
Betulinic Acid–Doxorubicin-Drug Combination Induced Apoptotic Death via ROS Stimulation in a Relapsed AML MOLM-13 Cell Model


Abstract
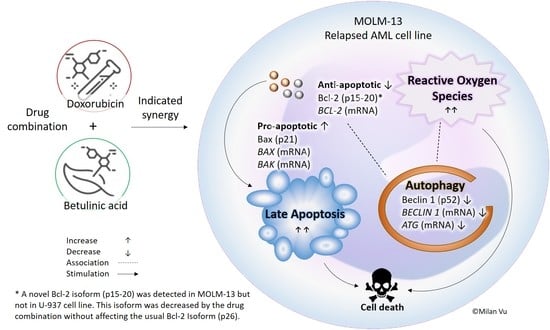
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drugs and Treatment Concentrations
2.3. Determination of Cell Viability and Cytotoxicity Using CyQUANT Direct® Assay
2.4. Cell Death Population Assays Using 488 Annexin V and PI: Flow Cytometry
2.5. Reactive Oxygen Species (ROS) Formation
2.6. Investigation of Proteins Involved in Cell Death: Western Blot Analysis
2.6.1. Cell Treatment
2.6.2. Antibodies
2.7. Investigating Gene Regulation via RT-PCR
2.7.1. Cell Treatment and RNA Isolation
2.7.2. Reverse Transcription–Polymerase Chain Reaction (RT-PCR)
2.8. Data and Statistical Analysis
3. Results
3.1. Combination of Betulinic Acid and Doxorubicin Synergistically Reduced Cell Viability in MOLM-13 AML Cell Line, but Did Not Significantly Affect the Viability of U-937 Cells
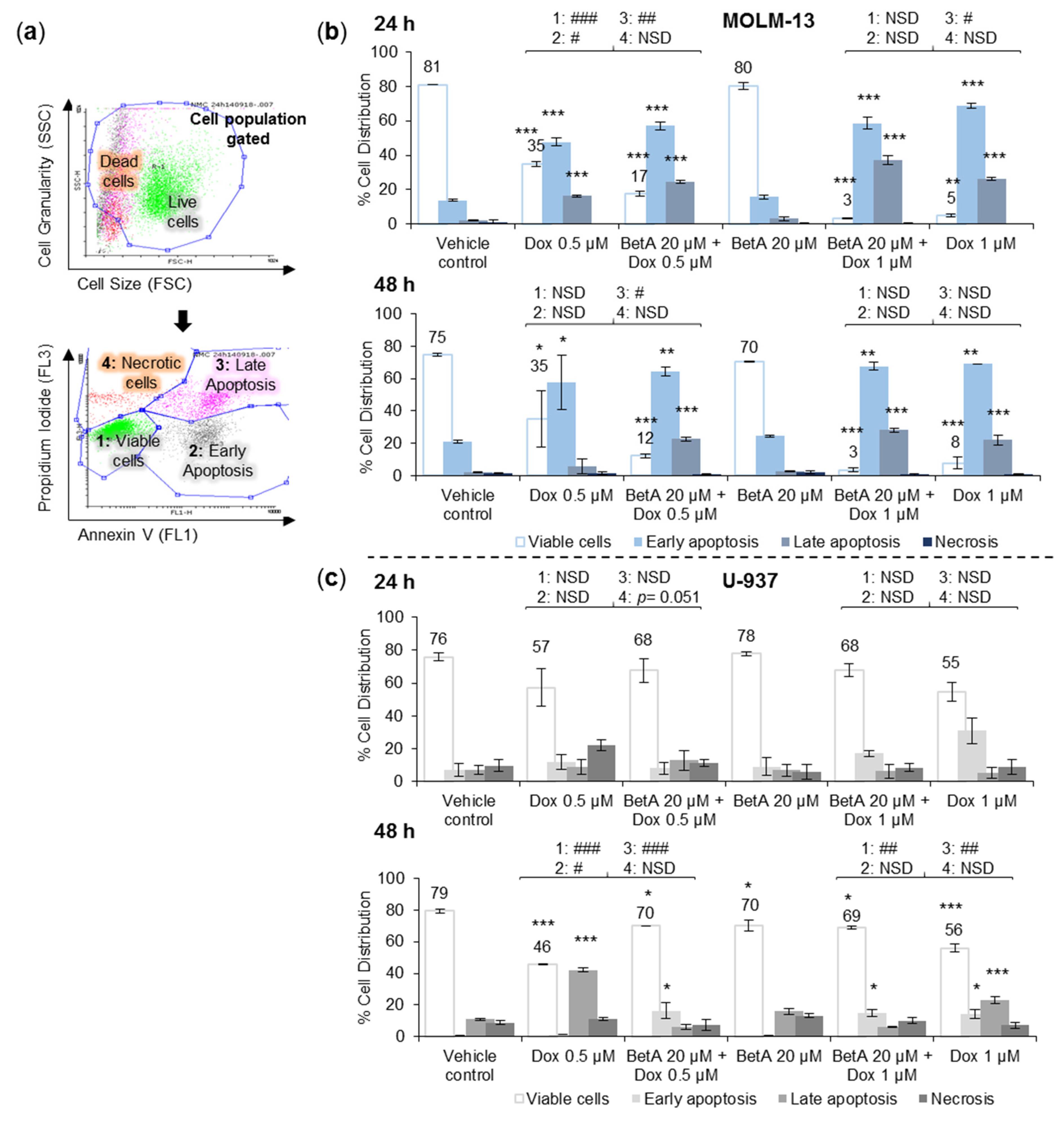
3.2. Combination Treatments Induced Apoptotic Death in MOLM-13 AML Cell Line, but Rescued U-937 Cells from Doxorubicin-Induced Cell Death
3.3. Betulinic Acid and Doxorubicin Combination Enhanced the Formation of Reactive Oxygen Species in MOLM-13 Cell Lines
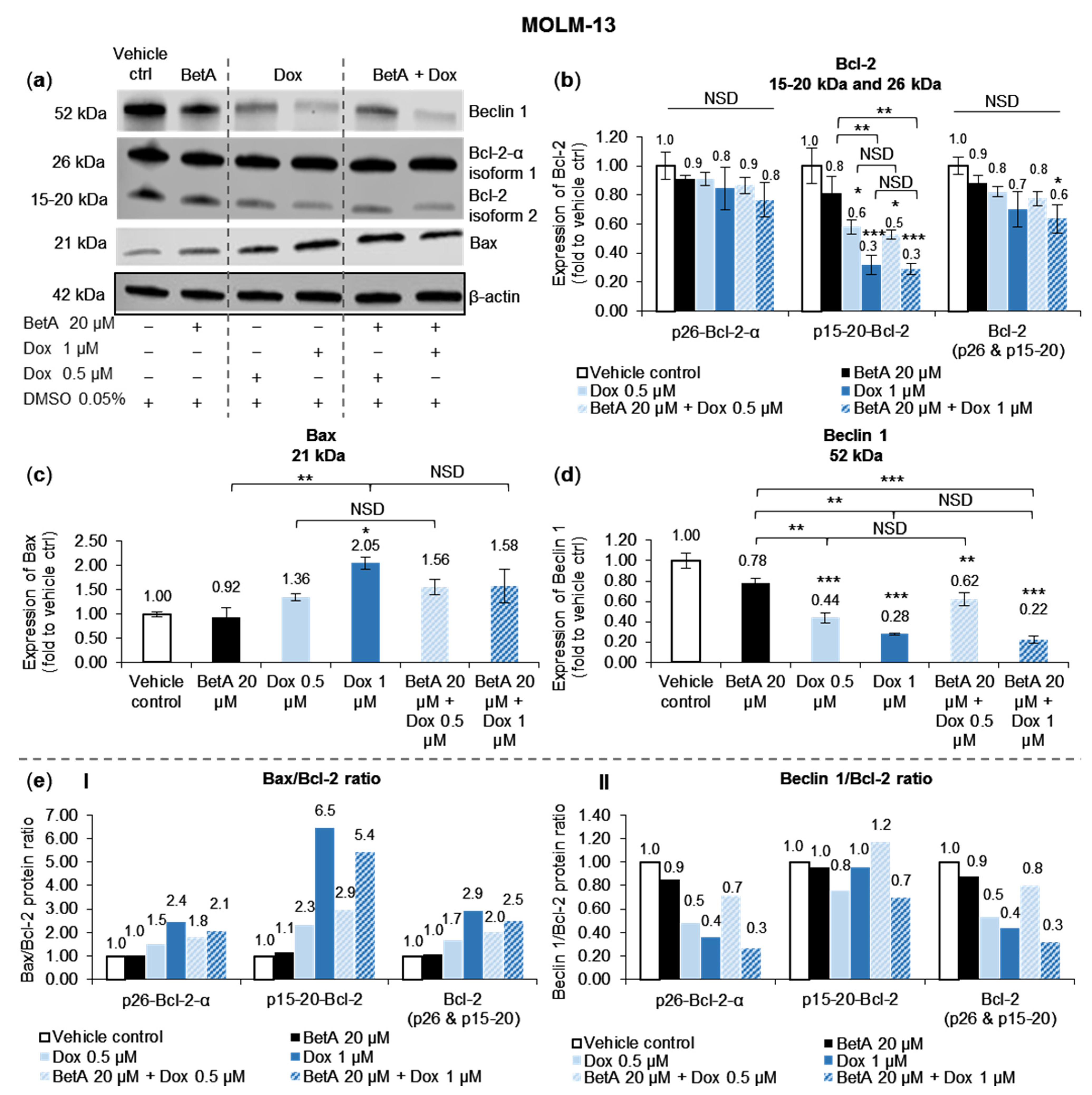
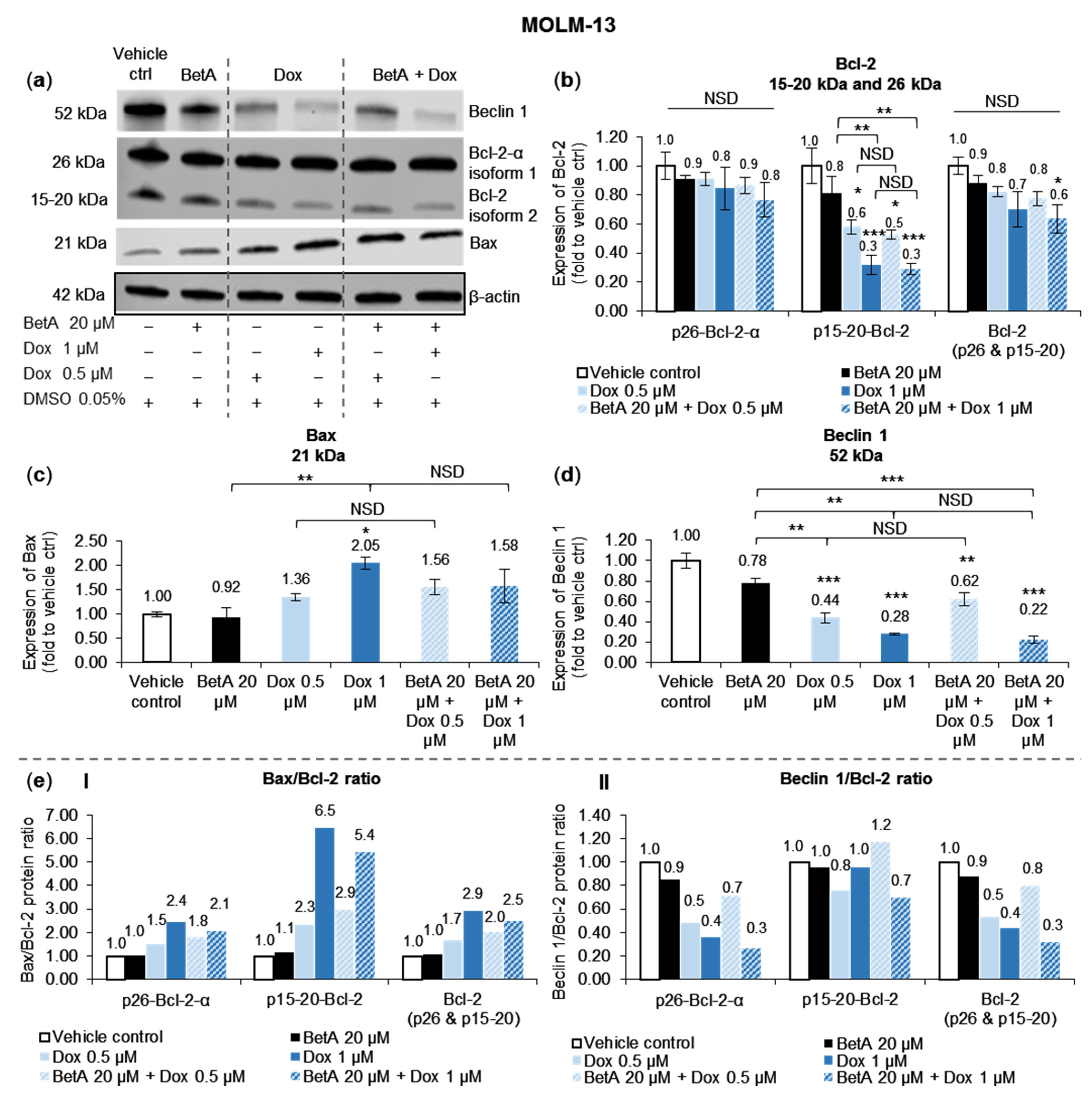
3.4. Doxorubicin, Alone and in Combination with Betulinic Acid, Inhibits a Novel Isoform of Bcl-2 in AML MOLM-13 Cells without a Potent Effect on the Main Bcl-2 Isoform
3.5. Autophagy Marker Beclin 1 Was Reduced by Doxorubicin and Betulinic Acid Co-Treatment in AML Cell Line
3.6. Apoptotic and Autophagy Signalling Protein Levels Were Not Altered by the Treatments in U-937 Cells
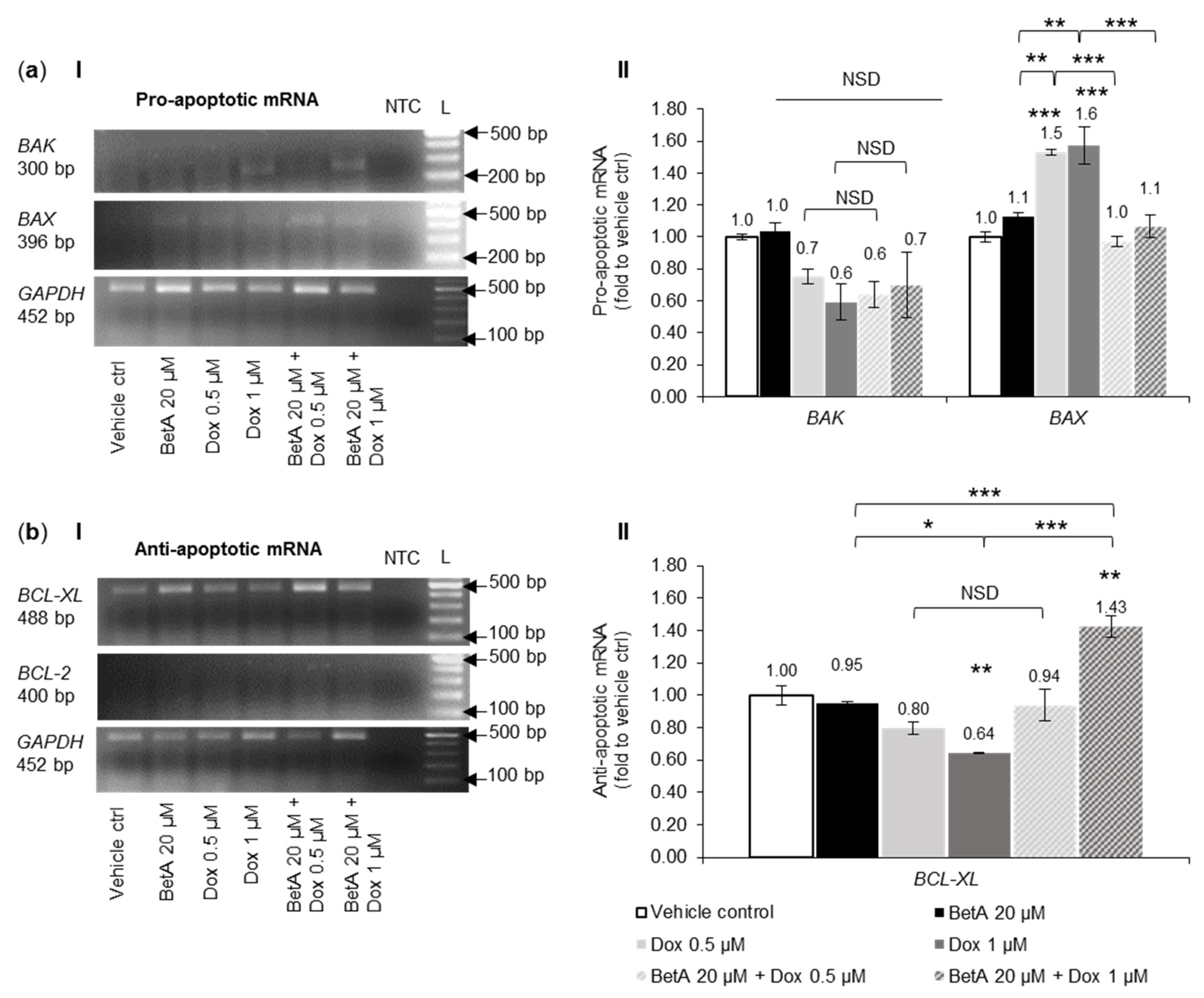
3.7. Combination Treatment Altered mRNA Expression of Bcl-2 Family Members and Autophagy towards Cell Death in MOLM-13 Cells, but Survival in U-937 Cells
4. Discussion
4.1. Cytotoxic Effect of Betulinic Acid on Leukaemia Cell Lines
4.2. The Effect of BetA-Dox Drug Combination on Cancer Cell Viability
4.3. Betulinic Acid Enhanced Anticancer Drug Activity of Doxorubicin by Sensitising the Cancer Cell Lines to Apoptosis and ROS Formation
4.4. Bcl-2 Protein Family Regulation by the Combination Treatment in Apoptotic Cell Death
4.5. Modulation of Autophagy upon Exposure by Betulinic Acid, Doxorubicin and Drug Combination
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ehrhardt, H.; Fulda, S.; Führer, M.; Debatin, K.M.; Jeremias, I. Betulinic Acid-Induced Apoptosis in Leukemia Cells. Leukemia 2004, 18, 1406–1412. [Google Scholar] [CrossRef]
- Selzer, E.; Pimentel, E.; Wacheck, V.; Schlegel, W.; Pehamberger, H.; Jansen, B.; Kodym, R. Effects of Betulinic Acid Alone and in Combination with Irradiation in Human Melanoma Cells. J. Investig. Dermatol. 2000, 114, 935–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aisha, A.F.A.; Abu-Salah, K.M.; Ismail, Z.; Majid, A.M.S.A. α-Mangostin Enhances Betulinic Acid Cytotoxicity and Inhibits Cisplatin Cytotoxicity on HCT 116 Colorectal Carcinoma Cells. Molecules 2012, 17, 2939–2954. [Google Scholar] [CrossRef] [Green Version]
- Faujan, H.N.; Alitheen, B.N.; Yeap, K.S.; Ali, A.; Muhajir, H.A.; Ahmad, F.H.B. Cytotoxic Effect of Betulinic Acid and Betulinic Acid Acetate Isolated from Melaleuca Cajuput on Human Myeloid Leukemia (HL-60) Cell Line. Afr. J. Biotechnol. 2010, 9, 6387–6396. [Google Scholar]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective Cytotoxicity of Betulinic Acid on Tumor Cell Lines, but Not on Normal Cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.-M. Sensitization for Anticancer Drug-Induced Apoptosis by Betulinic Acid. Neoplasia 2005, 7, 162–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Friesen, C.; Los, M.; Scaffidi, C.; Mier, W.; Benedict, M.; Nunez, G.; Krammer, P.H.; Peter, M.E.; Debatin, K.-M. Betulinic Acid Triggers CD95 (APO-1/Fas)- and P53-Independent Apoptosis via Activation of Caspases in Neuroectodermal Tumors. Cancer Res. 1997, 57, 4956. [Google Scholar] [PubMed]
- Liu, W.-K.; Ho, J.C.K.; Cheung, F.W.K.; Liu, B.P.L.; Ye, W.-C.; Che, C.-T. Apoptotic Activity of Betulinic Acid Derivatives on Murine Melanoma B16 Cell Line. Eur. J. Pharmacol. 2004, 498, 71–78. [Google Scholar] [CrossRef]
- Gopal, D.V.R.; Archana, A.N.; Badrinath, Y.; Mishra, K.P.; Joshi, D.S. Betulinic Acid Induces Apoptosis in Human Chronic Myelogenous Leukemia (CML) Cell Line K-562 without Altering the Levels of Bcr-Abl. Toxicol. Lett. 2005, 155, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Thurnher, D.; Turhani, D.; Pelzmann, M.; Wannemacher, B.; Knerer, B.; Formanek, M.; Wacheck, V.; Selzer, E. Betulinic Acid: A New Cytotoxic Compound against Malignant Head and Neck Cancer Cells. Head Neck 2003, 25, 732–740. [Google Scholar] [CrossRef]
- Mullauer, F.B.; Kessler, J.H.; Medema, J.P. Betulinic Acid Induces Cytochrome c Release and Apoptosis in a Bax/Bak-Independent, Permeability Transition Pore Dependent Fashion. Apoptosis 2009, 14, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Luo, W. Betulinic Acid Induces Bax/Bak-Independent Cytochrome c Release in Human Nasopharyngeal Carcinoma Cells. Mol. Cells 2012, 33, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Shankar, E.; Zhang, A.; Franco, D.; Gupta, S. Betulinic Acid-Mediated Apoptosis in Human Prostate Cancer Cells Involves P53 and Nuclear Factor-Kappa B (NF-ΚB) Pathways. Molecules 2017, 22, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panno, J. Cancer; Infobase Learning: New York, NY, USA, 2004. [Google Scholar]
- National Cancer Institute Drugs Approved for Leukemia. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/leukemia (accessed on 10 October 2020).
- Tallman, M.S.; Gilliland, D.G.; Rowe, J.M. Drug Therapy for Acute Myeloid Leukemia. Blood 2005, 106, 1154–1163. [Google Scholar] [CrossRef]
- Cools, J. Primetime for Chemotherapy in Acute Myeloid Leukemia. Haematologica 2012, 97, 1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamen, S.; Anel, A.; Pérez-Galán, P.; Lasierra, P.; Johnson, D.; Piñeiro, A.; Naval, J. Doxorubicin Treatment Activates a Z-VAD-Sensitive Caspase, Which Causes ΔΨm Loss, Caspase-9 Activity, and Apoptosis in Jurkat Cells. Exp. Cell Res. 2000, 258, 223–235. [Google Scholar] [CrossRef]
- Panaretakis, T.; Pokrovskaja, K.; Shoshan, C.M.; Grander, D. Activation of Bak, Bax, and BH3-Only Proteins in the Apoptotic Response to Doxorubicin. J. Biol. Chem. 2002, 277, 44317–44326. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Konorev, E.A.; Kotamraju, S.; Joseph, J.; Kalivendi, S.; Kalyanaraman, B. Doxorubicin Induces Apoptosis in Normal and Tumor Cells via Distinctly Different Mechanisms. J. Biol. Chem. 2004, 279, 25535–25543. [Google Scholar] [CrossRef] [Green Version]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Vu, M.; Kassouf, N.; Ofili, R.; Lund, T.; Bell, C.; Appiah, S. Doxorubicin Selectively Induces Apoptosis through the Inhibition of a Novel Isoform of Bcl-2 in Acute Myeloid Leukaemia MOLM-13 Cells with Reduced Beclin 1 Expression. Int. J. Oncol. 2020, 57, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Sellers, R.W.; Fisher, E.D. Apoptosis and Cancer Drug Targeting. J. Clin. Investig. 1999, 104, 1655–1661. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A. Targeting the Extrinsic Apoptosis Pathway in Cancer. Cytokine Growth Factor Rev. 2008, 19, 325–331. [Google Scholar] [CrossRef]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.-T.; Zhou, T.-T.; Liu, B.; Bao, J.-K. Programmed Cell Death Pathways in Cancer: A Review of Apoptosis, Autophagy and Programmed Necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Z.; Li, Y.; Xia, J.; Li, D.; Li, H.; Ren, M.; Liao, Y.; Yu, S.; Chen, Y.; et al. Cell Apoptosis, Autophagy and Necroptosis in Osteosarcoma Treatment. Oncotarget 2016, 7, 44763–44778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, A.; Katz, S.G. Non-Apoptotic Functions of BCL-2 Family Proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High Expression of Bcl-2 Protein in Acute Myeloid Leukemia Cells Is Associated with Poor Response to Chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensi, L.; Longo, R.; Vecchi, A.; Messora, C.; Garagnani, L.; Bernardi, S.; Tamassia, M.G.; Sacchi, S. Bcl-2 Oncoprotein Expression in Acute Myeloid Leukemia. Haematologica 1995, 80, 98–102. [Google Scholar] [PubMed]
- Zinkel, S.; Gross, A.; Yang, E. BCL2 Family in DNA Damage and Cell Cycle Control. Cell Death Differ. 2006, 13, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Sohn, S.K.; Lee, M.-H.; Jang, J.H.; Kim, K.; Jung, C.W.; Kim, D.H. BCL2 Gene Polymorphism Could Predict the Treatment Outcomes in Acute Myeloid Leukemia Patients. Leuk. Res. 2009, 34, 166–172. [Google Scholar] [CrossRef]
- Radogna, F.; Dicato, M.; Diederich, M. Cancer-Type-Specific Crosstalk between Autophagy, Necroptosis and Apoptosis as a Pharmacological Target. Biochem. Pharmacol. 2015, 94, 1–11. [Google Scholar] [CrossRef]
- Marquez, R.T.; Xu, L. Bcl-2:Beclin 1 Complex: Multiple, Mechanisms Regulating Autophagy/Apoptosis Toggle Switch. Am. J. Cancer Res. 2012, 2, 214–221. [Google Scholar]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between Apoptosis, Necrosis and Autophagy. BBA-Mol. Cell Res. 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Yao, Z.; Klionsky, D.J. How to Control Self-Digestion: Transcriptional, Post-Transcriptional, and Post-Translational Regulation of Autophagy. Trends Cell Biol. 2015, 25, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Viji, V.; Shobha, B.; Kavitha, S.K.; Ratheesh, M.; Kripa, K.; Helen, A. Betulinic Acid Isolated from Bacopa Monniera (L.) Wettst Suppresses Lipopolysaccharide Stimulated Interleukin-6 Production through Modulation of Nuclear Factor-ΚB in Peripheral Blood Mononuclear Cells. Int. Immunopharmacol. 2010, 10, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Pisha, E.; Kinghorn, A.D.; Lee, I.-S.; Pezzuto, J.M.; Hieken, T.J.; Brown, D.M.; Wall, M.E.; das Gupta, T.K.; Chagwedera, T.E.; Wani, M.C.; et al. Discovery of Betulinic Acid as a Selective Inhibitor of Human Melanoma That Functions by Induction of Apoptosis. Nat. Med. 1995, 1, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- McHowat, J.; Swift, L.M.; Arutunyan, A.; Sarvazyan, N. Clinical Concentrations of Doxorubicin Inhibit Activity of Myocardial Membrane-Associated, Calcium-Independent Phospholipase A(2). Cancer Res. 2001, 61, 4024–4029. [Google Scholar]
- Jones, L.J.; Gray, M.; Yue, S.T.; Haugland, R.P.; Singer, V.L. Sensitive Determination of Cell Number Using the CyQUANT® Cell Proliferation Assay. J. Immunol. Methods 2001, 254, 85–98. [Google Scholar] [CrossRef]
- Invitrogen CyQUANT® Direct Cell Proliferation Assay Kit. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/mp35011.pdf (accessed on 3 March 2020).
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C. The Mass-Action Law Based Algorithms for Quantitative Econo-Green Bio-Research. Integr. Biol. Quant. Biosci. Nano Macro 2011, 3, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal Evolution in Relapsed Acute Myeloid Leukaemia Revealed by Whole-Genome Sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Chintharlapalli, S.; Papineni, S.; Ramaiah, S.K.; Safe, S. Betulinic Acid Inhibits Prostate Cancer Growth through Inhibition of Specificity Protein Transcription Factors. Cancer Res. 2007, 67, 2816–2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, J.H.; Mullauer, F.B.; de Roo, G.M.; Medema, J.P. Broad in Vitro Efficacy of Plant-Derived Betulinic Acid against Cell Lines Derived from the Most Prevalent Human Cancer Types. Cancer Lett. 2007, 251, 132–145. [Google Scholar] [CrossRef]
- Fulda, S. Betulinic Acid for Cancer Treatment and Prevention. Int. J. Mol. Sci. 2008, 9, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Pang, Q.; Zhou, D.; Zhang, A.; Luo, S.; Wang, Y.; Yan, X. Proteomic Investigation into Betulinic Acid-Induced Apoptosis of Human Cervical Cancer HeLa Cells. PLoS ONE 2014, 9, e105768. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.; Castilho, R.O.; da Costa, M.R.; Wagner-Souza, K.; Coelho Kaplan, M.A.; Gattass, C.R. Pentacyclic Triterpenes from Chrysobalanaceae Species: Cytotoxicity on Multidrug Resistant and Sensitive Leukemia Cell Lines. Cancer Lett. 2003, 190, 165–169. [Google Scholar] [CrossRef]
- Hata, K.; Hori, K.; Ogasawara, H.; Takahashi, S. Anti-Leukemia Activities of Lup-28-al-20(29)-En-3-One, a Lupane Triterpene. Toxicol. Lett. 2003, 143, 1–7. [Google Scholar] [CrossRef]
- Li, Q.; Li, Y.; Wang, X.; Fang, X.; He, K.; Guo, X.; Zhan, Z.; Sun, C.; Jin, Y. Co-treatment with Ginsenoside Rh2 and Betulinic Acid Synergistically Induces Apoptosis in Human Cancer Cells in Association with Enhanced Capsase-8 Activation, Bax Translocation, and Cytochrome c Release. Mol. Carcinog. 2011, 50, 760–769. [Google Scholar] [CrossRef]
- Fulda, S.; Jeremias, I.; Debatin, K.-M. Cooperation of Betulinic Acid and TRAIL to Induce Apoptosis in Tumor Cells. Oncogene 2004, 23, 7611–7620. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Margolin, A.A.; Haery, L.; Brown, E.; Cucolo, L.; Julian, B.; Shehata, S.; Kung, A.L.; Beroukhim, R.; Golub, T.R. Chemical Genomics Identifies Small-Molecule MCL1 Repressors and BCL-XL as a Predictor of MCL1 Dependency. Cancer Cell 2012, 21, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Poon, I.K.H.; Hulett, M.D.; Parish, C.R. Molecular Mechanisms of Late Apoptotic Necrotic Cell Clearance. Cell Death Differ. 2010, 17, 381–397. [Google Scholar] [CrossRef] [Green Version]
- Chou, D.; Adamson, B.; Dephoure, N.; Tan, X.; Nottke, A.; Hurov, K.; Gygi, S.; Colaiácov, M.; Elledge, S. A Chromatin Localization Screen Reveals Poly (ADP Ribose)-Regulated Recruitment of the Repressive Polycomb and NuRD Complexes to Sites of DNA Damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, A.; Das, B.; Roy, A.; Sen, N.; Dasgupta, S.B.; Mukhopadhayay, S.; Majumder, H.K. Betulinic Acid, a Catalytic Inhibitor of Topoisomerase I, Inhibits Reactive Oxygen Species Mediated Apoptotic Topoisomerase I DNA Cleavable Complex Formation in Prostate Cancer Cells but Does Not Affect the Process of Cell Death. Cancer Res. 2007, 67, 11848–11858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive Oxygen Species and Cancer Paradox: To Promote or to Suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial Oxidative Stress: Implications for Cell Death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Wang, S.X.; Wen, X.; Bell, C.; Appiah, S. Liposome-Delivered Baicalein Induction of Myeloid Leukemia K562 Cell Death via Reactive Oxygen Species Generation. Mol. Med. Rep. 2018, 17, 4524–4530. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Yu, R.; Pezzuto, J. Betulinic Acid-Induced Programmed Cell Death in Human Melanoma Cells Involves Mitogen-Activated Protein Kinase Activation. Clin. Cancer Res. 2003, 9, 2866. [Google Scholar]
- Fulda, S.; Kroemer, G. Targeting Mitochondrial Apoptosis by Betulinic Acid in Human Cancers. Drug Discov. Today 2009, 14, 885–890. [Google Scholar] [CrossRef]
- Xu, T.; Pang, Q.; Wang, Y.; Yan, X. Betulinic Acid Induces Apoptosis by Regulating PI3K/Akt Signaling and Mitochondrial Pathways in Human Cervical Cancer Cells. Int. J. Mol. Med. 2017, 40, 1669–1678. [Google Scholar] [CrossRef] [Green Version]
- Fornari, F.; Randolph, J.; Yalowich, J.; Ritke, M.; Gewirtz, D. Interference by Doxorubicin with DNA Unwinding in MCF-7 Breast Tumor Cells. Mol. Pharmacol. 1994, 45, 649–656. [Google Scholar]
- Gewirtz, D. A Critical Evaluation of the Mechanisms of Action Proposed for the Antitumor Effects of the Anthracycline Antibiotics Adriamycin and Daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Acésio, N.; Oliveira, P.; Mastrocola, D.; Lima, I.; Munari, C.; Sato, V.; Souza, A.; Faluzino, L.; Cunha, W.; Tavares, D. Modulatory Effect of Betulinic Acid on the Genotoxicity Induced by Different Mutagens in V79 Cells. Evid.-Based Complementary Altern. Med. ECAM 2016, 2016, 8942730–8942736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.M.; Cory, S. The Bcl-2 Apoptotic Switch in Cancer Development and Therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [Green Version]
- Rzeski, W.; Stepulak, A.; Szymański, M.; Sifringer, M.; Kaczor, J.; Wejksza, K.; Zdzisińska, B.; Kandefer-Szerszeń, M. Betulinic Acid Decreases Expression of Bcl-2 and Cyclin D1, Inhibits Proliferation, Migration and Induces Apoptosis in Cancer Cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2006, 374, 11–20. [Google Scholar] [CrossRef]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of Doxorubicin on Apoptosis and Oxidative Stress in Breast Cancer Cell Lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Bien, S.; Rimmbach, C.; Neumann, H.; Niessen, J.; Reimer, E.; Ritter, C.A.; Rosskopf, D.; Cinatl, J.; Michaelis, M.; Schroeder, H.W.S.; et al. Doxorubicin-Induced Cell Death Requires Cathepsin B in HeLa Cells. Biochem. Pharmacol. 2010, 80, 1466–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, T.; Güell, M.; Serrano, L. Correlation of MRNA and Protein in Complex Biological Samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [Green Version]
- Yip, K.W.; Reed, J.C. Bcl-2 Family Proteins and Cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of Action of Bcl-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C. The Dynamic Nature of Autophagy in Cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise Protects against Doxorubicin-Induced Markers of Autophagy Signaling in Skeletal Muscle. J. Appl. Physiol. 2011, 111, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizarro, M.; Troncoso, R.; Martínez, G.J.; Chiong, M.; Castro, P.F.; Lavandero, S. Basal Autophagy Protects Cardiomyocytes from Doxorubicin-Induced Toxicity. Toxicology 2016, 370, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Schott, C.R.; Ludwig, L.; Mutsaers, A.J.; Foster, R.A.; Wood, G.A. The Autophagy Inhibitor Spautin-1, Either Alone or Combined with Doxorubicin, Decreases Cell Survival and Colony Formation in Canine Appendicular Osteosarcoma Cells. PLoS ONE 2018, 13, e0206427. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, Y.; He, J.; Yi, S.; Wen, L.; Zhao, J.; Zhang, P.B.; Cui, H.G. Betulinic Acid Inhibits Autophagic Flux and Induces Apoptosis in Human Multiple Myeloma Cells in Vitro. Acta Pharmacol. Sin. 2012, 33, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Hseu, Y.-C.; Tsai, T.-J.; Korivi, M.; Liu, J.-Y.; Chen, H.-J.; Lin, C.-M.; Shen, Y.-C.; Yang, H.-L. Antitumor Properties of Coenzyme Q0 against Human Ovarian Carcinoma Cells via Induction of ROS-Mediated Apoptosis and Cytoprotective Autophagy. Sci. Rep. 2017, 7, 1–21. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, M.-Y.; Wang, T.; Fan, Y.-Y.; Yu, L.-S.; Ye, G.-H.; Wang, Z.-F.; Gao, C.; Wang, H.-C.; Luo, C.-L.; et al. IL-33/ST2L Signaling Provides Neuroprotection Through Inhibiting Autophagy, Endoplasmic Reticulum Stress, and Apoptosis in a Mouse Model of Traumatic Brain Injury. Front. Cell. Neurosci. 2018, 12, 95. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, L.; Zheng, H.; Mao, C.; Hu, W.; Xiong, K.; Wang, F.; Liu, C. Application and Interpretation of Current Autophagy Inhibitors and Activators. Acta Pharm. Sin 2013, 34, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Primer Sequence (5′–3′) | Product Size (bp) |
|---|---|---|---|
| Pro-apoptotic Bcl-2 family | |||
| BAK | NM_001188.4 | Fwd: CTGTTTTTACCGCCATCAGCAGG | 249 |
| Rev: CTCTCAAACAGGCTGGTGGCAATC | |||
| BAX | NM_001291428.2 | Fwd: CCGTTCATCTCAGTCCCCTG | 396 |
| Rev: GAAGTGTGTCCCGAAGGAGG | |||
| Anti-apoptotic Bcl-2 family | |||
| BCL-2 | NM_000633.3 | Fwd: GACTTCTTCCGCCGCTACCG | 341 |
| Rev: GACAGCCAGGAGAAATGAAAC | |||
| BCL-XL | NM_138578.3 | Fwd: CCCAGAAAGGATACAGCTGG | 488 |
| Rev: GCGATCCGACTCACCAATAC | |||
| Autophagy marker | |||
| ATG5 | NM_004849.4 | Fwd: TCTAAGGATGCAATTGAAGCTCA | 153 |
| Rev: GGCCCAAAACTGGTCAAATCT | |||
| BECLIN 1 | NM_003766.5 | Fwd: GCTGGAAGACGTGGAAAAGA | 135 |
| Rev: TCCAGCTGCTGTCGTTTAAATT | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, M.; Kassouf, N.; Appiah, S. Betulinic Acid–Doxorubicin-Drug Combination Induced Apoptotic Death via ROS Stimulation in a Relapsed AML MOLM-13 Cell Model. Antioxidants 2021, 10, 1456. https://doi.org/10.3390/antiox10091456
Vu M, Kassouf N, Appiah S. Betulinic Acid–Doxorubicin-Drug Combination Induced Apoptotic Death via ROS Stimulation in a Relapsed AML MOLM-13 Cell Model. Antioxidants. 2021; 10(9):1456. https://doi.org/10.3390/antiox10091456
Chicago/Turabian StyleVu, Milan, Nick Kassouf, and Sandra Appiah. 2021. "Betulinic Acid–Doxorubicin-Drug Combination Induced Apoptotic Death via ROS Stimulation in a Relapsed AML MOLM-13 Cell Model" Antioxidants 10, no. 9: 1456. https://doi.org/10.3390/antiox10091456
APA StyleVu, M., Kassouf, N., & Appiah, S. (2021). Betulinic Acid–Doxorubicin-Drug Combination Induced Apoptotic Death via ROS Stimulation in a Relapsed AML MOLM-13 Cell Model. Antioxidants, 10(9), 1456. https://doi.org/10.3390/antiox10091456