Experientia 1994, 50, 602–605. [Google Scholar] [CrossRef]
- Van Bennekom, C.A.; Oerlemans, F.T.; Kulakowski, S.; De Bruyn, C.H. Enzymes of purine metabolism in muscle specimens from patients with Duchenne-type muscular dystrophy. Adv. Exp. Med. Biol. 1984, 165(Pt. B), 447–450. [Google Scholar]
- Ge, Y.; Molloy, M.P.; Chamberlain, J.S.; Andrews, P.C. Proteomic analysis of mdx skeletal muscle: Great reduction of adenylate kinase 1 expression and enzymatic activity. Proteomics 2003, 3, 1895–1903. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.R.; von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell. Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.F.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef] [Green Version]
- Gaglianone, R.B.; Bloise, F.F.; Ortiga-Carvalho, T.M.; Quirico-Santos, T.; Costa, M.L.; Mermelstein, C. Comparative study of calcium and calcium-related enzymes with differentiation markers in different ages and muscle types in mdx mice. Histol. Histopathol. 2020, 35, 203–216. [Google Scholar]
- Blanchet, E.; Annicotte, J.S.; Pradelli, L.A.; Hugon, G.; Matecki, S.; Mornet, D.; Rivier, F.; Fajas, L. E2F transcription factor-1 deficiency reduces pathophysiology in the mouse model of Duchenne muscular dystrophy through increased muscle oxidative metabolism. Hum. Mol. Genet. 2012, 21, 3910–3917. [Google Scholar] [CrossRef]
- Strakova, J.; Kamdar, F.; Kulhanek, D.; Razzoli, M.; Garry, D.J.; Ervasti, J.M.; Bartolomucci, A.; Townsend, D. Integrative effects of dystrophin loss on metabolic function of the mdx mouse. Sci. Rep. 2018, 8, 13624. [Google Scholar] [CrossRef] [Green Version]
- Cole, M.A.; Rafael, J.A.; Taylor, D.J.; Lodi, R.; Davies, K.E.; Styles, P. A quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscul. Disord. 2002, 12, 247–257. [Google Scholar] [CrossRef]
- Percival, J.M.; Whitehead, N.P.; Adams, M.E.; Adamo, C.M.; Beavo, J.A.; Froehner, S.C. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J. Pathol. 2012, 228, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of Inherited Muscular Dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar] [PubMed] [Green Version]
- Powers, S.K.; Talbert, E.E.; Adhihetty, P.J. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J. Physiol. 2011, 589, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J. On the mechanisms underlying attenuated redox responses to exercise in older individuals: A hypothesis. Free Radic. Biol. Med. 2020, 161, 326–338. [Google Scholar] [CrossRef]
- Scicchitano, B.M.; Pelosi, L.; Sica, G.; Musaro, A. The physiopathologic role of oxidative stress in skeletal muscle. Mech. Ageing Dev. 2018, 170, 37–44. [Google Scholar] [CrossRef]
- Disatnik, M.H.; Dhawan, J.; Yu, Y.; Beal, M.F.; Whirl, M.M.; Franco, A.A.; Rando, T.A. Evidence of oxidative stress in mdx mouse muscle: Studies of the pre-necrotic state. J. Neurol. Sci. 1998, 161, 77–84. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Yeung, E.W.; Allen, D.G. Muscle damage in mdx (dystrophic) mice: Role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2006, 33, 657–662. [Google Scholar] [CrossRef]
- Shkryl, V.M.; Martins, A.S.; Ullrich, N.D.; Nowycky, M.C.; Niggli, E.; Shirokova, N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflügers Arch. 2009, 458, 915–928. [Google Scholar] [CrossRef]
- Renjini, R.; Gayathri, N.; Nalini, A.; Srinivas Bharath, M.M. Oxidative damage in muscular dystrophy correlates with the severity of the pathology: Role of glutathione metabolism. Neurochem. Res. 2012, 37, 885–898. [Google Scholar] [CrossRef]
- Guiraud, S.; Davies, K.E. Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.H.; Ow, J.R.; Yang, N.D.; Taneja, R. Oxidative Stress-Mediated Skeletal Muscle Degeneration: Molecules, Mechanisms, and Therapies. Oxid. Med. Cell. Longev. 2016, 2016, 6842568. [Google Scholar] [CrossRef] [Green Version]
- Kar, N.C.; Pearson, C.M. Catalase, superoxide dismutase, glutathione reductase and thiobarbituric acid-reactive products in normal and dystrophic human muscle. Clin. Chim. Acta 1979, 94, 277–280. [Google Scholar] [CrossRef]
- Rodriguez, M.C.; Tarnopolsky, M.A. Patients with dystrophinopathy show evidence of increased oxidative stress. Free Radic. Biol. Med. 2003, 34, 1217–1220. [Google Scholar] [CrossRef]
- Almeida-Becerril, T.; Rodriguez-Cruz, M.; Sanchez-Gonzalez, J.R.; Villaldama-Soriano, M.A.; Atilano-Miguel, S.; Villa-Morales, J.; Cardenas-Conejo, A.; Cardenas-Vazquez, R. Circulating markers of oxidative stress are associated with a muscle injury in patients with muscular dystrophy Duchenne. Brain Dev. 2021, 43, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Souza, L.B.; Maziero, C.; Lazzarin, M.C.; Quintana, H.T.; Tome, T.C.; Baptista, V.I.A.; de Oliveira, F. Presence of metalloproteinases 2 and 9 and 8-OHdG in the fibrotic process in skeletal muscle of Mdx mice. Acta Histochem. 2020, 122, 151458. [Google Scholar] [CrossRef]
- El-Shafey, A.F.; Armstrong, A.E.; Terrill, J.R.; Grounds, M.D.; Arthur, P.G. Screening for increased protein thiol oxidation in oxidatively stressed muscle tissue. Free Radic. Res. 2011, 45, 991–999. [Google Scholar] [CrossRef]
- Terrill, J.R.; Radley-Crabb, H.G.; Iwasaki, T.; Lemckert, F.A.; Arthur, P.G.; Grounds, M.D. Oxidative stress and pathology in muscular dystrophies: Focus on protein thiol oxidation and dysferlinopathies. FEBS J. 2013, 280, 4149–4164. [Google Scholar] [CrossRef]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef]
- Percival, J.M.; Siegel, M.P.; Knowels, G.; Marcinek, D.J. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet. 2013, 22, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [Green Version]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; Van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef] [Green Version]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Ramadasan-Nair, R.; Gayathri, N.; Mishra, S.; Sunitha, B.; Mythri, R.B.; Nalini, A.; Subbannayya, Y.; Harsha, H.C.; Kolthur-Seetharam, U.; Srinivas Bharath, M.M. Mitochondrial alterations and oxidative stress in an acute transient mouse model of muscle degeneration: Implications for muscular dystrophy and related muscle pathologies. J. Biol. Chem. 2014, 289, 485–509. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Menazza, S.; Blaauw, B.; Tiepolo, T.; Toniolo, L.; Braghetta, P.; Spolaore, B.; Reggiani, C.; Di Lisa, F.; Bonaldo, P.; Canton, M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet. 2010, 19, 4207–4215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, T.M.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in mdx Mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Edgett, B.A.; Huber, J.S.; Polidovitch, N.; Schlattner, U.; Backx, P.H.; Simpson, J.A.; Perry, C.G.R. Impairments in left ventricular mitochondrial bioenergetics precede overt cardiac dysfunction and remodelling in Duchenne muscular dystrophy. J. Physiol. 2020, 598, 1377–1392. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, V.E.; Van Der Meulen, J.H.; Johnston, H.K.; Ghimbovschi, S.; Partridge, T.; Hoffman, E.P.; Nagaraju, K. Metabolic remodeling agents show beneficial effects in the dystrophin-deficient mdx mouse model. Skelet Muscle 2012, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selsby, J.T.; Morine, K.J.; Pendrak, K.; Barton, E.R.; Sweeney, H.L. Rescue of dystrophic skeletal muscle by PGC-1alpha involves a fast to slow fiber type shift in the mdx mouse. PLoS ONE 2012, 7, e30063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.C.; Rowe, G.C.; Raghuram, S.; Patten, I.S.; Farrell, C.; Arany, Z. Post-natal induction of PGC-1alpha protects against severe muscle dystrophy independently of utrophin. Skelet Muscle 2014, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases in mechano-transduction: Mechanisms and consequences. Antioxid. Redox Signal. 2014, 20, 887–898. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.F.; Laitano, O. Regulation of NADPH oxidases in skeletal muscle. Free Radic. Biol. Med. 2016, 98, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Sakellariou, G.K.; Jackson, M.J.; Vasilaki, A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic. Res. 2014, 48, 12–29. [Google Scholar] [CrossRef]
- Ward, C.W.; Prosser, B.L.; Lederer, W.J. Mechanical stretch-induced activation of ROS/RNS signaling in striated muscle. Antioxid. Redox Signal. 2014, 20, 929–936. [Google Scholar] [CrossRef]
- Cherednichenko, G.; Zima, A.V.; Feng, W.; Schaefer, S.; Blatter, L.A.; Pessah, I.N. NADH oxidase activity of rat cardiac sarcoplasmic reticulum regulates calcium-induced calcium release. Circ. Res. 2004, 94, 478–486. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, C.; Sanchez, G.; Barrientos, G.; Aracena-Parks, P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S -glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Bey, E.A.; Wientjes, F.B.; Cathcart, M.K. Cytosolic phospholipase A2 (cPLA2) regulation of human monocyte NADPH oxidase activity. cPLA2 affects translocation but not phosphorylation of p67(phox) and p47(phox). J. Biol. Chem. 2002, 277, 25385–25392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, E.; Sestili, P. Reactive oxygen species in skeletal muscle signaling. J. Signal Transduct. 2012, 2012, 982794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef]
- Prosser, B.L.; Khairallah, R.J.; Ziman, A.P.; Ward, C.W.; Lederer, W.J. X-ROS signaling in the heart and skeletal muscle: Stretch-dependent local ROS regulates [Ca(2+)]i. J. Mol. Cell. Cardiol. 2013, 58, 172–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsay, A.; McCourt, P.M.; Karachunski, P.; Lowe, D.A.; Ervasti, J.M. Xanthine oxidase is hyper-active in Duchenne muscular dystrophy. Free Radic. Biol. Med. 2018, 129, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, N.P.; Kim, M.J.; Bible, K.L.; Adams, M.E.; Froehner, S.C. A new therapeutic effect of simvastatin revealed by functional improvement in muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 12864–12869. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv294. [Google Scholar] [CrossRef] [Green Version]
- Tidball, J.G. Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol. 2011, 1, 2029–2062. [Google Scholar]
- Kozakowska, M.; Pietraszek-Gremplewicz, K.; Jozkowicz, A.; Dulak, J. The role of oxidative stress in skeletal muscle injury and regeneration: Focus on antioxidant enzymes. J. Muscle Res. Cell Motil. 2015, 36, 377–393. [Google Scholar] [CrossRef] [Green Version]
- Williams, I.A.; Allen, D.G. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1969–H1977. [Google Scholar] [CrossRef] [Green Version]
- Damiano, S.; Muscariello, E.; La Rosa, G.; Di Maro, M.; Mondola, P.; Santillo, M. Dual Role of Reactive Oxygen Species in Muscle Function: Can Antioxidant Dietary Supplements Counteract Age-Related Sarcopenia? Int. J. Mol. Sci. 2019, 20, 3815. [Google Scholar] [CrossRef] [Green Version]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef] [Green Version]
- Loehr, J.A.; Abo-Zahrah, R.; Pal, R.; Rodney, G.G. Sphingomyelinase promotes oxidant production and skeletal muscle contractile dysfunction through activation of NADPH oxidase. Front. Physiol. 2014, 5, 530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, R.; Basu Thakur, P.; Li, S.; Minard, C.; Rodney, G.G. Real-time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS ONE 2013, 8, e63989. [Google Scholar] [CrossRef]
- Powers, S.K. Can antioxidants protect against disuse muscle atrophy? Sports Med. 2014, 44 (Suppl. 2), S155–S165. [Google Scholar] [CrossRef] [Green Version]
- Hollander, J.; Fiebig, R.; Gore, M.; Bejma, J.; Ookawara, T.; Ohno, H.; Ji, L.L. Superoxide dismutase gene expression in skeletal muscle: Fiber-specific adaptation to endurance training. Am. J. Physiol. 1999, 277, R856–R862. [Google Scholar] [CrossRef]
- Powers, S.K.; Criswell, D.; Lawler, J.; Ji, L.L.; Martin, D.; Herb, R.A.; Dudley, G. Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am. J. Physiol. 1994, 266, R375–R380. [Google Scholar] [CrossRef] [PubMed]
- Burr, I.M.; Asayama, K.; Fenichel, G.M. Superoxide dismutases, glutathione peroxidase, and catalase in neuromuscular disease. Muscle Nerve 1987, 10, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Mechler, F.; Imre, S.; Dioszeghy, P. Lipid peroxidation and superoxide dismutase activity in muscle and erythrocytes in Duchenne muscular dystrophy. J. Neurol. Sci. 1984, 63, 279–283. [Google Scholar] [CrossRef]
- Ragusa, R.J.; Chow, C.K.; St Clair, D.K.; Porter, J.D. Extraocular, limb and diaphragm muscle group-specific antioxidant enzyme activity patterns in control and mdx mice. J. Neurol. Sci. 1996, 139, 180–186. [Google Scholar] [CrossRef]
- Dudley, R.W.; Khairallah, M.; Mohammed, S.; Lands, L.; Des Rosiers, C.; Petrof, B.J. Dynamic responses of the glutathione system to acute oxidative stress in dystrophic mouse (mdx) muscles. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R704–R710. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Yang, C.; Xue, R.; Li, S.; Zhang, T.; Pan, L.; Ma, X.; Wang, L.; Li, D. Sulforaphane alleviates muscular dystrophy in mdx mice by activation of Nrf2. J. Appl. Physiol. 2015, 118, 224–237. [Google Scholar] [CrossRef]
- Sun, C.; Li, S.; Li, D. Sulforaphane mitigates muscle fibrosis in mdx mice via Nrf2-mediated inhibition of TGF-beta/Smad signaling. J. Appl. Physiol. 2016, 120, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Mendoza, N.; Morales-Gonzalez, A.; Madrigal-Santillan, E.O.; Madrigal-Bujaidar, E.; Alvarez-Gonzalez, I.; Garcia-Melo, L.F.; Anguiano-Robledo, L.; Fregoso-Aguilar, T.; Morales-Gonzalez, J.A. Antioxidant and Adaptative Response Mediated by Nrf2 during Physical Exercise. Antioxidants 2019, 8, 196. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Rando, T.A.; Disatnik, M.H.; Yu, Y.; Franco, A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul. Disord. 1998, 8, 14–21. [Google Scholar] [CrossRef]
- Choi, S.J.; Kim, H.S. Deregulation of Nrf2/ARE signaling pathway causes susceptibility of dystrophin-deficient myotubes to menadione-induced oxidative stress. Exp. Cell Res. 2018, 364, 224–233. [Google Scholar] [CrossRef]
- Afroze, D.; Kumar, A. ER stress in skeletal muscle remodeling and myopathies. FEBS J. 2019, 286, 379–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauly, M.; Angebault-Prouteau, C.; Dridi, H.; Notarnicola, C.; Scheuermann, V.; Lacampagne, A.; Matecki, S.; Fauconnier, J. ER stress disturbs SR/ER-mitochondria Ca(2+) transfer: Implications in Duchenne muscular dystrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Steinbacher, P.; Eckl, P. Impact of oxidative stress on exercising skeletal muscle. Biomolecules 2015, 5, 356–377. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, R.H.; Levada-Pires, A.C.; Rossoni, L.V.; Curi, R.; Pithon-Curi, T.C. Effects of aerobic exercise training on antioxidant enzyme activities and mRNA levels in soleus muscle from young and aged rats. Mech. Ageing Dev. 2007, 128, 267–275. [Google Scholar] [CrossRef]
- Simioni, C.; Zauli, G.; Martelli, A.M.; Vitale, M.; Sacchetti, G.; Gonelli, A.; Neri, L.M. Oxidative stress: Role of physical exercise and antioxidant nutraceuticals in adulthood and aging. Oncotarget 2018, 9, 17181–17198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finaud, J.; Lac, G.; Filaire, E. Oxidative stress: Relationship with exercise and training. Sports Med. 2006, 36, 327–358. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Domenech, E.; Romagnoli, M.; Arduini, A.; Borras, C.; Pallardo, F.V.; Sastre, J.; Vina, J. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am. J. Clin. Nutr. 2008, 87, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, G.; Cumming, K.T.; Holden, G.; Hallen, J.; Ronnestad, B.R.; Sveen, O.; Skaug, A.; Paur, I.; Bastani, N.E.; Ostgaard, H.N.; et al. Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: A double-blind, randomised, controlled trial. J. Physiol. 2014, 592, 1887–1901. [Google Scholar] [CrossRef]
- Kang, C.; Chung, E.; Diffee, G.; Ji, L.L. Exercise training attenuates aging-associated mitochondrial dysfunction in rat skeletal muscle: Role of PGC-1alpha. Exp. Gerontol. 2013, 48, 1343–1350. [Google Scholar] [CrossRef]
- Kang, C.; Li Ji, L. Role of PGC-1alpha signaling in skeletal muscle health and disease. Ann. N. Y. Acad. Sci. 2012, 1271, 110–117. [Google Scholar] [CrossRef]
- Tanaka, T.; Nishimura, A.; Nishiyama, K.; Goto, T.; Numaga-Tomita, T.; Nishida, M. Mitochondrial dynamics in exercise physiology. Pflügers Arch. 2020, 472, 137–153. [Google Scholar] [CrossRef]
- Moore, T.M.; Zhou, Z.; Cohn, W.; Norheim, F.; Lin, A.J.; Kalajian, N.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; et al. The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Mol. Metab. 2019, 21, 51–67. [Google Scholar] [CrossRef]
- Trewin, A.J.; Berry, B.J.; Wojtovich, A.P. Exercise and Mitochondrial Dynamics: Keeping in Shape with ROS and AMPK. Antioxidants 2018, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Balan, E.; Schwalm, C.; Naslain, D.; Nielens, H.; Francaux, M.; Deldicque, L. Regular Endurance Exercise Promotes Fission, Mitophagy, and Oxidative Phosphorylation in Human Skeletal Muscle Independently of Age. Front. Physiol. 2019, 10, 1088. [Google Scholar] [CrossRef] [Green Version]
- Vainshtein, A.; Tryon, L.D.; Pauly, M.; Hood, D.A. Role of PGC-1alpha during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiol. Cell Physiol. 2015, 308, C710–C719. [Google Scholar] [CrossRef] [Green Version]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- McCormick, R.; Vasilaki, A. Age-related changes in skeletal muscle: Changes to life-style as a therapy. Biogerontology 2018, 19, 519–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, B.S.; Delgado Diaz, D.C.; Kostek, M.C. Resveratrol decreases inflammation and increases utrophin gene expression in the mdx mouse model of Duchenne muscular dystrophy. Clin. Nutr. 2013, 32, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Mastaloudis, A.; Leonard, S.W.; Traber, M.G. Oxidative stress in athletes during extreme endurance exercise. Free Radic. Biol. Med. 2001, 31, 911–922. [Google Scholar] [CrossRef]
- Knez, W.L.; Jenkins, D.G.; Coombes, J.S. The effect of an increased training volume on oxidative stress. Int. J. Sports Med. 2014, 35, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Domenech, E.; Vina, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenburgh, C.; Fiebig, R.; Chandwaney, R.; Ji, L.L. Aging and exercise training in skeletal muscle: Responses of glutathione and antioxidant enzyme systems. Am. J. Physiol. 1994, 267, R439–R445. [Google Scholar] [CrossRef] [PubMed]
- Oh-ishi, S.; Kizaki, T.; Nagasawa, J.; Izawa, T.; Komabayashi, T.; Nagata, N.; Suzuki, K.; Taniguchi, N.; Ohno, H. Effects of endurance training on superoxide dismutase activity, content and mRNA expression in rat muscle. Clin. Exp. Pharmacol. Physiol. 1997, 24, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Criswell, D.; Lawler, J.; Martin, D.; Ji, L.L.; Herb, R.A.; Dudley, G. Regional training-induced alterations in diaphragmatic oxidative and antioxidant enzymes. Respir. Physiol. 1994, 95, 227–237. [Google Scholar] [CrossRef]
- Vincent, H.K.; Powers, S.K.; Stewart, D.J.; Demirel, H.A.; Shanely, R.A.; Naito, H. Short-term exercise training improves diaphragm antioxidant capacity and endurance. Eur. J. Appl. Physiol. 2000, 81, 67–74. [Google Scholar] [CrossRef]
- Ji, L.L. Modulation of skeletal muscle antioxidant defense by exercise: Role of redox signaling. Free Radic. Biol. Med. 2008, 44, 142–152. [Google Scholar] [CrossRef]
- Pereira, B.; Costa Rosa, L.F.; Safi, D.A.; Medeiros, M.H.; Curi, R.; Bechara, E.J. Superoxide dismutase, catalase, and glutathione peroxidase activities in muscle and lymphoid organs of sedentary and exercise-trained rats. Physiol. Behav. 1994, 56, 1095–1099. [Google Scholar] [CrossRef]
- Nakao, C.; Ookawara, T.; Kizaki, T.; Oh-Ishi, S.; Miyazaki, H.; Haga, S.; Sato, Y.; Ji, L.L.; Ohno, H. Effects of swimming training on three superoxide dismutase isoenzymes in mouse tissues. J. Appl. Physiol. 2000, 88, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ohno, H.; Oh-ishi, S.; Kizaki, T.; Ookawara, T.; Fujii, J.; Radák, Z.; Taniguchi, N. Chapter 11—Superoxide dismutases in exercise and disease. In Handbook of Oxidants and Antioxidants in Exercise; Part IV; Sen, C.K., Packer, L., Hänninen, O.O.P., Eds.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2000; pp. 243–295. [Google Scholar] [CrossRef]
- Hitomi, Y.; Watanabe, S.; Kizaki, T.; Sakurai, T.; Takemasa, T.; Haga, S.; Ookawara, T.; Suzuki, K.; Ohno, H. Acute exercise increases expression of extracellular superoxide dismutase in skeletal muscle and the aorta. Redox Rep. 2008, 13, 213–216. [Google Scholar] [CrossRef] [Green Version]
- Criswell, D.; Powers, S.; Dodd, S.; Lawler, J.; Edwards, W.; Renshler, K.; Grinton, S. High intensity training-induced changes in skeletal muscle antioxidant enzyme activity. Med. Sci. Sports Exerc. 1993, 25, 1135–1140. [Google Scholar] [CrossRef]
- Hellsten, Y.; Apple, F.S.; Sjodin, B. Effect of sprint cycle training on activities of antioxidant enzymes in human skeletal muscle. J. Appl. Physiol. 1996, 81, 1484–1487. [Google Scholar] [CrossRef]
- Leeuwenburgh, C.; Hollander, J.; Leichtweis, S.; Griffiths, M.; Gore, M.; Ji, L.L. Adaptations of glutathione antioxidant system to endurance training are tissue and muscle fiber specific. Am. J. Physiol. 1997, 272, R363–R369. [Google Scholar] [CrossRef]
- Venditti, P.; Di Meo, S. Effect of training on antioxidant capacity, tissue damage, and endurance of adult male rats. Int. J. Sports Med. 1997, 18, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Rius-Perez, S.; Torres-Cuevas, I.; Millan, I.; Ortega, A.L.; Perez, S. PGC-1alpha, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Lawler, J.M. Amplification of proinflammatory phenotype, damage, and weakness by oxidative stress in the diaphragm muscle of mdx mice. Free Radic. Biol. Med. 2012, 52, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Selsby, J.T. Increased catalase expression improves muscle function in mdx mice. Exp. Physiol. 2011, 96, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Faist, V.; Konig, J.; Hoger, H.; Elmadfa, I. Decreased mitochondrial oxygen consumption and antioxidant enzyme activities in skeletal muscle of dystrophic mice after low-intensity exercise. Ann. Nutr. Metab. 2001, 45, 58–66. [Google Scholar] [CrossRef]
- Hulmi, J.J.; Oliveira, B.M.; Silvennoinen, M.; Hoogaars, W.M.; Pasternack, A.; Kainulainen, H.; Ritvos, O. Exercise restores decreased physical activity levels and increases markers of autophagy and oxidative capacity in myostatin/activin-blocked mdx mice. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E171–E182. [Google Scholar] [CrossRef]
- Handschin, C.; Kobayashi, Y.M.; Chin, S.; Seale, P.; Campbell, K.P.; Spiegelman, B.M. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007, 21, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulmi, J.J.; Hentila, J.; DeRuisseau, K.C.; Oliveira, B.M.; Papaioannou, K.G.; Autio, R.; Kujala, U.M.; Ritvos, O.; Kainulainen, H.; Korkmaz, A.; et al. Effects of muscular dystrophy, exercise and blocking activin receptor IIB ligands on the unfolded protein response and oxidative stress. Free Radic. Biol. Med. 2016, 99, 308–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gali Ramamoorthy, T.; Laverny, G.; Schlagowski, A.I.; Zoll, J.; Messaddeq, N.; Bornert, J.M.; Panza, S.; Ferry, A.; Geny, B. The transcriptional coregulator PGC-1beta controls mitochondrial function and anti-oxidant defence in skeletal muscles. Nat. Commun. 2015, 6, 10210. [Google Scholar] [CrossRef] [Green Version]
- Booth, F.W.; Ruegsegger, G.N.; Toedebusch, R.G.; Yan, Z. Endurance Exercise and the Regulation of Skeletal Muscle Metabolism. Prog. Mol. Biol. Transl. Sci. 2015, 135, 129–151. [Google Scholar] [PubMed]
- Debold, E.P. Potential molecular mechanisms underlying muscle fatigue mediated by reactive oxygen and nitrogen species. Front. Physiol. 2015, 6, 239. [Google Scholar] [CrossRef]
- Ostrowski, K.; Rohde, T.; Asp, S.; Schjerling, P.; Pedersen, B.K. Pro- and anti-inflammatory cytokine balance in strenuous exercise in humans. J. Physiol. 1999, 515(Pt. 1), 287–291. [Google Scholar] [CrossRef]
- Cerqueira, E.; Marinho, D.A.; Neiva, H.P.; Lourenco, O. Inflammatory Effects of High and Moderate Intensity Exercise—A Systematic Review. Front. Physiol. 2019, 10, 1550. [Google Scholar] [CrossRef]
- Rose, G.L.; Skinner, T.L.; Mielke, G.I.; Schaumberg, M.A. The effect of exercise intensity on chronic inflammation: A systematic review and meta-analysis. J. Sci. Med. Sport 2020, 24, 345–351. [Google Scholar] [CrossRef]
- Kramer, H.F.; Goodyear, L.J. Exercise, MAPK, and NF-kappaB signaling in skeletal muscle. J. Appl. Physiol. 2007, 103, 388–395. [Google Scholar] [CrossRef]
- Cuevas, M.J.; Almar, M.; Garcia-Glez, J.C.; Garcia-Lopez, D.; De Paz, J.A.; Alvear-Ordenes, I.; Gonzalez-Gallego, J. Changes in oxidative stress markers and NF-kappaB activation induced by sprint exercise. Free Radic. Res. 2005, 39, 431–439. [Google Scholar] [CrossRef]
- Scicchitano, B.M.; Faraldi, M.; Musaro, A. The Proteolytic Systems of Muscle Wasting. Recent Adv. DNA Gene Seq. 2015, 9, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Judge, A.R.; Dodd, S.L. Xanthine oxidase and activated neutrophils cause oxidative damage to skeletal muscle after contractile claudication. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H252–H256. [Google Scholar] [CrossRef] [Green Version]
- Radak, Z.; Taylor, A.W.; Ohno, H.; Goto, S. Adaptation to exercise-induced oxidative stress: From muscle to brain. Exerc. Immunol. Rev. 2001, 7, 90–107. [Google Scholar]
- Peake, J.; Suzuki, K. Neutrophil activation, antioxidant supplements and exercise-induced oxidative stress. Exerc. Immunol. Rev. 2004, 10, 129–141. [Google Scholar]
- Moylan, J.S.; Reid, M.B. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 2007, 35, 411–429. [Google Scholar] [CrossRef]
- Gomes, E.C.; Silva, A.N.; de Oliveira, M.R. Oxidants, antioxidants, and the beneficial roles of exercise-induced production of reactive species. Oxid. Med. Cell. Longev. 2012, 2012, 756132. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 2008, 454, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Boriek, A.M. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: A possible role in Duchenne muscular dystrophy. FASEB J. 2003, 17, 386–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyatake, S.; Shimizu-Motohashi, Y.; Takeda, S.; Aoki, Y. Anti-inflammatory drugs for Duchenne muscular dystrophy: Focus on skeletal muscle-releasing factors. Drug Des. Devel. Ther. 2016, 10, 2745–2758. [Google Scholar] [PubMed] [Green Version]
- Monici, M.C.; Aguennouz, M.; Mazzeo, A.; Messina, C.; Vita, G. Activation of nuclear factor-kappaB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology 2003, 60, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.; Carathers, M.; Li, Z.W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Invest. 2007, 117, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, C.G.; Samadi, A.; Siegel, A. Chronic treatment with agents that stabilize cytosolic IkappaB-alpha enhances survival and improves resting membrane potential in MDX muscle fibers subjected to chronic passive stretch. Neurobiol. Dis. 2005, 20, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, C.; Shen, Y.; Zhu, C.H.; Wang, G.; Wang, X.C.; Chen, H.Q.; Zhu, M.S. Curcumin alleviates dystrophic muscle pathology in mdx mice. Mol. Cells 2008, 25, 531–537. [Google Scholar] [PubMed]
- Manzur, A.Y.; Kuntzer, T.; Pike, M.; Swan, A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2004, CD003725. [Google Scholar] [CrossRef]
- Andrews, J.G.; Wahl, R.A. Duchenne and Becker muscular dystrophy in adolescents: Current perspectives. Adolesc. Health Med. Ther. 2018, 9, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef]
- Song, T.J.; Lee, K.A.; Kang, S.W.; Cho, H.; Choi, Y.C. Three cases of manifesting female carriers in patients with Duchenne muscular dystrophy. Yonsei Med. J. 2011, 52, 192–195. [Google Scholar] [CrossRef]
- Finsterer, J.; Stollberger, C.; Freudenthaler, B.; Simoni, D.; Hoftberger, R.; Wagner, K. Muscular and cardiac manifestations in a Duchenne-carrier harboring a dystrophin deletion of exons 12-29. Intractable Rare Dis. Res. 2018, 7, 120–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakim, C.H.; Duan, D. Gender differences in contractile and passive properties of mdx extensor digitorum longus muscle. Muscle Nerve 2012, 45, 250–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostick, B.; Yue, Y.; Duan, D. Gender influences cardiac function in the mdx model of Duchenne cardiomyopathy. Muscle Nerve 2010, 42, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Dodd, S.L.; Jackson, E.M. Total Fitness & Wellness, 6th ed.; Pearson: Boston, MA, USA, 2013. [Google Scholar]

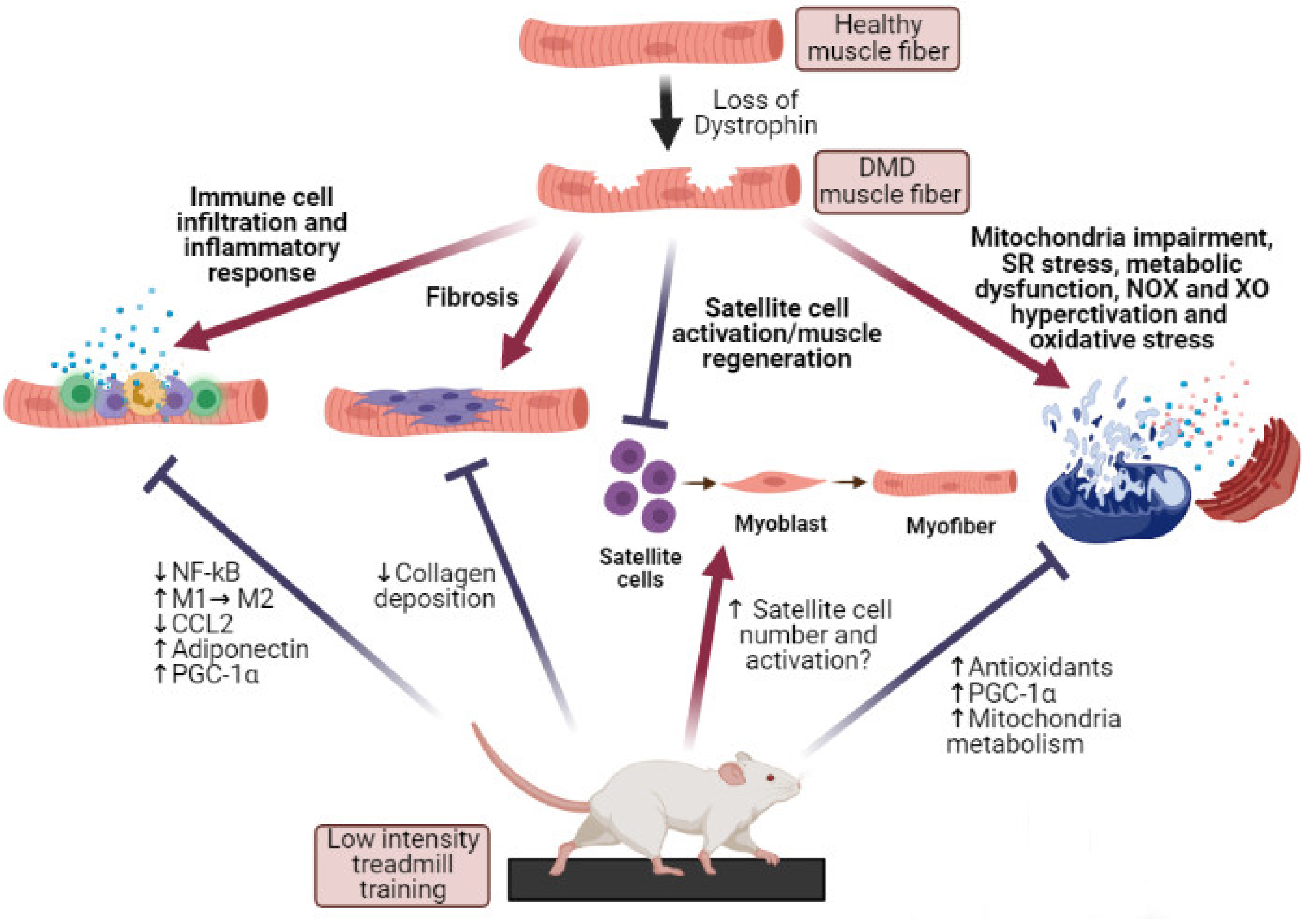
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frinchi, M.; Morici, G.; Mudó, G.; Bonsignore, M.R.; Di Liberto, V. Beneficial Role of Exercise in the Modulation of mdx Muscle Plastic Remodeling and Oxidative Stress. Antioxidants 2021, 10, 558. https://doi.org/10.3390/antiox10040558
Frinchi M, Morici G, Mudó G, Bonsignore MR, Di Liberto V. Beneficial Role of Exercise in the Modulation of mdx Muscle Plastic Remodeling and Oxidative Stress. Antioxidants. 2021; 10(4):558. https://doi.org/10.3390/antiox10040558
Chicago/Turabian StyleFrinchi, Monica, Giuseppe Morici, Giuseppa Mudó, Maria R. Bonsignore, and Valentina Di Liberto. 2021. "Beneficial Role of Exercise in the Modulation of mdx Muscle Plastic Remodeling and Oxidative Stress" Antioxidants 10, no. 4: 558. https://doi.org/10.3390/antiox10040558