
Activation of Adenosine A1 Receptor in Ischemic Stroke: Neuroprotection by Tetrahydroxy Stilbene Glycoside as an Agonist

Abstract
:
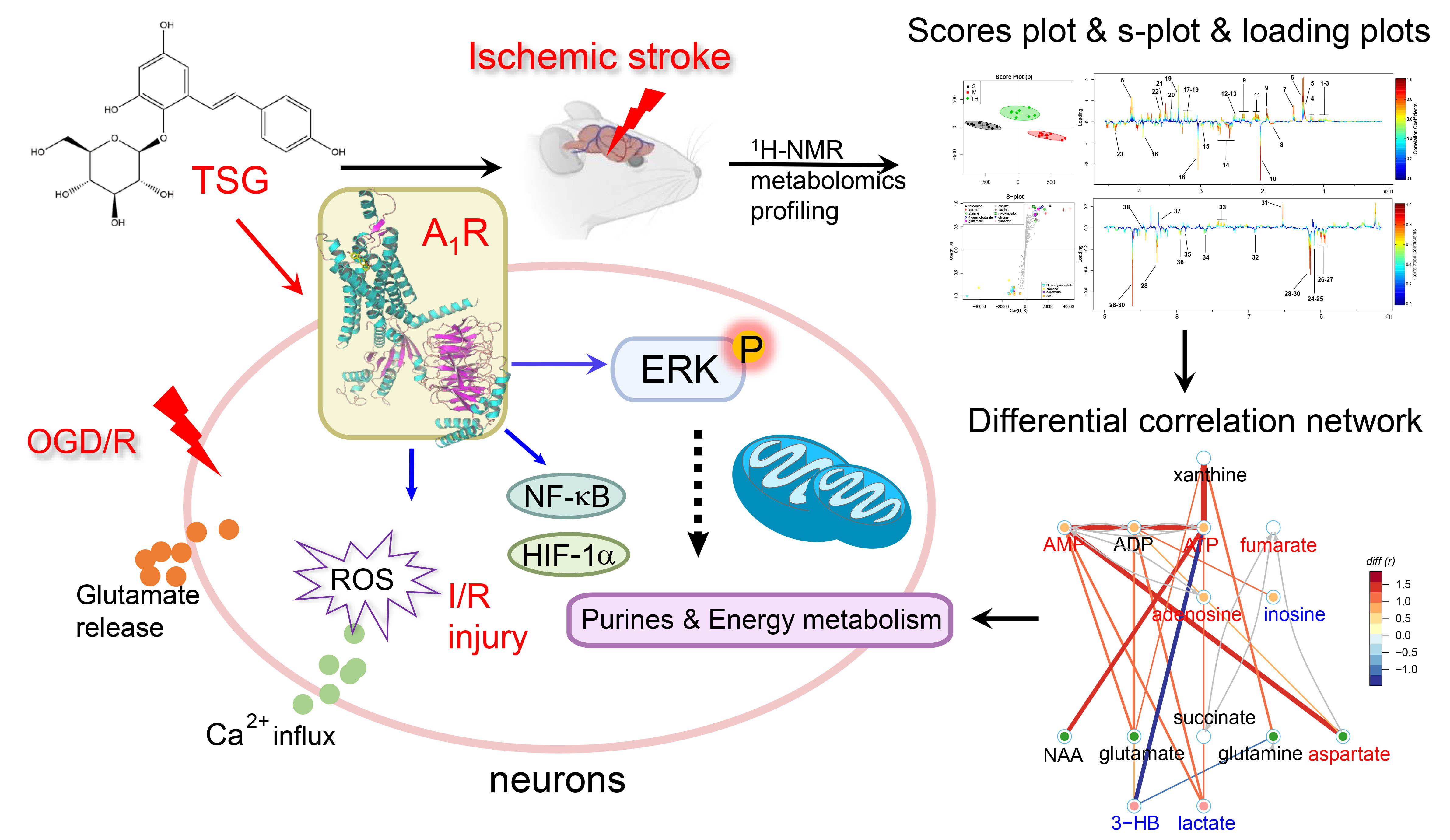
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
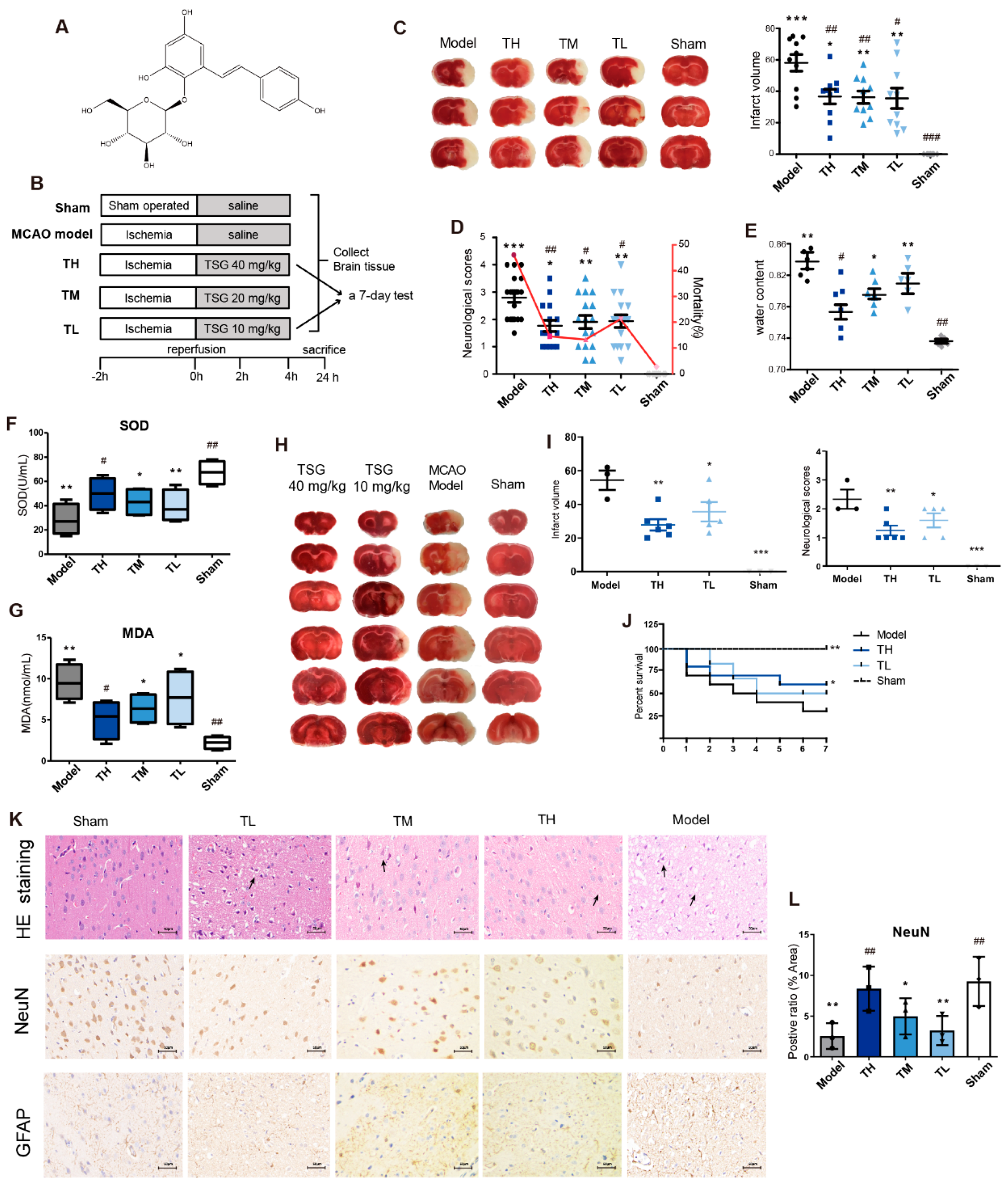
2.2. MCAO Model Establishment and Drug Administration
2.3. TTC, HE Staining and Immunohistochemistry
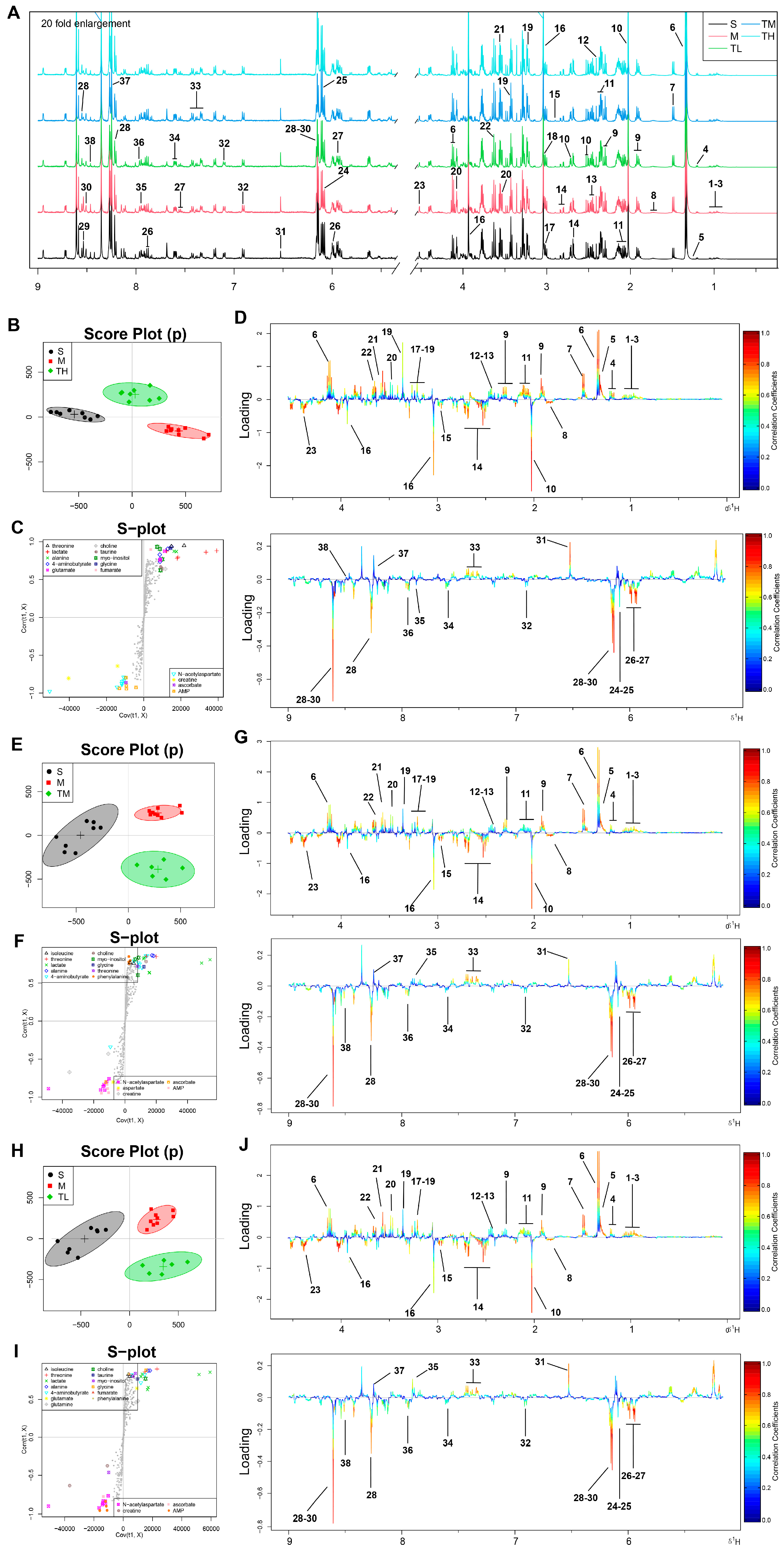
2.4. Sample Preparation, Acquisition of 1H NMR Spectra and Data Processing
2.5. Metabolomic Analysis and Correlation Network Analysis
2.6. Target Prediction and Pathway Enrichment Analysis
2.7. Molecular Docking Experiments
2.8. Constructiontion of HEK293T Cell Lines Stably Expressing Human A1R
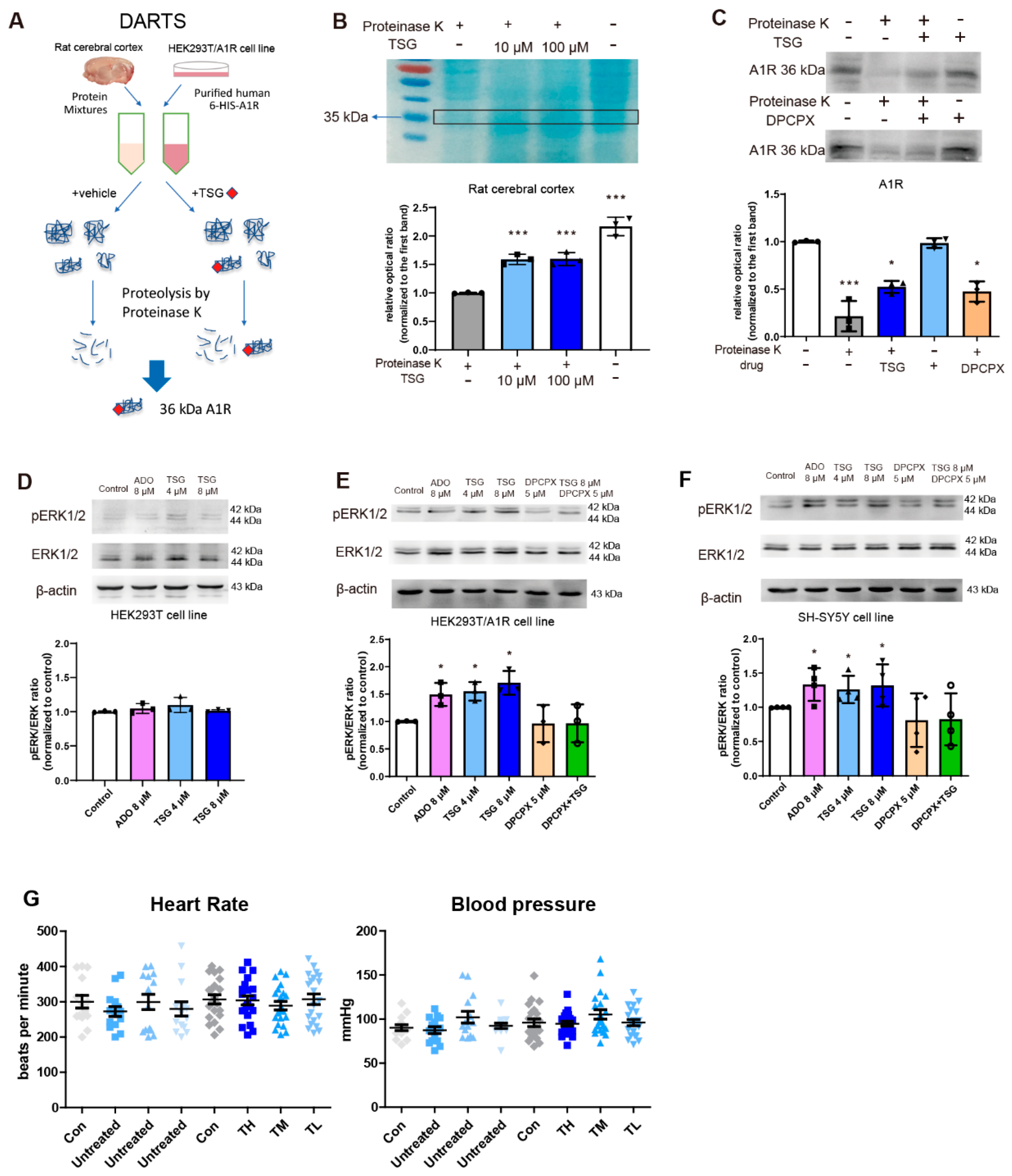
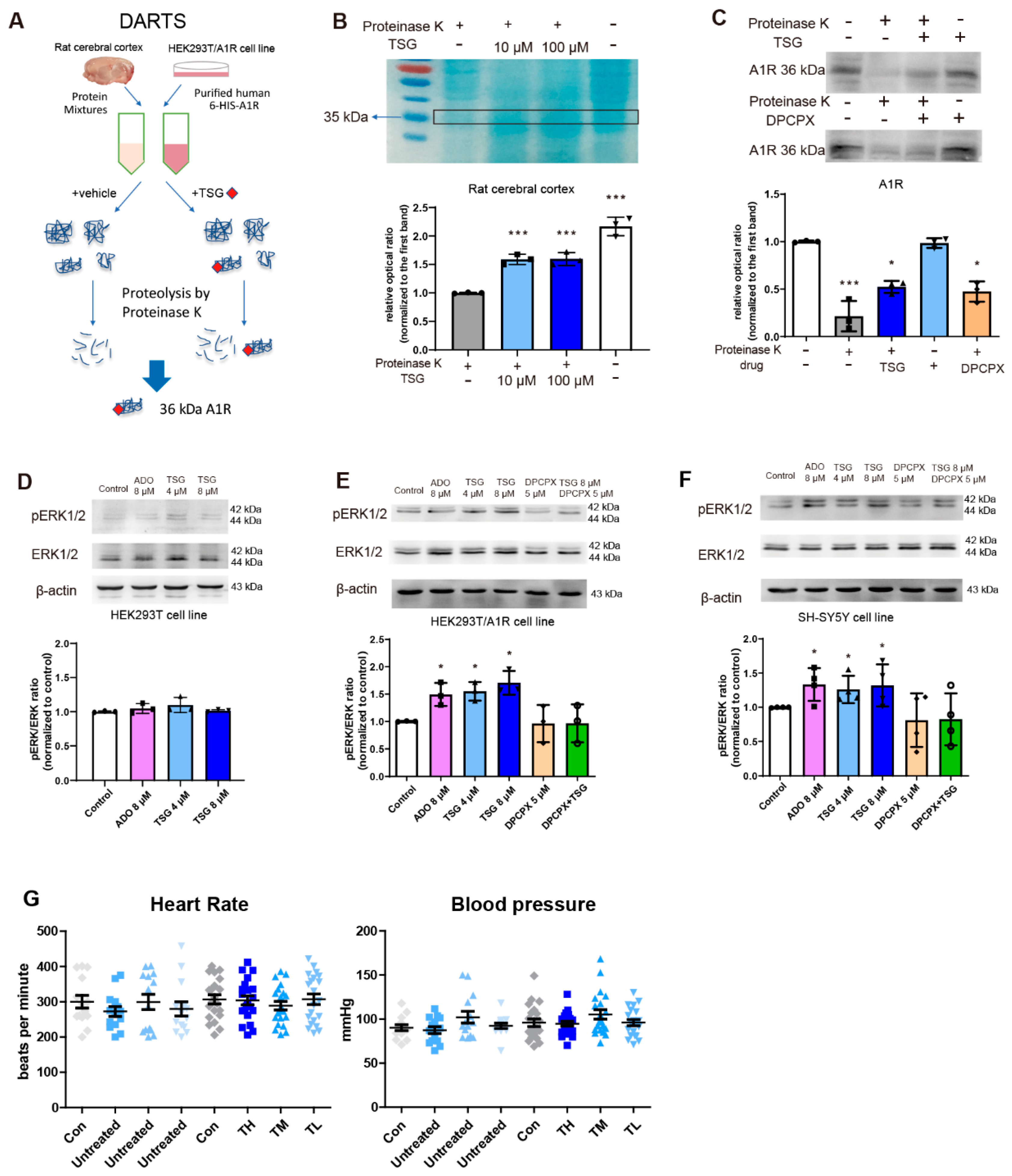
2.9. DARTS Assay
2.10. OGD/R Model
2.11. Biochemical Indexes Assays
2.12. Western Blotting (WB) and Q-RT-PCR
2.13. Determination of Blood Pressure and Heart Rate
2.14. Data Analysis
3. Results
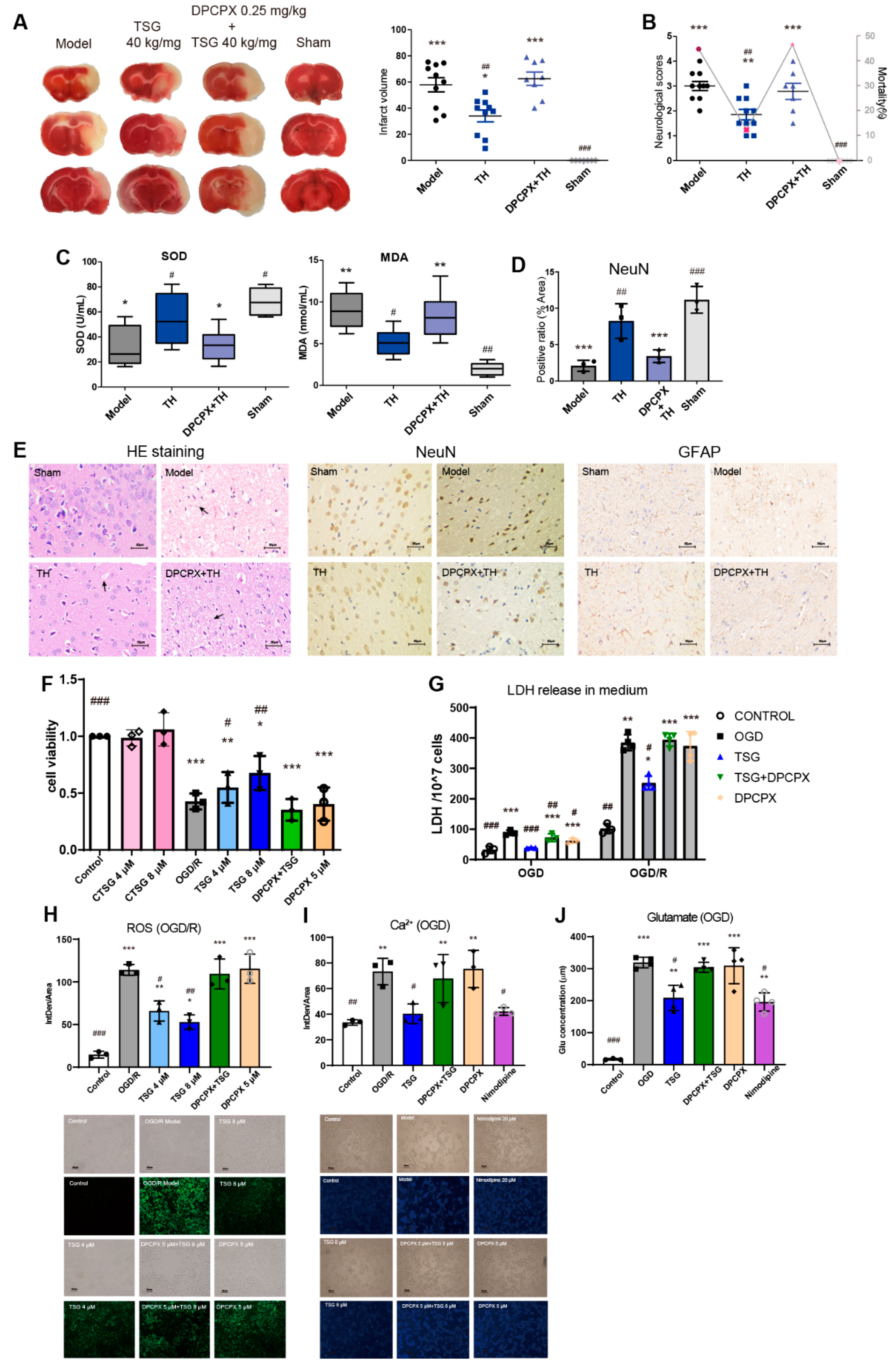
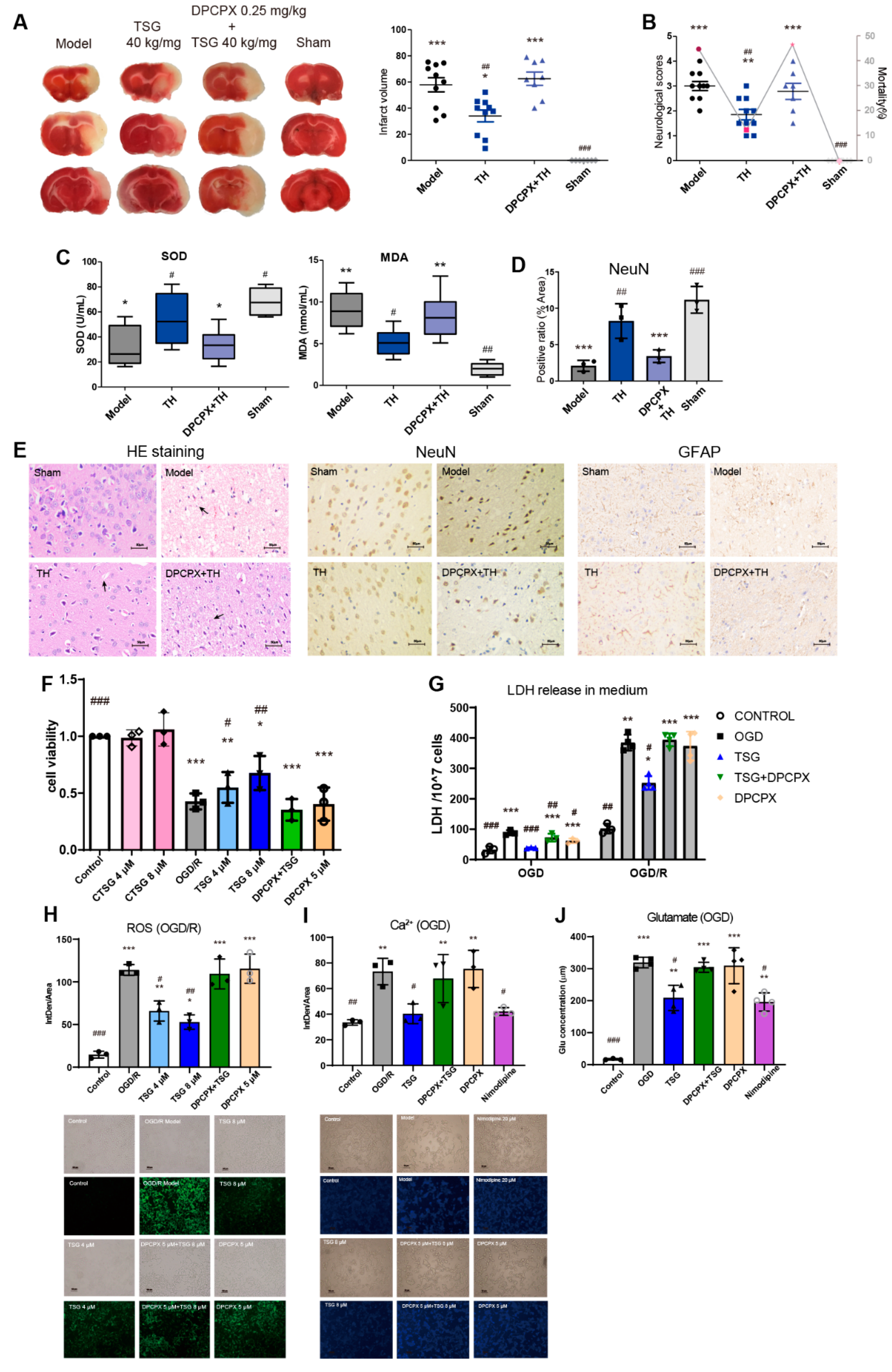
3.1. TSG Ameliorated Cerebral I/R Injury in MCAO Rats
3.2. TSG Administration Relieved Brain Cell Injury Induced by MCAO
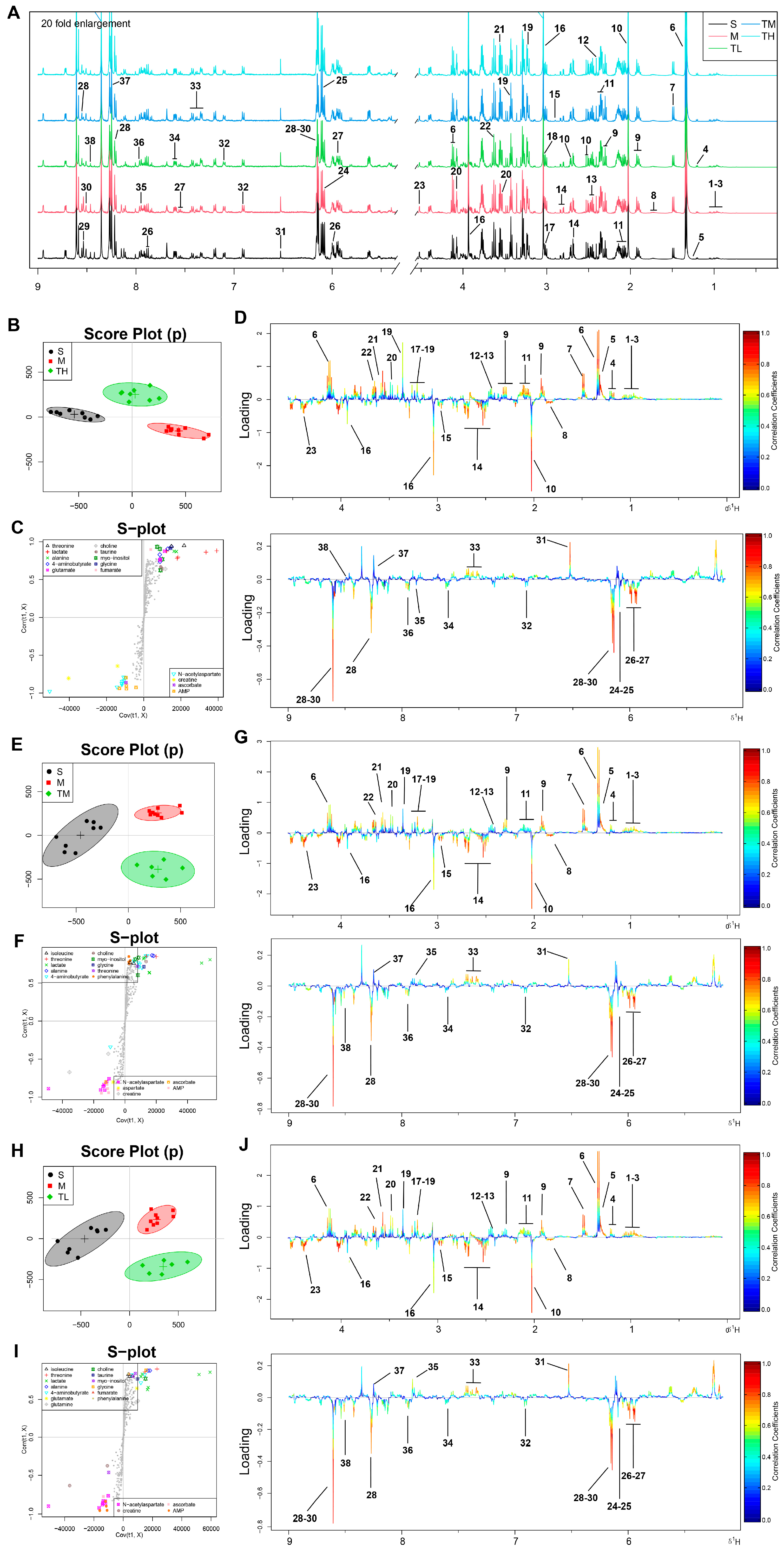
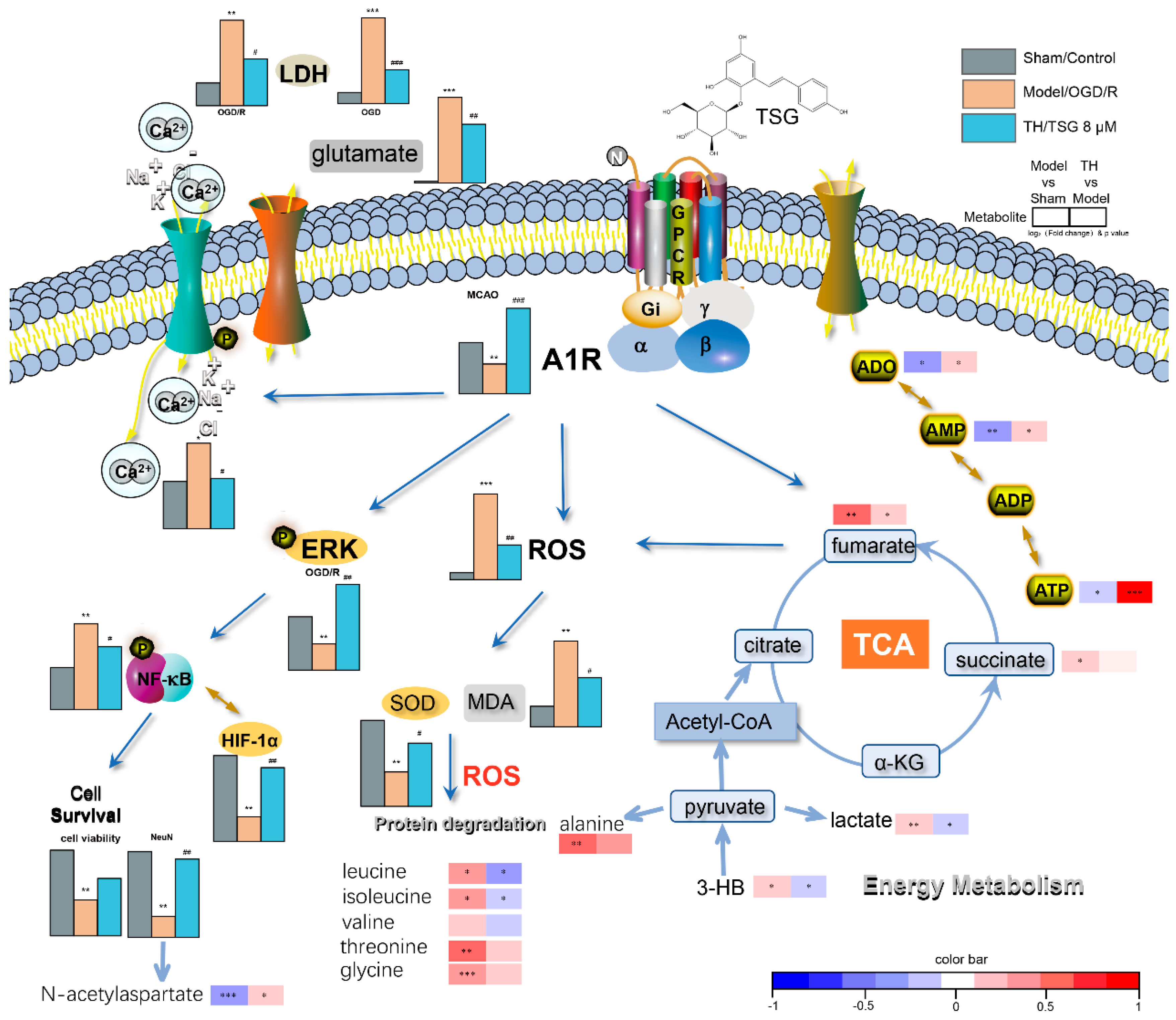
3.3. TSG Ameliorated the Disturbed Metabolic Profiles of MCAO Rats towards Normal Status
3.4. Pathway Enrichment Analysis of TSG Affected Metabolites and Predicted Targets
3.5. Differential Correlation Network Analysis of TSG Affected Metabolites
3.6. DPCPX Abolished the Neuroprotection of TSG in Stroke Rats
3.7. TSG-Increased Cell Survival during OGD/R was Abolished by DPCPX
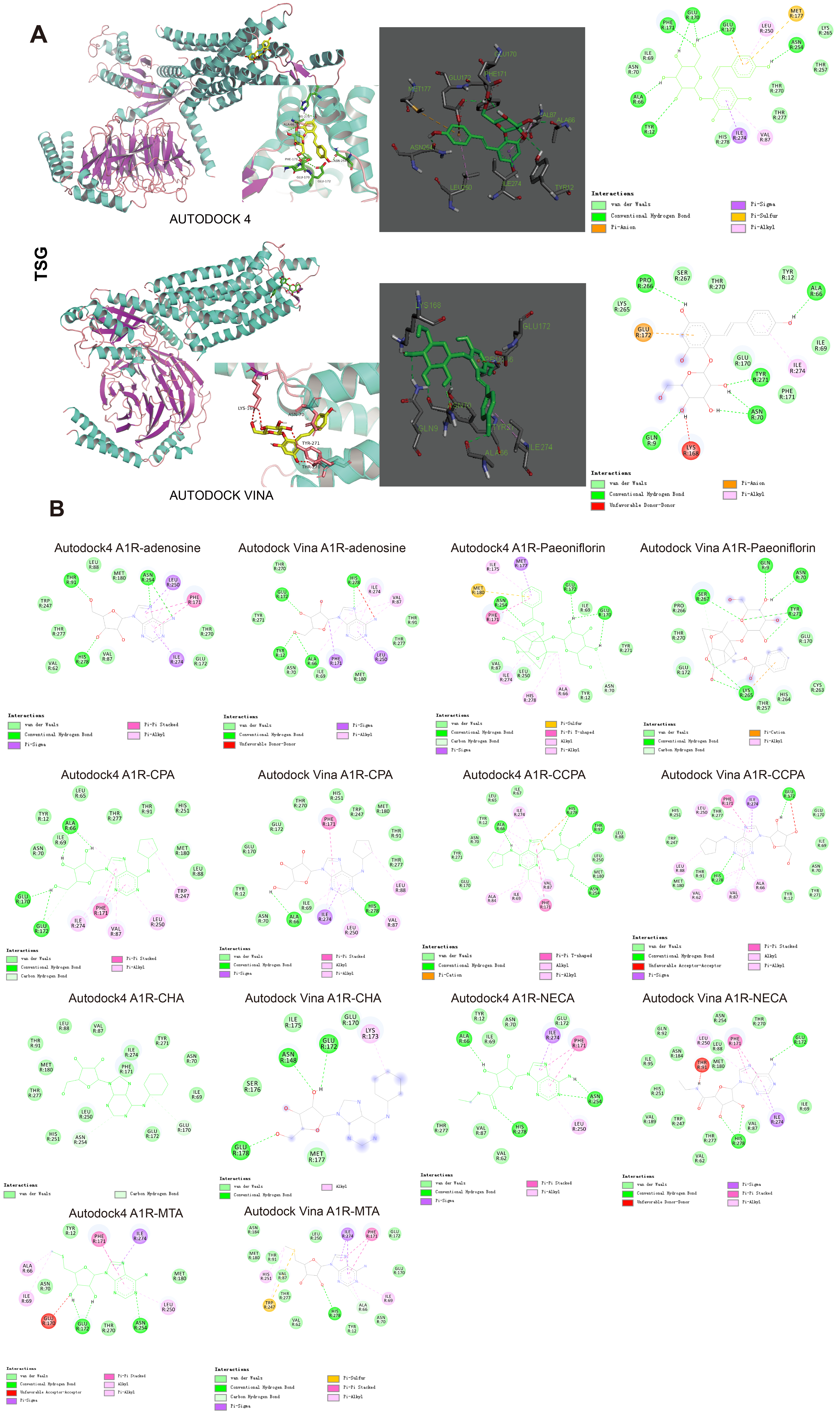
3.8. Docking Results Using AutoDock4 and AutoDock Vina
3.9. TSG Bound with A1R and Activated ERK1/2 Phosphorylation
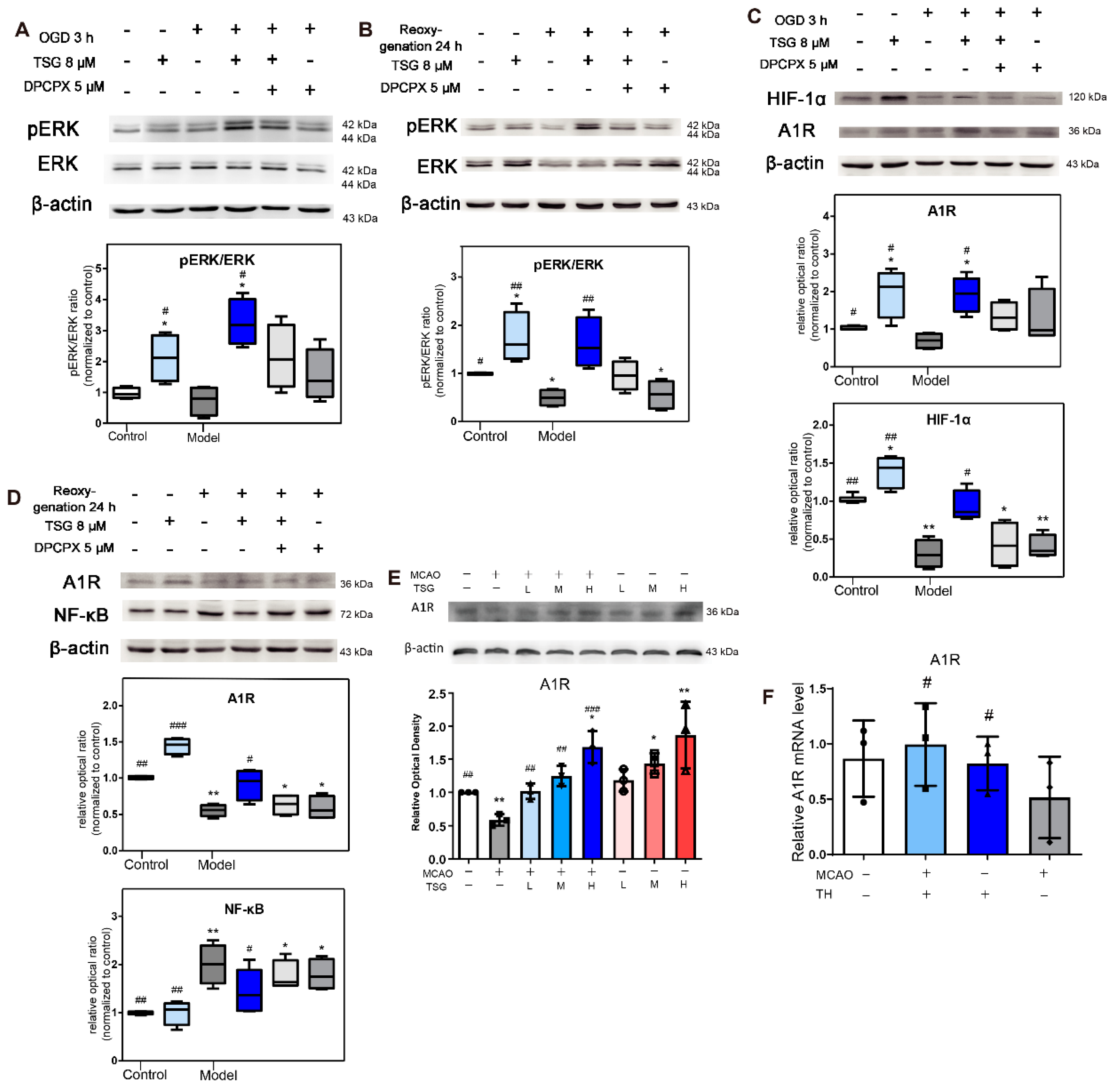
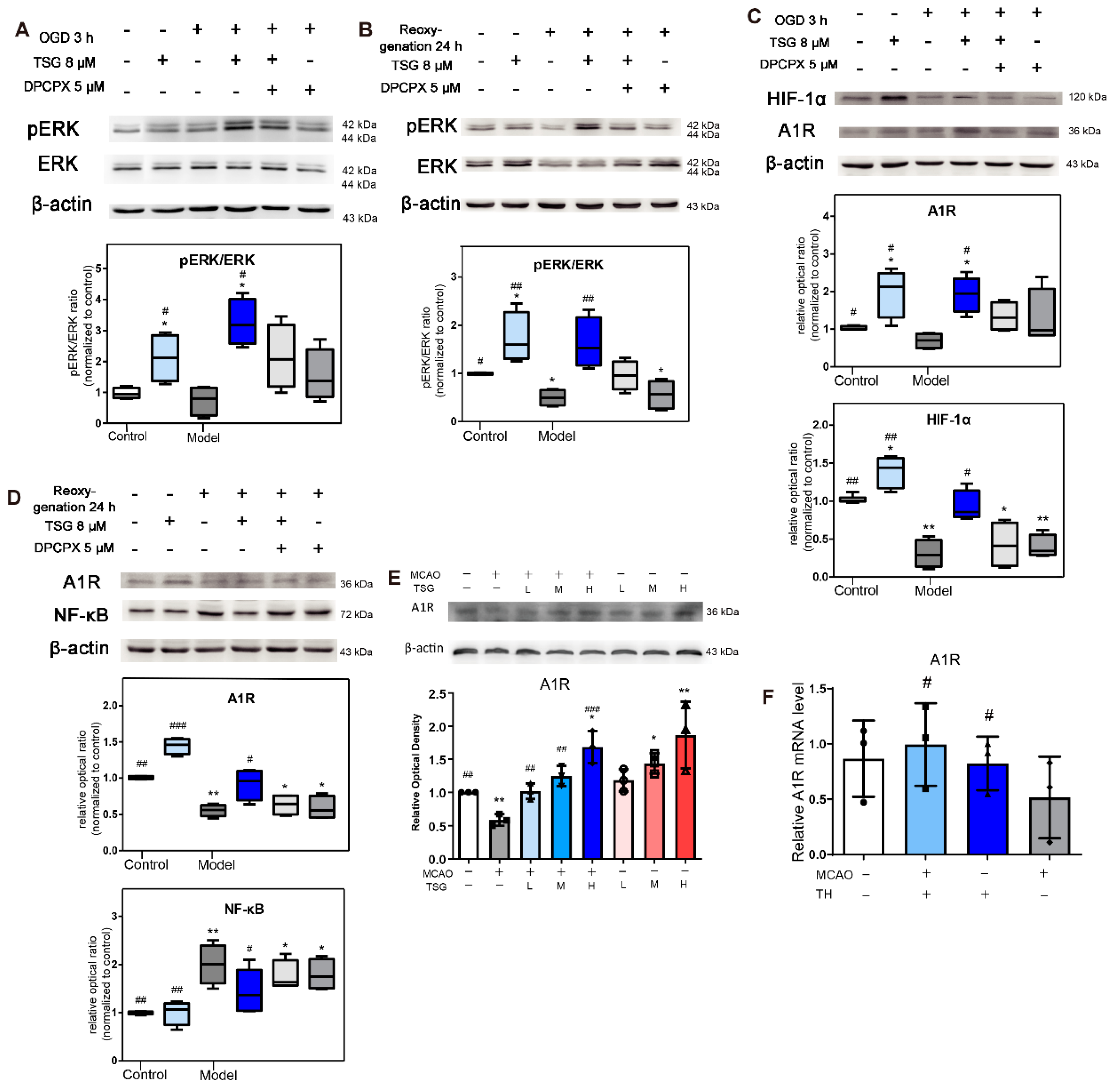
3.10. Activation of ERK1/2 Signaling Pathway Contributed to TSG’s Neuroprotection
3.11. TSG Increased A1R Expression
3.12. HIF-1α and NF-κB Were Involved in the Neuroprotection of TSG
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Hankey, G.J. Neuroprotection for Acute Ischaemic Stroke: Hope Reignited. Lancet. Neurol. 2006, 5, 287–288. [Google Scholar] [CrossRef]
- Karsy, M.; Brock, A.; Guan, J.; Taussky, P.; Kalani, M.Y.S.; Park, M.S. Neuroprotective Strategies and the Underlying Molecular Basis of Cerebrovascular Stroke. Neurosurg. Focus 2017, 42, E3. [Google Scholar] [CrossRef] [PubMed]
- Tomai, F.; Crea, F.; Chiariello, L.; Gioffrè, P.A. Ischemic Preconditioning in Humans: Models, Mediators, and Clinical Relevance. Circulation 1999, 100, 559–563. [Google Scholar] [CrossRef]
- Murry, C.E.; Richard, V.J.; Reimer, K.A.; Jennings, R.B. Ischemic Preconditioning Slows Energy Metabolism and Delays Ultrastructural Damage during a Sustained Ischemic Episode. Circ. Res. 1990, 66, 913–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugliese, A.M.; Latini, S.; Corradetti, R.; Pedata, F. Brief, Repeated, Oxygen-Glucose Deprivation Episodes Protect Neurotransmission from a Longer Ischemic Episode in the In Vitro Hippocampus: Role of Adenosine Receptors. Br. J. Pharmacol. 2003, 140, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Guo, M.; Wang, X.; Zhao, Y.; Zhao, Q.; Ding, H.; Dong, Q.; Cui, M. Ischemic Preconditioning with a Ketogenic Diet Improves Brain Ischemic Tolerance through Increased Extracellular Adenosine Levels and Hypoxia-Inducible Factors. Brain Res. 2017, 1667, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Latini, S.; Pedata, F. Adenosine in the Central Nervous System: Release Mechanisms and Extracellular Concentrations. J. Neurochem. 2001, 79, 463–484. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Pinzón, M.A.; Born, J.G. Rapid Preconditioning Neuroprotection Following Anoxia in Hippocampal Slices: Role of the K+ATP Channel and Protein Kinase C. Neuroscience 1999, 89, 453–459. [Google Scholar] [CrossRef]
- Burnstock, G.; Fredholm, B.B.; Verkhratsky, A. Adenosine and ATP Receptors in the Brain. Curr. Top. Med. Chem. 2011, 11, 973–1011. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Chen, J.F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.M. Adenosine and Brain Function. Int. Rev. Neurobiol. 2005, 63, 191–270. [Google Scholar]
- Li, M.H.; Ruan, L.Y.; Chen, C.; Xing, Y.X.; Hong, W.; Du, R.H.; Wang, J.S. Protective Effects of Polygonum Multiflorum on Ischemic Stroke Rat Model Analysed by (1)H NMR Metabolic Profiling. J. Pharm. Biomed. Anal. 2018, 155, 91–103. [Google Scholar] [CrossRef]
- Lee, S.Y.; Ahn, S.M.; Wang, Z.; Choi, Y.W.; Shin, H.K.; Choi, B.T. Neuroprotective Effects pf 2,3,5,4′-Tetrahydoxystilbene-2-O-Β-D-Glucoside from Polygonum Multiflorum against Glutamate-Induced Oxidative Toxicity in HT22 Cells. J. Ethnopharmacol. 2017, 195, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Matsumoto, K.; Murakami, Y.; Tezuka, Y.; Wu, Y.; Kadota, S. Neuroprotective Effects of Polygonum Multiflorum on Nigrostriatal Dopaminergic Degeneration Induced by Paraquat and Maneb in Mice. Pharm. Biochem. Behav. 2005, 82, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gu, J.; Wu, P.F.; Wang, F.; Xiong, Z.; Yang, Y.J.; Wu, W.N.; Dong, L.D.; Chen, J.G. Protection by Tetrahydroxystilbene Glucoside against Cerebral Ischemia: Involvement of JNK, SIRT1, and NF-Kappab Pathways and Inhibition of Intracellular ROS/RNS Generation. Free. Radic. Biol. Med. 2009, 47, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liang, Y.; Song, F.; Xu, S.; Nian, L.; Zhou, X.; Wang, S. TSG attenuates LPC-Induced Endothelial Cells Inflammatory Damage through Notch Signaling Inhibition. IUBMB Life 2016, 68, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Hu, W.; Gong, Y.; Wang, P.; Tong, L.; Chen, X.; Chen, Z.; Xu, X.; Yao, W.; Zhang, W.; et al. Current Pharmacological Developments in 2,3,4’,5-Tetrahydroxystilbene 2-O-β-D-Glucoside (TSG). Eur. J. Pharm. 2017, 811, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Büchter, C.; Zhao, L.; Havermann, S.; Honnen, S.; Fritz, G.; Proksch, P.; Wätjen, W. TSG (2,3,5,4′-Tetrahydroxystilbene-2-O-β-D-glucoside) from the Chinese Herb Polygonum Multiflorum Increases Life Span and Stress Resistance of Caenorhabditis Elegans. Oxid. Med. Cell Longev. 2015, 2015, 124357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xing, Y.; Ye, C.F.; Ai, H.X.; Wei, H.F.; Li, L. Learning-Memory Deficit with Aging in APP Transgenic Mice of Alzheimer’s Disease and Intervention by Using Tetrahydroxystilbene Glucoside. Behav. Brain Res. 2006, 173, 246–254. [Google Scholar] [CrossRef]
- He, H.; Wang, S.; Tian, J.; Chen, L.; Zhang, W.; Zhao, J.; Tang, H.; Zhang, X.; Chen, J. Protective Effects of 2,3,5,4′-Tetrahydroxystilbene-2-O-β-D-Glucoside in the MPTP-Induced Mouse Model of Parkinson’s Disease: Involvement of Reactive Oxygen Species-Mediated JNK, P38 and Mitochondrial Pathways. Eur. J. Pharmacol. 2015, 767, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, W.; Lu, X.; Jiang, B.; Wang, J.; Zhang, W.; Huang, C. Mechanism of 2,3,4′,5-Tetrahydroxystilbene 2-O-β-D-Glucoside-Induced Upregulation of Glutamate Transporter 1 Protein Expression in Mouse Primary Astrocytes. Pharmacology 2017, 99, 153–159. [Google Scholar] [CrossRef]
- Xiang, K.; Liu, G.; Zhou, Y.J.; Hao, H.Z.; Yin, Z.; He, A.D.; Da, X.W.; Xiang, J.Z.; Wang, J.L.; Ming, Z.Y. 2,3,5,4′-Tetrahydroxystilbene-2-O-β-D-Glucoside (THSG) Attenuates Human Platelet Aggregation, Secretion and Spreading In Vitro. Thromb. Res. 2014, 133, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Xu, J.W. Biological Activities of 2,3,5,4′-Tetrahydroxystilbene-2-O-beta-D-Glucoside in Antiaging and Antiaging-Related Disease Treatments. Oxid. Med. Cell Longev. 2016, 2016, 4973239. [Google Scholar] [CrossRef] [Green Version]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible Middle Cerebral Artery Occlusion without Craniectomy in Rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Ruan, L.Y.; Li, M.H.; Xing, Y.X.; Hong, W.; Chen, C.; Chen, J.F.; Xu, H.; Zhao, W.L.; Wang, J.S. Hepatotoxicity and Hepatoprotection of Polygonum Multiflorum Thund. as Two Sides of the Same Biological Coin. J. Ethnopharmacol. 2019, 230, 81–94. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A Web Server for Target Prediction of Bioactive Small Molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A Web Server for Metabolomic Data Analysis and Interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Sun, W.C.; Cao, Y.; Jin, L.; Wang, L.Z.; Meng, F.; Zhu, X.Z. Modulating Effect of Adenosine Deaminase on Function of Adenosine A1 Receptors. Acta Pharmacol. Sin. 2005, 26, 160–165. [Google Scholar] [CrossRef]
- Cao, Y.; Sun, W.C.; Jin, L.; Xie, K.Q.; Zhu, X.Z. Activation of Adenosine A1 Receptor Modulates Dopamine D1 Receptor Activity in Stably Cotransfected Human Embryonic Kidney 293 Cells. Eur. J. Pharmacol. 2006, 548, 29–35. [Google Scholar] [CrossRef]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S.; Wang, J.; Wu, R.P.; Gomez, F.; Loo, J.A.; et al. Target Identification Using Drug Affinity Responsive Target Stability (DARTS). Proc. Natl. Acad. Sci. USA 2009, 106, 21984–21989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabata, K.; Matsumoto, K.; Murakami, Y.; Watanabe, H. Ameliorative Effects of Paeoniflorin, a Major Constituent of Peony Root, on Adenosine A1Receptor-Mediated Impairment of Passive Avoidance Performance and Long-Term Potentiation in the Hippocampus. Biol. Pharm. Bull. 2001, 24, 496–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Liang, C.L.; Liliang, P.C.; Yang, C.H.; Cho, C.L.; Weng, H.C.; Tsai, Y.D.; Wang, K.W.; Chen, H.J. Inhibition of Extracellular Signal-Regulated Kinases 1/2 Provides Neuroprotection in Spinal Cord Ischemia/Reperfusion Injury in Rats: Relationship with the Nuclear Factor-Κb-Regulated Anti-Apoptotic Mechanisms. J. Neurochem. 2010, 114, 237–246. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-Inducible Factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Ridder, D.A.; Schwaninger, M. NF-KappaB Signaling in Cerebral Ischemia. Neuroscience 2009, 158, 995–1006. [Google Scholar] [CrossRef]
- Dixon, A.K.; Gubitz, A.K.; Sirinathsinghji, D.J.; Richardson, P.J.; Freeman, T.C. Tissue Distribution of Adenosine Receptor Mrnas in the Rat. Br. J. Pharmacol. 1996, 118, 1461–1468. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Song, W.L.; Xu, Y.C.; Ye, Y.; Feng, L.Y. Paeoniflorin Ameliorates Ischemic Neuronal Damage In Vitro via Adenosine A1 Receptor-Mediated Transactivation of Epidermal Growth Factor Receptor. Acta Pharmacol. Sin. 2015, 36, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Jorg, M.; Glukhova, A.; Abdul-Ridha, A.; Vecchio, E.A.; Nguyen, A.T.; Sexton, P.M.; White, P.J.; May, L.T.; Christopoulos, A.; Scammells, P.J. Novel Irreversible Agonists Acting at the A1 Adenosine Receptor. J. Med. Chem. 2016, 59, 11182–11194. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Tabrizi, M.A.; Gessi, S.; Borea, P.A. Adenosine Receptor Antagonists: Translating Medicinal Chemistry and Pharmacology into Clinical Utility. Chem. Rev. 2008, 108, 238–263. [Google Scholar] [CrossRef]
- Adén, U.; Lindström, K.; Bona, E.; Hagberg, H.; Fredholm, B.B. Changes in Adenosine Receptors in the Neonatal Rat Brain Following Hypoxic Ischemia. Brain Res. Mol. Brain Res. 1994, 23, 354–358. [Google Scholar] [CrossRef]
- Bischofberger, N.; Jacobson, K.A.; von Lubitz, D.K. Adenosine A1 Receptor Agonists as Clinically Viable Agents for Treatment of Ischemic Brain Disorders. Ann. N. Y. Acad. Sci. 1997, 825, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Daval, J.L.; Von Lubitz, D.K.; Deckert, J.; Redmond, D.J.; Marangos, P.J. Protective Effect of Cyclohexyladenosine on Adenosine A1-Receptors, Guanine Nucleotide and Forskolin Binding Sites Following Transient Brain Ischemia: A Quantitative Autoradiographic Study. Brain Res. 1989, 491, 212–226. [Google Scholar] [CrossRef]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the Adenosine A Receptor Reveals the Basis for Subtype Selectivity. Cell 2017, 168, 867–877.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melani, A.; Pugliese, A.M.; Pedata, F. Adenosine Receptors in Cerebral Ischemia. Int. Rev. Neurobiol. 2014, 119, 309–348. [Google Scholar]
- Baltos, J.-A.; Gregory, K.J.; White, P.J.; Sexton, P.M.; Christopoulos, A.; May, L.T. Quantification of Adenosine A(1) Receptor Biased Agonism: Implications for Drug Discovery. Biochem. Pharmacol. 2016, 99, 101–112. [Google Scholar] [CrossRef]
- Wu, P.F.; Zhang, Z.; Wang, F.; Chen, J.G. Natural Compounds from Traditional Medicinal Herbs in the Treatment of Cerebral Ischemia/Reperfusion Injury. Acta Pharmacol. Sin. 2010, 31, 1523–1531. [Google Scholar] [CrossRef]
- Canals, M.; Angulo, E.; Casadó, V.; Canela, E.I.; Mallol, J.; Viñals, F.; Staines, W.; Tinner, B.; Hillion, J.; Agnati, L.; et al. Molecular Mechanisms Involved in the Adenosine A1 and A2A Receptor-Induced Neuronal Differentiation in Neuroblastoma Cells and Striatal Primary Cultures. J. Neurochem. 2005, 92, 337–348. [Google Scholar] [CrossRef]
- Lage, R.; Diéguez, C.; Vidal-Puig, A.; López, M. AMPK: A Metabolic Gauge Regulating Whole-Body Energy Homeostasis. Trends Mol. Med. 2008, 14, 539–549. [Google Scholar] [CrossRef]
- Small, D.L.; Buchan, A.M. Mechanisms of Cerebral Ischemia: Intracellular Cascades and Therapeutic Interventions. J. Cardiothorac. Vasc. Anesth. 1996, 10, 139–146. [Google Scholar] [CrossRef]
- Varani, K.; Vincenzi, F.; Merighi, S.; Gessi, S.; Borea, P.A. Biochemical and Pharmacological Role of A Adenosine Receptors and Their Modulation as Novel Therapeutic Strategy. Adv. Exp. Med Biol. 2017, 1051, 193–232. [Google Scholar] [PubMed]
- Heron, A.; Lasbennes, F.; Seylaz, J. Adenosine Modulation of Amino Acid Release in Rat Hippocampus during Ischemia and Veratridine Depolarization. Brain Res. 1993, 608, 27–32. [Google Scholar] [CrossRef]
- Phillis, J.W.; O’Regan, M.H.; Estevez, A.Y.; Song, D.; VanderHeide, S.J. Cerebral Energy Metabolism during Severe Ischemia of Varying Duration and Following Reperfusion. J. Neurochem. 1996, 67, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Hoffmann, C.; Cattabeni, F.; Abbracchio, M.P. Adenosine-Induced Cell Death: Evidence for Receptor-Mediated Signalling. Apoptosis 1999, 4, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Kristián, T.; Siesjö, B.K. Calcium in Ischemic Cell Death. Stroke 1998, 29, 705–718. [Google Scholar] [CrossRef]
- Chambers, D.E.; Parks, D.A.; Patterson, G.; Roy, R.; McCord, J.M.; Yoshida, S.; Parmley, L.F.; Downey, J.M. Xanthine Oxidase as a Source of Free Radical Damage in Myocardial Ischemia. J. Mol. Cell Cardiol. 1985, 17, 145–152. [Google Scholar] [CrossRef]
- Marcillat, O.; Zhang, Y.; Davies, K.J. Oxidative and Non-Oxidative Mechanisms in the Inactivation of Cardiac Mitochondrial Electron Transport Chain Components by Doxorubicin. Biochem. J. 1989, 259, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, H.F.; Poonit, N.; Zhang, Y.C.; Ye, C.Y.; Cai, H.L.; Yu, C.Y.; Zhou, Y.H.; Wu, B.B.; Cai, J.; Cai, X.H. Activating Adenosine A1 Receptor Accelerates PC12 Cell Injury via ADORA1/PKC/KATP Pathway after Intermittent Hypoxia Exposure. Mol. Cell Biochem. 2018, 446, 161–170. [Google Scholar] [CrossRef]
- Milton, S.L.; Nayak, G.; Kesaraju, S.; Kara, L.; Prentice, H.M. Suppression of Reactive Oxygen Species Production Enhances Neuronal Survival In Vitro and In Vivo in the Anoxia-Tolerant Turtle Trachemys Scripta. J. Neurochem. 2007, 101, 993–1001. [Google Scholar] [CrossRef]
- Lambert, C.M.; Roy, M.; Robitaille, G.A.; Richard, D.E.; Bonnet, S. HIF-1 Inhibition Decreases Systemic Vascular Remodelling Diseases by Promoting Apoptosis through a Hexokinase 2-Dependent Mechanism. Cardiovasc. Res. 2010, 88, 196–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, D.; Estlin, E.J.; Dive, C.; Makin, G.W.J. Chronic Hypoxia Promotes Hypoxia-Inducible Factor-1alpha-Dependent Resistance to Etoposide and Vincristine in Neuroblastoma Cells. Mol. Cancer Ther. 2006, 5, 2241–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Du, L. PERK Pathway Is Involved in Oxygen-Glucose-Serum Deprivation-Induced NF-Kb Activation via ROS Generation in Spinal Cord Astrocytes. Biochem. Biophys. Res. Commun. 2015, 467, 197–203. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
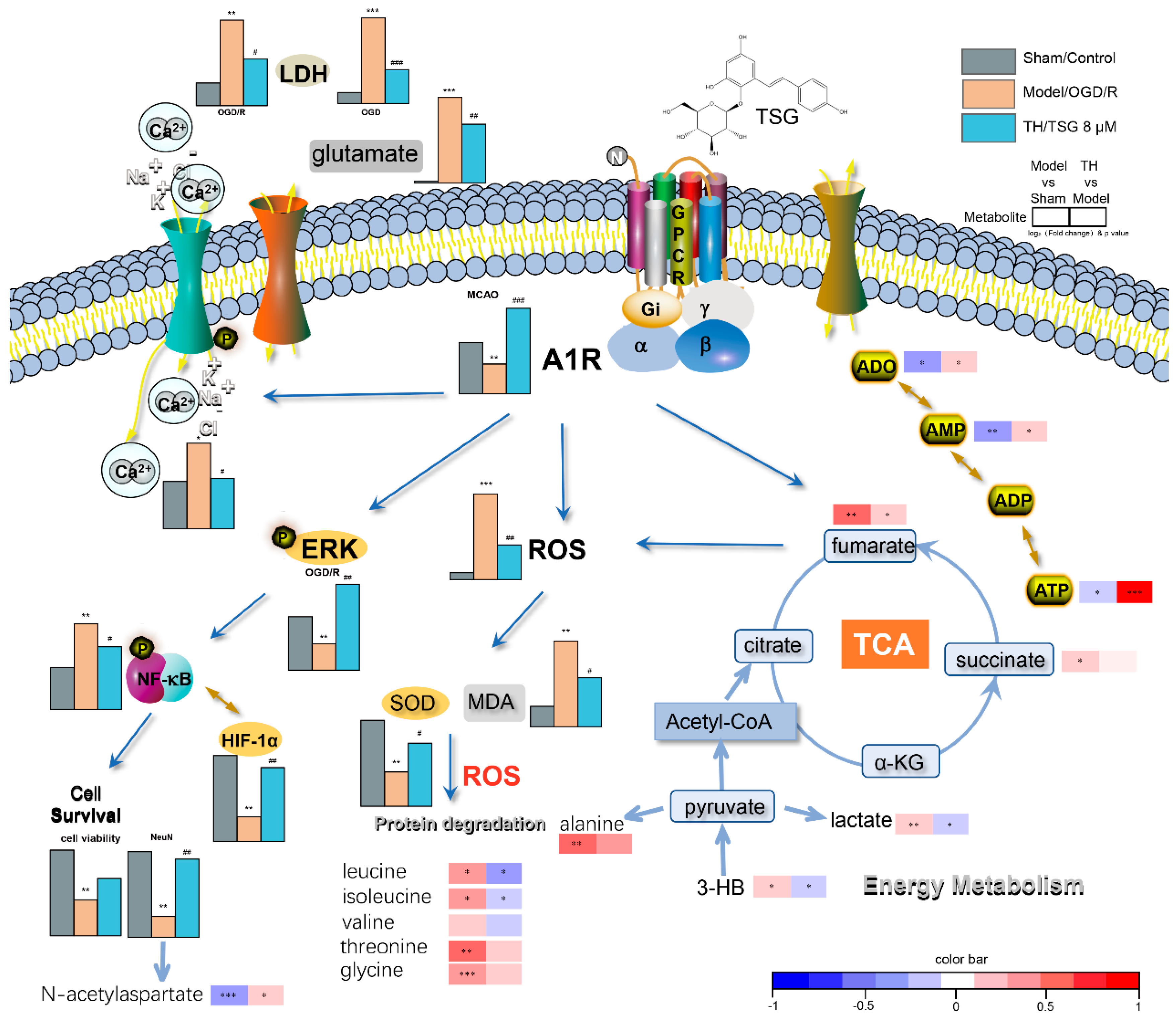{kind=link}
| Brain Metabolites | Model vs. Sham | TH vs. Model | TM vs. Model | TL vs. Model | ||||
|---|---|---|---|---|---|---|---|---|
| log2 (FC) | p | log2 (FC) | p | log2 (FC) | p | log2 (FC) | p | |
| Isoleucine | 0.41 | * | −0.12 | * | −0.18 | * | −0.28 | * |
| Leucine | 0.30 | * | −0.34 | * | −0.29 | * | −0.43 | * |
| Valine | 0.20 | −0.11 | −0.22 | −0.39 | ||||
| 3-hydroxybutyrate | 0.24 | * | −0.10 | * | 0.02 | −0.37 | * | |
| 2-hydroxyisobutyrate | 0.78 | *** | 0.22 | 0.28 | * | 0.09 | ||
| Lactate | 0.24 | ** | −0.14 | * | −0.28 | * | −0.29 | * |
| Alanine | 0.63 | ** | 0.39 | 0.31 | 0.12 | |||
| Lysine | 0.05 | −0.09 | −0.03 | −0.24 | ||||
| 4-Aminobutyrate | −0.18 | * | 0.25 | *** | 0.19 | ** | 0.22 | *** |
| N-acetyl aspartate | −0.37 | *** | 0.14 | * | −0.13 | 0.04 | ||
| Glutamate | 0.07 | −0.05 | 0.09 | * | 0.01 | |||
| Succinate | 0.15 | * | 0.09 | −0.04 | 0.39 | * | ||
| Glutamine | 0.11 | −0.12 | −0.03 | −0.11 | ||||
| Aspartate | −0.18 | ** | 0.14 | * | 0.07 | −0.06 | ||
| Trimethylamine | -0.09 | −0.32 | *** | −0.28 | ** | −0.28 | * | |
| Creatine | −0.18 | * | 0.05 | −0.10 | * | −0.14 | ||
| Choline | 0.19 | * | −0.06 | −0.05 | −0.12 | |||
| Phosphocholine | −0.01 | 0.09 | 0.16 | −0.07 | ||||
| Taurine | 0.00 | 0.02 | 0.13 | 0.09 | ||||
| Myo-inositol | 0.04 | 0.01 | 0.05 | 0.10 | ||||
| Glycine | 0.48 | *** | 0.21 | 0.30 | 0.18 | |||
| Threonine | 0.52 | ** | 0.21 | 0.17 | 0.21 | |||
| Ascorbate | −0.20 | *** | 0.15 | 0.44 | * | 0.01 | ||
| Adenosine | −0.50 | * | 0.17 | * | 0.10 | * | 0.05 | |
| Inosine | 0.06 | −0.29 | * | −0.47 | * | −0.37 | * | |
| Uridine | −0.02 | 0.08 | −0.15 | −0.09 | ||||
| Uracil | 0.22 | 0.19 | 0.13 | 0.20 | ||||
| AMP | −0.38 | ** | 0.20 | * | 0.56 | * | 0.52 | * |
| ADP | 0.09 | 0.27 | 0.39 | 0.01 | ||||
| ATP | −0.21 | * | 1.33 | *** | 1.34 | ** | 0.97 | * |
| Fumarate | 0.67 | ** | 0.24 | * | 0.29 | * | 0.09 | |
| Tyrosine | −0.10 | −0.06 | 0.03 | 0.02 | ||||
| Phenylalanine | 0.25 | * | −0.07 | −0.13 | −0.05 | |||
| Niacinamide | −0.09 | 0.05 | −0.13 | 0.05 | ||||
| Histidine | 0.01 | 0.09 | −0.05 | 0.97 | ||||
| Xanthine | −0.29 | ** | −0.06 | −0.08 | −0.19 | |||
| Oxypurinol | 0.38 | 0.09 | 0.33 | * | 0.27 | |||
| Formate | 0.40 | * | 0.73 | ** | 0.82 | ** | 0.92 | *** |
 . p-Values corrected by Benjamini–Hochberg methods were calculated based on a parametric Student’s t test or a nonparametric Mann–Whitney test (dependent on the conformity to normal distribution). * p < 0.05, ** p < 0.01 and *** p < 0.001.
. p-Values corrected by Benjamini–Hochberg methods were calculated based on a parametric Student’s t test or a nonparametric Mann–Whitney test (dependent on the conformity to normal distribution). * p < 0.05, ** p < 0.01 and *** p < 0.001.| Drug | Binding Energy Calculated by Autodock4 (kcal/mol) | Binding Energy Calculated by Autodock Vina (kcal/mol) |
|---|---|---|
| CCPA | −7.24 | −7.9 |
| CPA | −7.1 | −8.1 |
| CHA | −5.1 | −5.4 |
| MTA | −5.62 | −6.4 |
| NECA | −5.51 | −7.6 |
| Paeoniflorin | −7.39 | −6.9 |
| TSG | −7.71 | −7.0 |
| Adenosine | −6.49 | −6.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, L.; Li, G.; Zhao, W.; Meng, H.; Zheng, Q.; Wang, J. Activation of Adenosine A1 Receptor in Ischemic Stroke: Neuroprotection by Tetrahydroxy Stilbene Glycoside as an Agonist. Antioxidants 2021, 10, 1112. https://doi.org/10.3390/antiox10071112
Ruan L, Li G, Zhao W, Meng H, Zheng Q, Wang J. Activation of Adenosine A1 Receptor in Ischemic Stroke: Neuroprotection by Tetrahydroxy Stilbene Glycoside as an Agonist. Antioxidants. 2021; 10(7):1112. https://doi.org/10.3390/antiox10071112
Chicago/Turabian StyleRuan, Lingyu, Guanghui Li, Wenlong Zhao, Huihui Meng, Qi Zheng, and Junsong Wang. 2021. "Activation of Adenosine A1 Receptor in Ischemic Stroke: Neuroprotection by Tetrahydroxy Stilbene Glycoside as an Agonist" Antioxidants 10, no. 7: 1112. https://doi.org/10.3390/antiox10071112