
Iron–Sulfur Cluster Biogenesis as a Critical Target in Cancer


Abstract
:1. Introduction
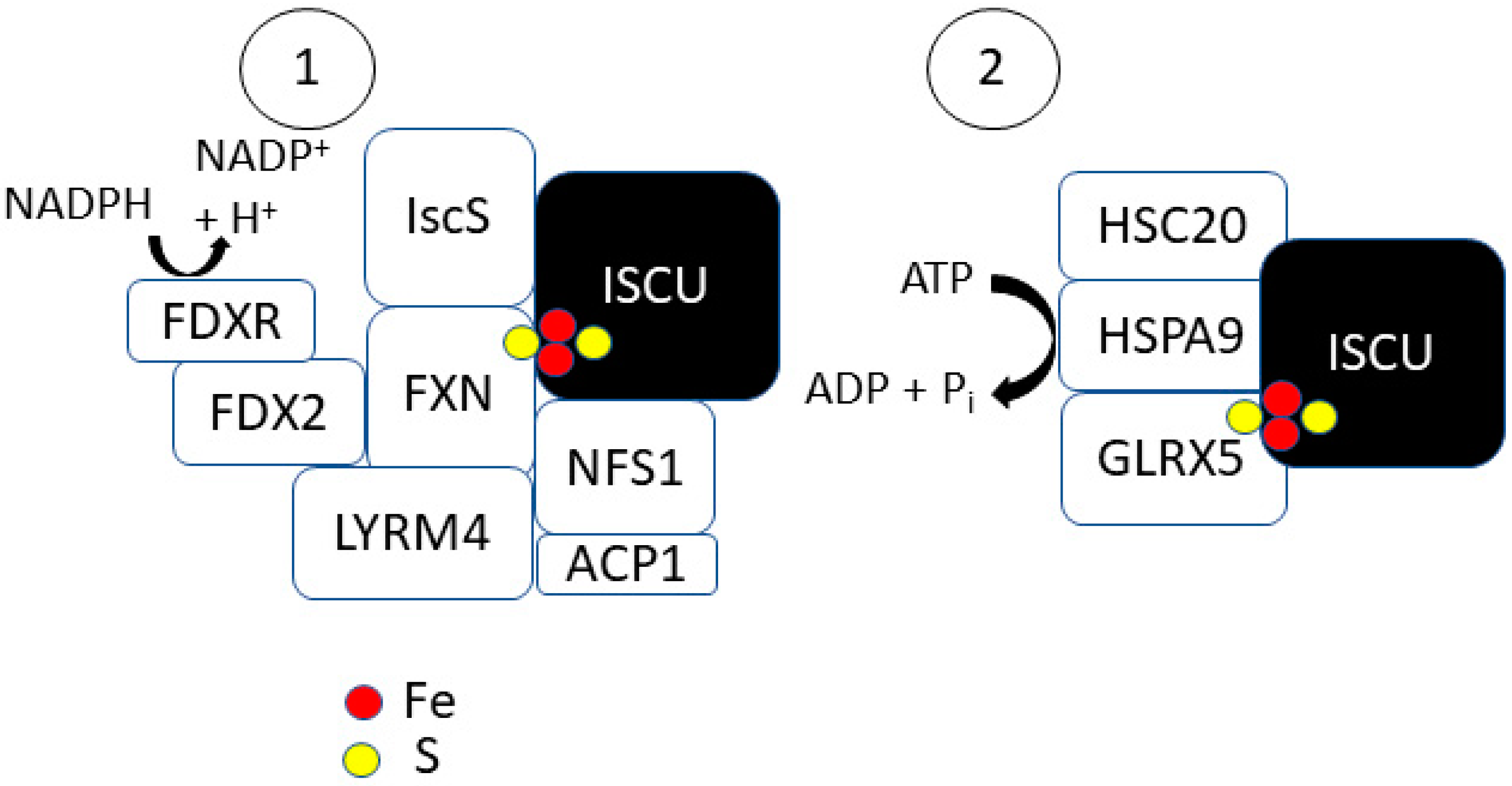
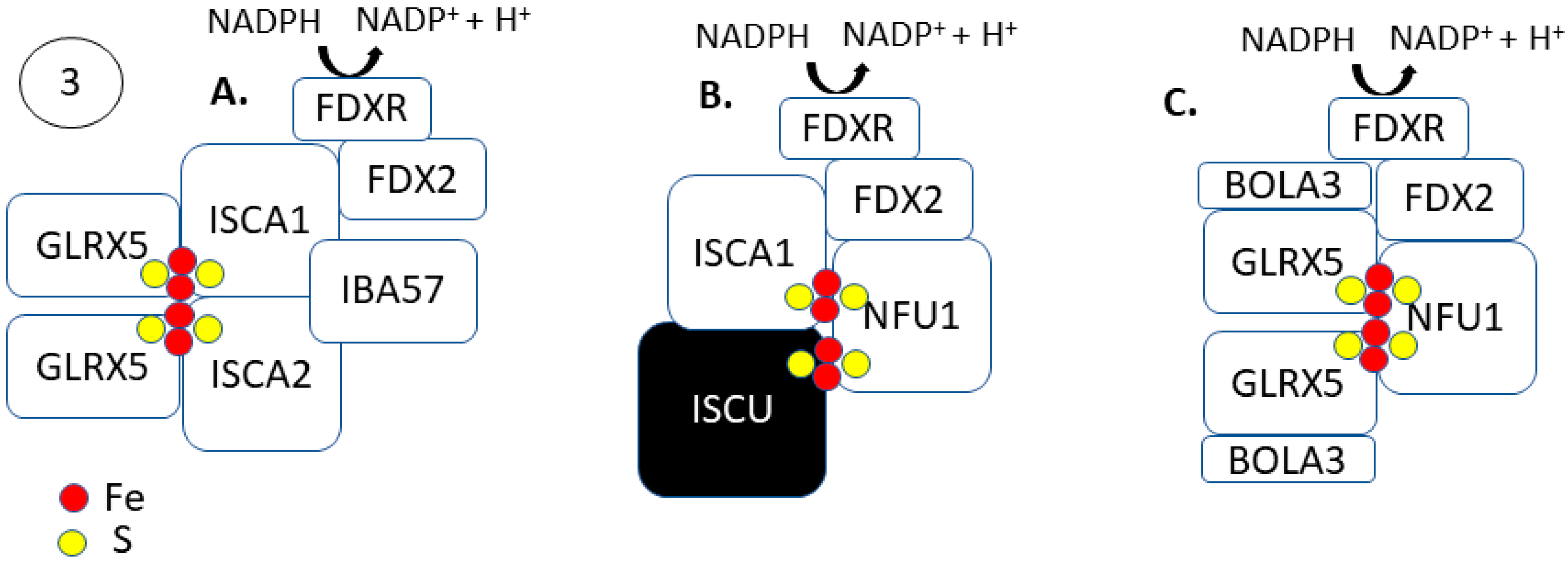
2. Overview of Fe–S Biogenesis
- [2Fe-2S] cluster formation on a scaffold protein ISCU;
- Chaperone protein-mediated release and trafficking of the newly formed [2Fe-2S] cluster to target proteins;
- Conversion of [2Fe-2S] clusters to [4Fe-4S] clusters;
- Incorporation of newly formed [4Fe-4S] clusters into apo-proteins.
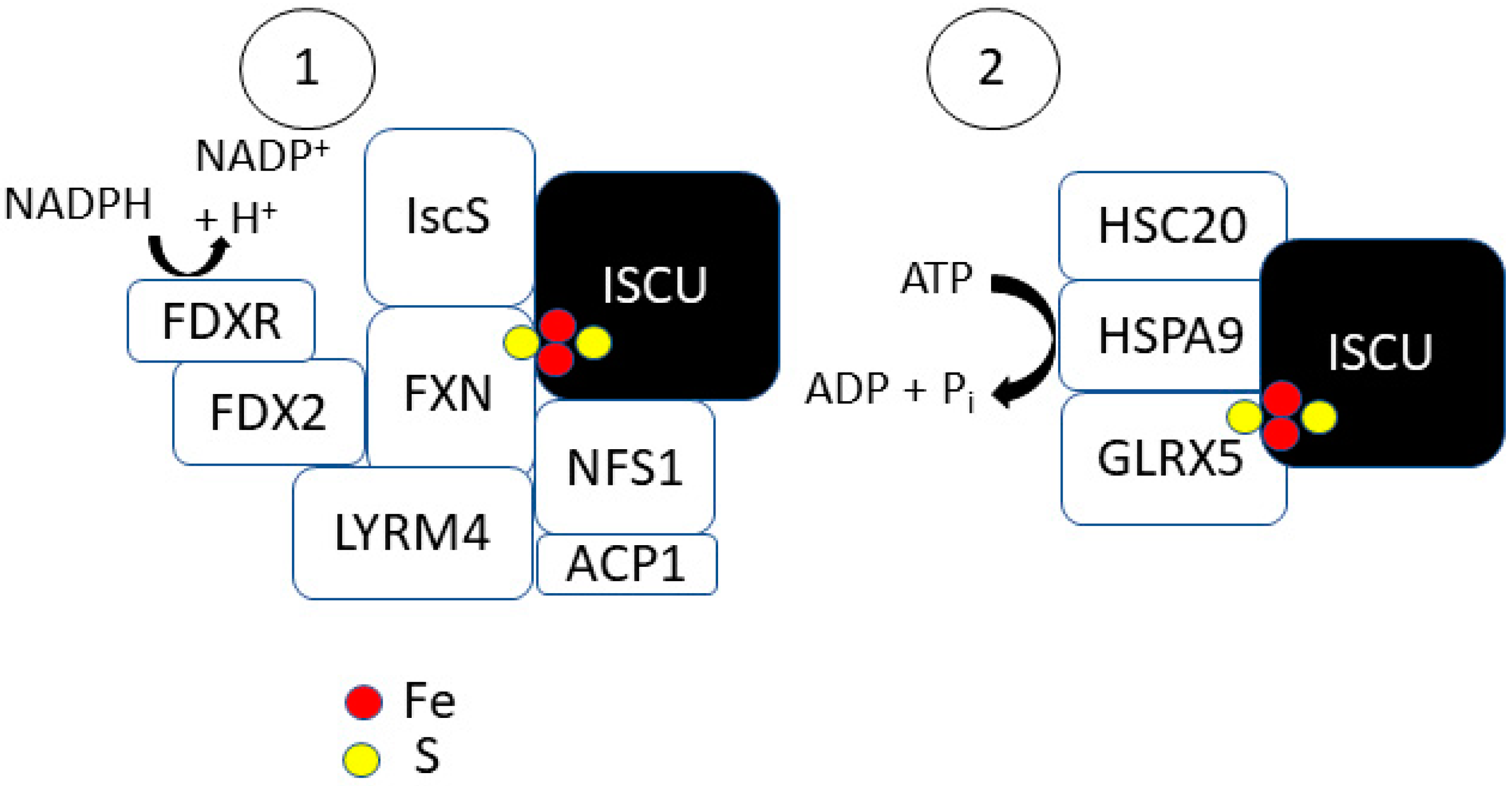
2.1. Step 1: [2Fe-2S] Synthesis on ISCU
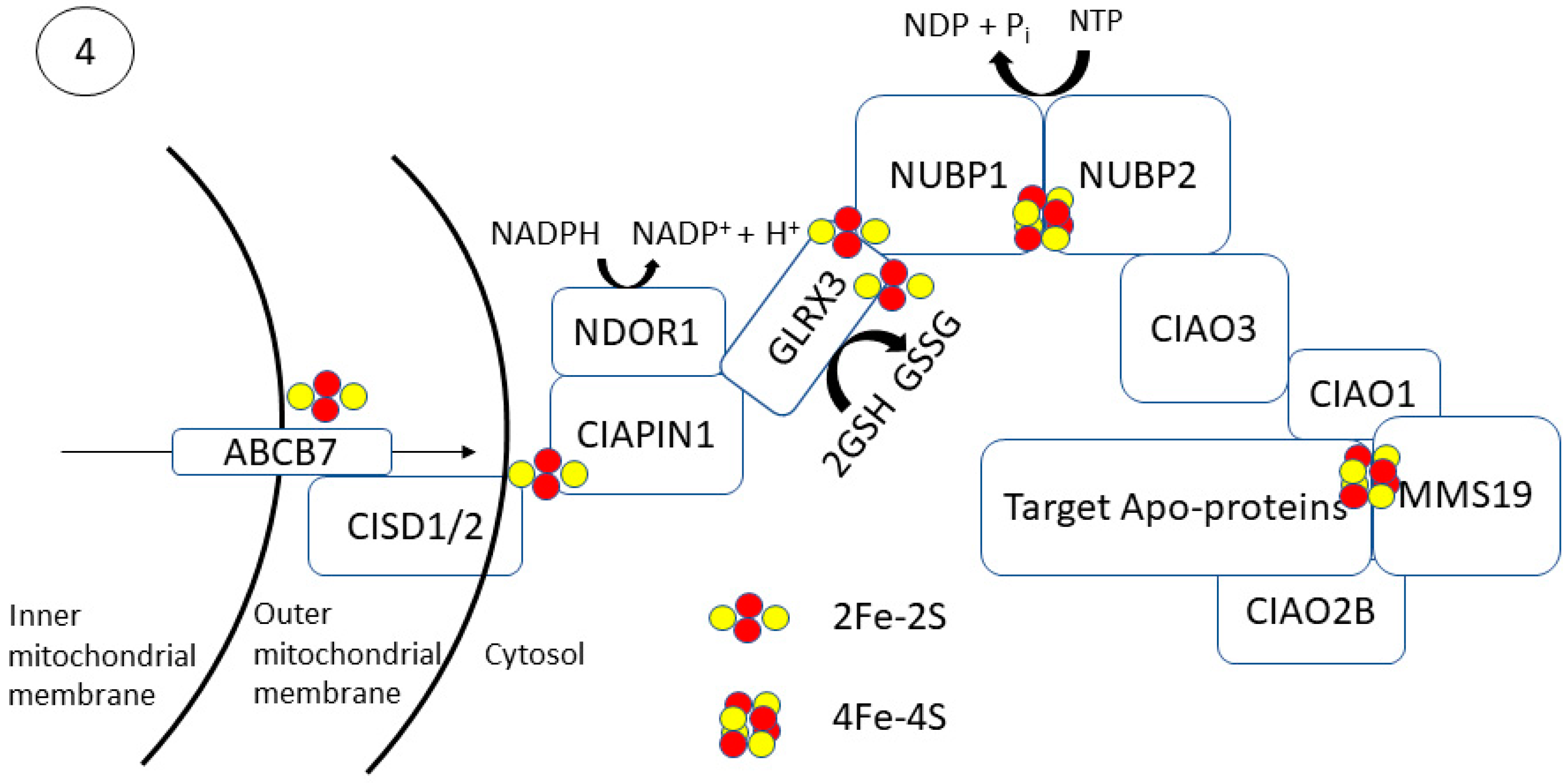
2.2. Step 2: [2Fe-2S] Trafficking
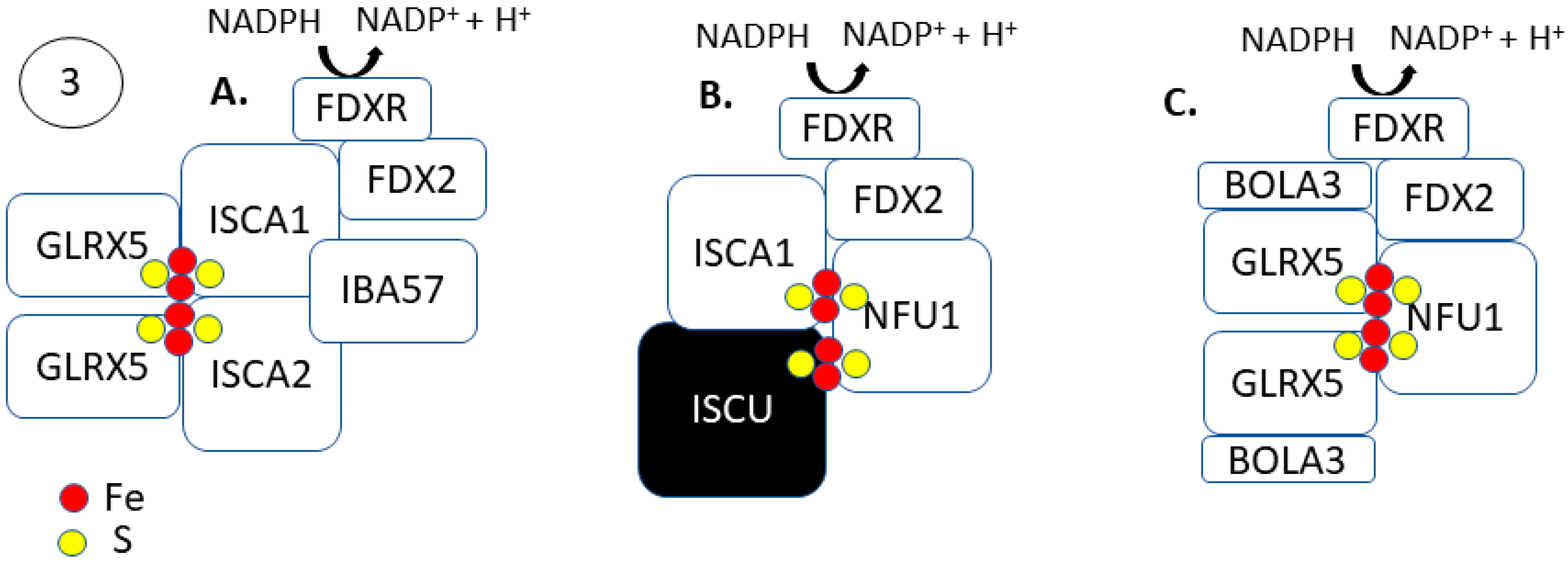
2.3. Steps 3 and 4: [4Fe-4S] Formation and Trafficking to Apo-Proteins
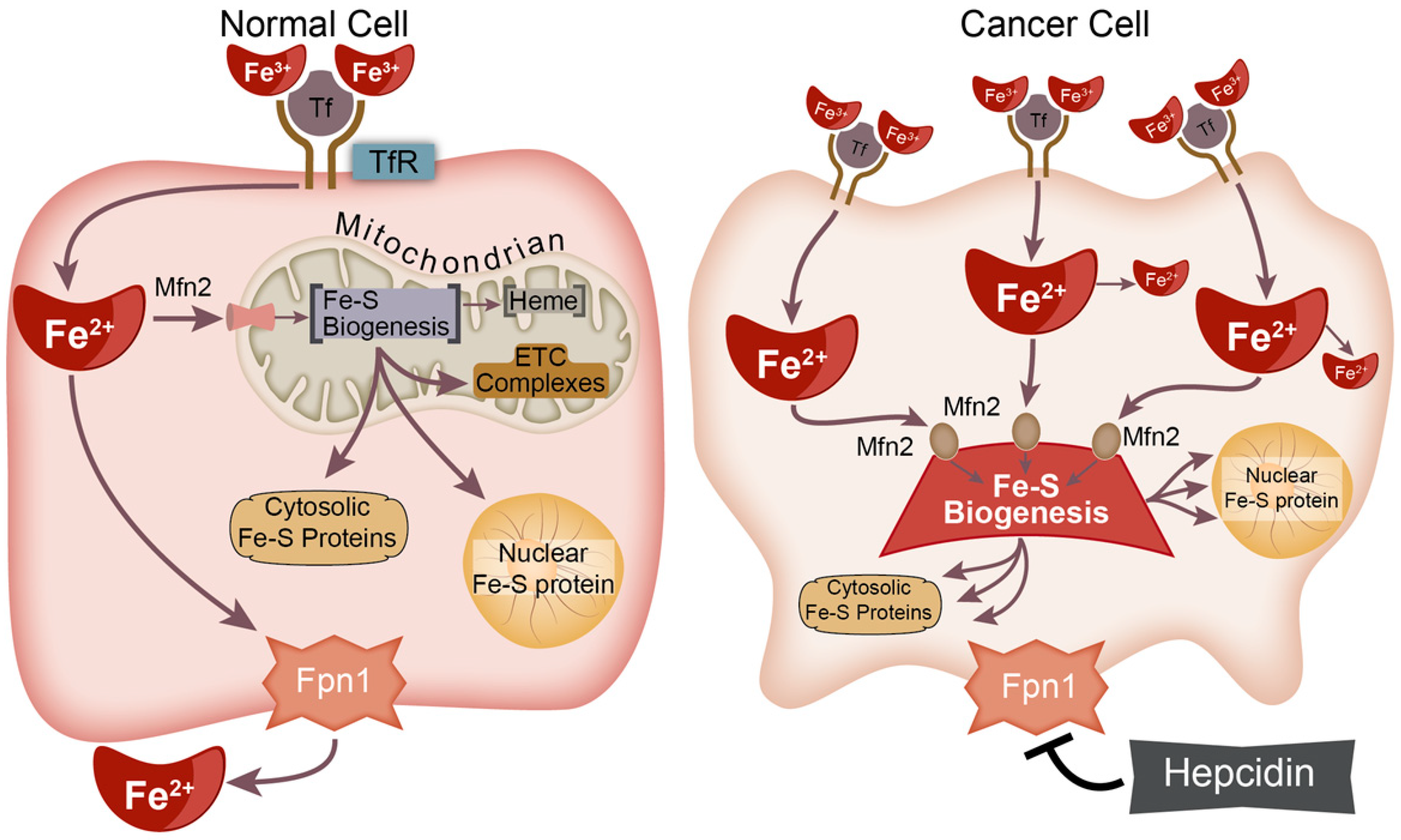
3. Fe–S Biogenesis and Cancer Initiation
4. Potential Therapeutic Strategies to Target Fe–S Clusters in Cancer
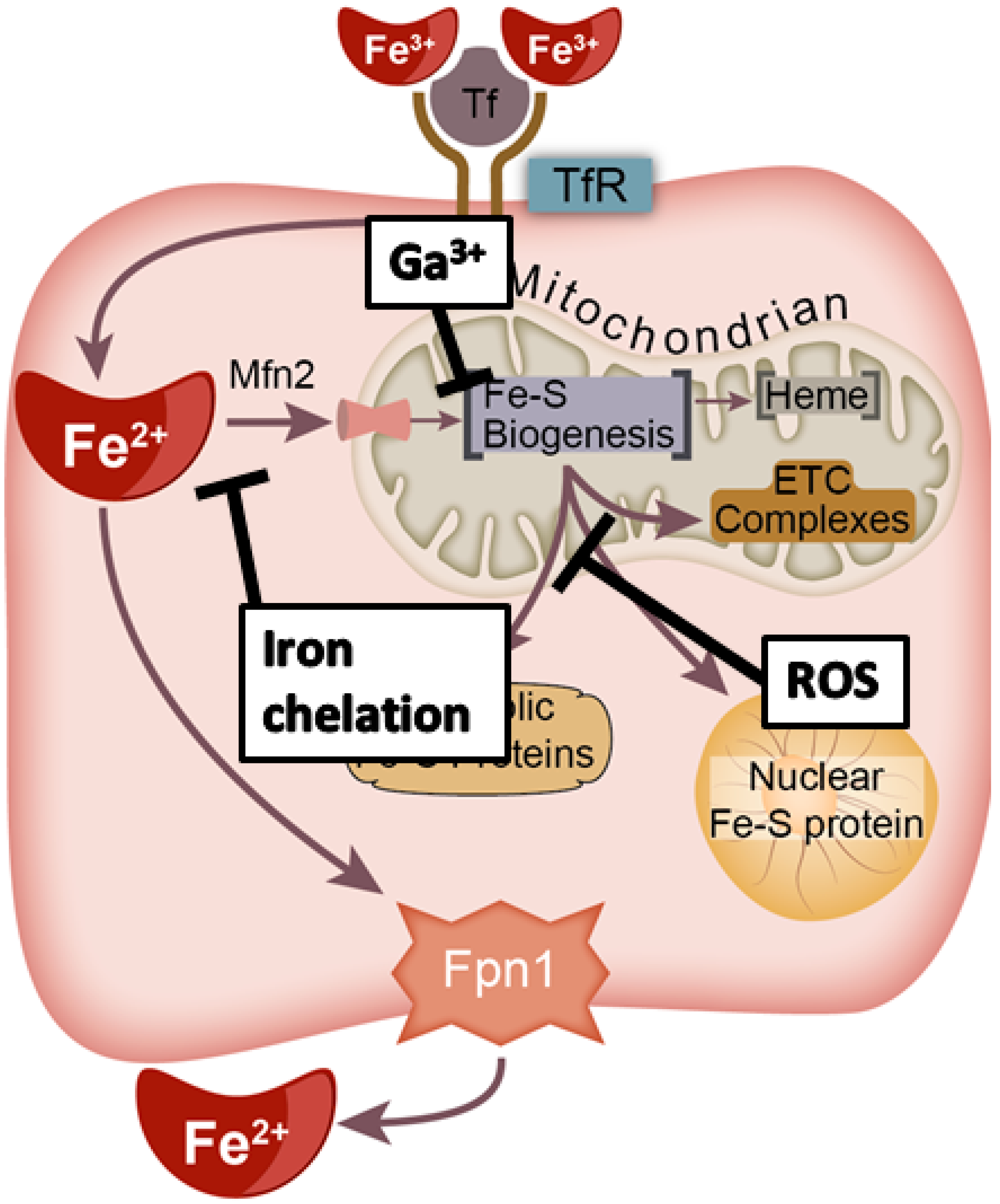
4.1. Redox Manipulation
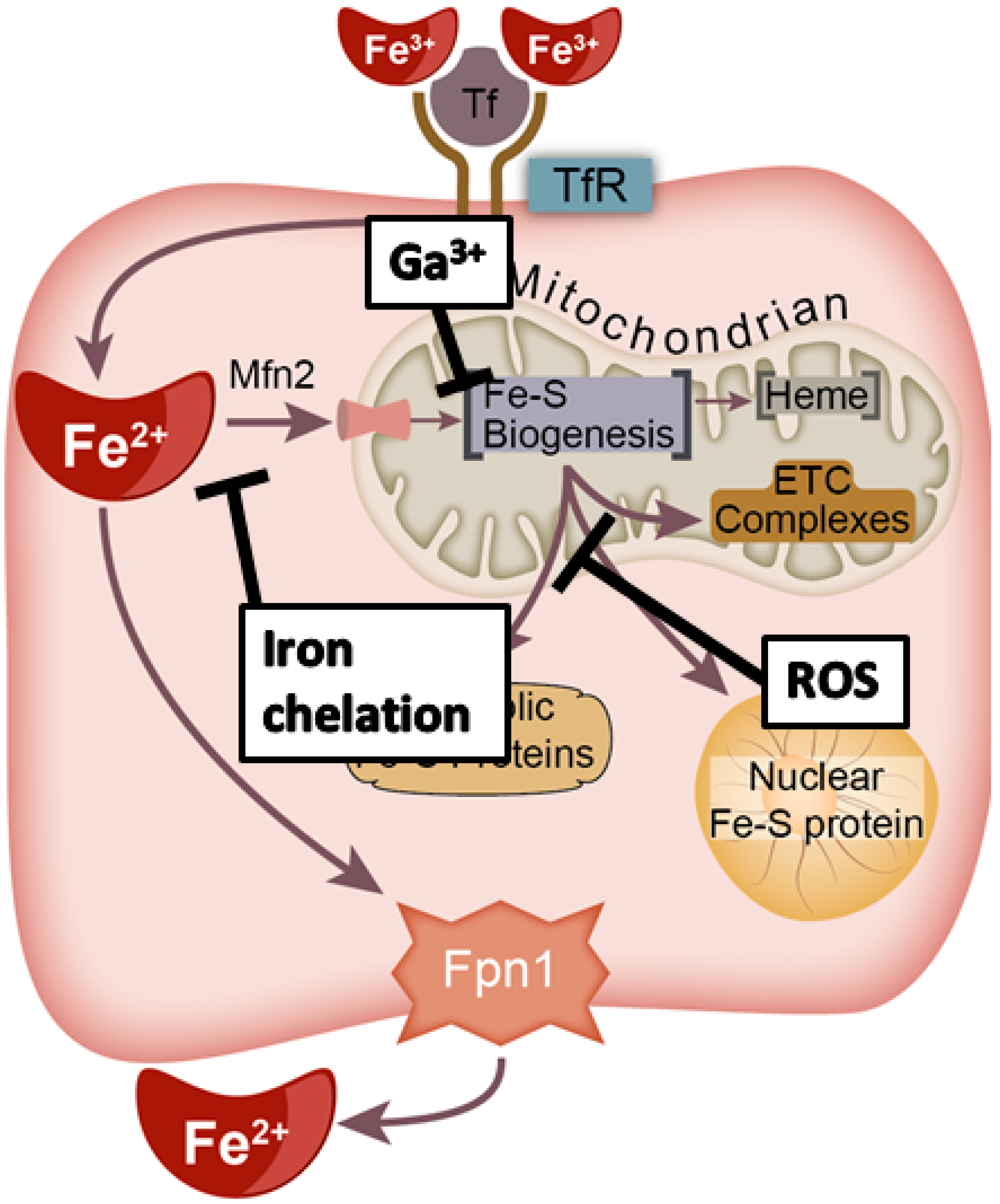
4.2. Iron Chelation
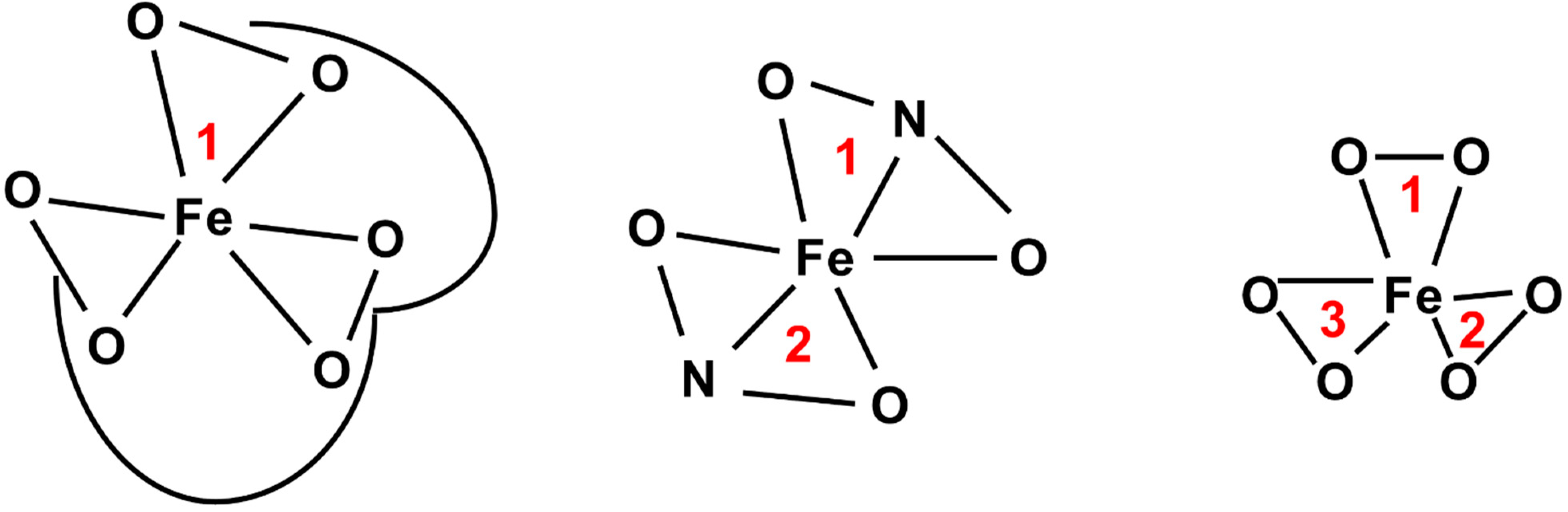
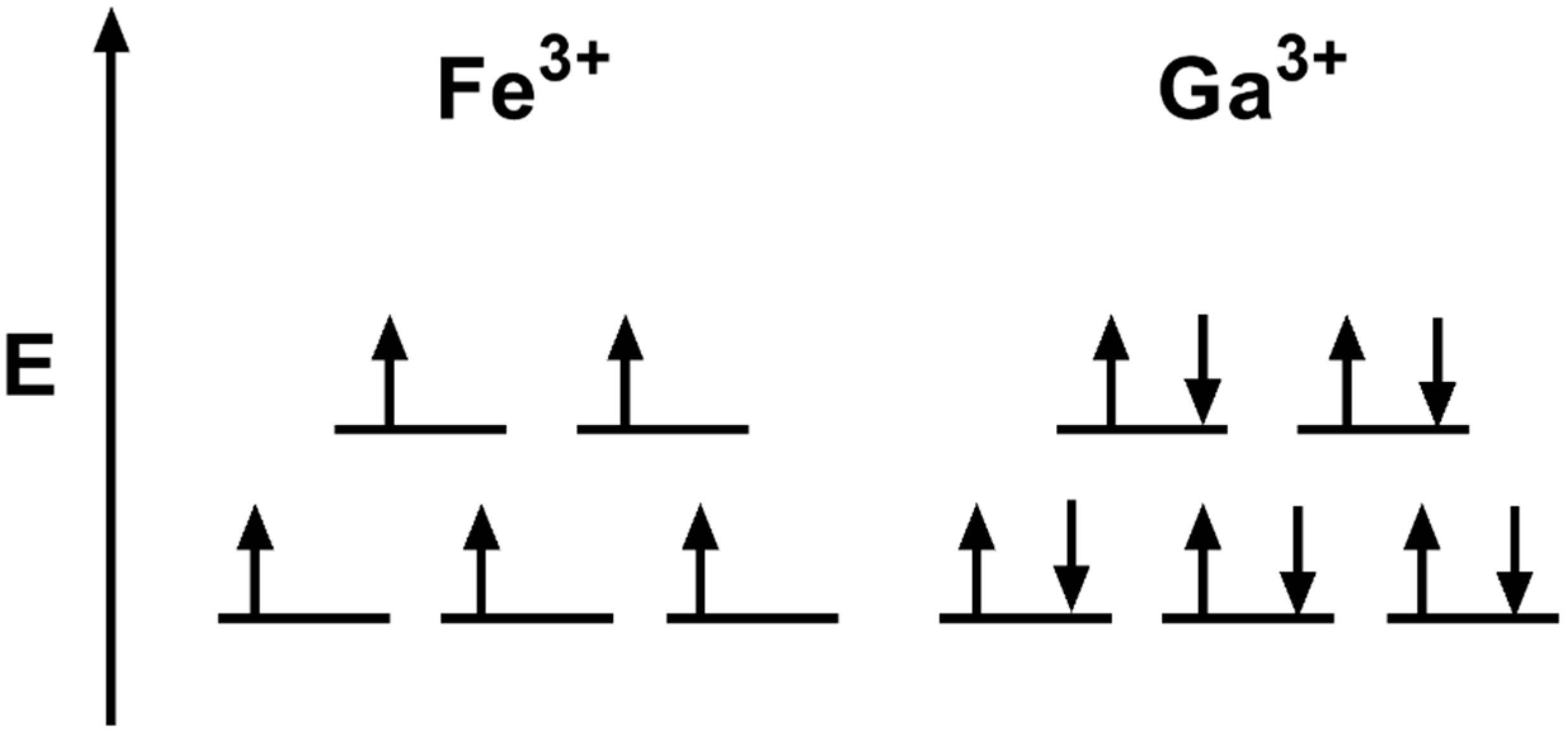
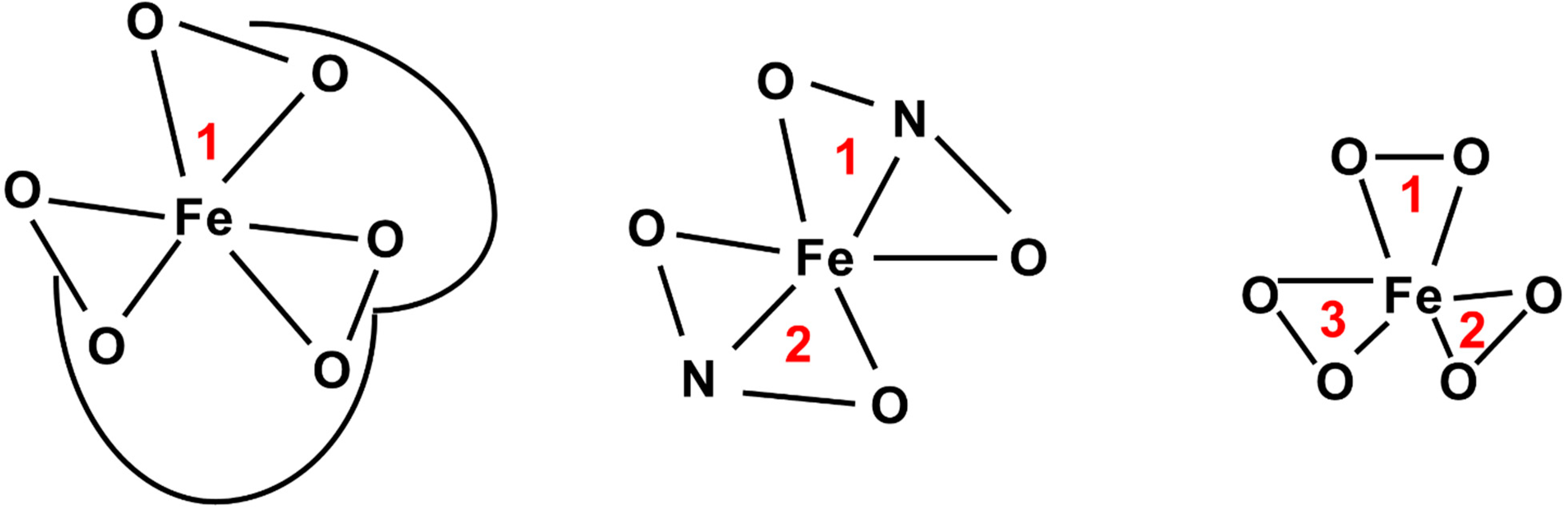
4.3. Iron Mimicry
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCB7: | ATP-binding cassette sub-family B member 7 |
| ACP1: | Low-molecular-weight phosphotyrosine protein phosphatase |
| AML: | Acute myeloid leukemia |
| AscH−: | Ascorbic acid, vitamin C |
| BOLA3: | BolA Family Member 3 |
| CIAO1: | Cytosolic Iron–Sulfur Assembly Component 1 |
| CIAO2B: | Cytosolic iron–Sulfur Assembly Component 2B |
| CIAO3: | Cytosolic Iron–Sulfur Assembly Component 3 |
| CIAPIN1: | Cytokine Induced Apoptosis Inhibitor 1, anamorsin |
| CISD1: | CDGSH Iron Sulfur Domain 1 |
| CISD2: | CDGSH Iron Sulfur Domain 2 |
| DFO: | Desferrioxamine |
| DFX: | Deferasirox |
| FDX1: | Ferrodoxin-1 |
| FDX2: | Ferrodoxin-2 |
| FDXR: | Ferrodoxin reductase |
| Fpn-1: | Ferroportin |
| FXN: | Frataxin |
| FancJ: | Fanconi anemia group J protein |
| GLRX3: | Glutaredoxin-3 |
| GLRX5: | Glutaredoxin-5 |
| HIF-1: | Hypoxia inducible factor-1 |
| HSPA9: | Heat Shock Protein Family A (Hsp70) Member 9 |
| HSC20: | HscB Mitochondrial Iron–Sulfur Cluster Cochaperone |
| IBA57: | Iron–Sulfur Cluster Assembly Factor IBA57 |
| ISCA1/2: | Iron–Sulfur Cluster Assembly 1/2 |
| ISCU: | Iron–Sulfur Cluster Assembly Enzyme |
| IscS: | Cysteine desulfurase (prokaryotic) |
| LYRM4: | LYR Motif Containing 4 |
| Mfn2: | Mitoferrin-2 |
| MMS19: | Cytosolic Iron–Sulfur Assembly Component |
| NDOR1: | NADPH-Dependent Diflavin Oxidoreductase 1 |
| NFS1: | Cysteine Desulfurase (eukaryotic) |
| NFU1: | Iron–Sulfur Cluster Scaffold Protein |
| NUBP1/2: | Nucleotide-Binding Protein 1/2 |
| XPD: | Xeroderma pigmentosum group D |
References
- Petronek, M.S.; Spitz, D.R.; Buettner, G.R.; Allen, B.G. Linking Cancer Metabolic Dysfunction and Genetic Instability through the Lens of Iron Metabolism. Cancers 2019, 11, 1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar] [PubMed]
- Lok, C.N.; Ponka, P. Identification of a Hypoxia Response Element in the Transferrin Receptor Gene. J. Biol. Chem. 1999, 274, 24147–24152. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, K.A.; Yu, D.; Zeller, K.I.; Kim, J.-W.; Racke, F.; Thomas-Tikhonenko, A.; Dang, C.V. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol. Cell Biol. 2006, 26, 2373–2386. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, A.J.; Goyert, J.W.; Solanki, S.; Kerk, S.A.; Chen, B.; Castillo, C.; Hsu, P.P.; Do, B.T.; Singhal, R.; Dame, M.K.; et al. Hepcidin sequesters iron to sustain nucleotide metabolism and mitochondrial function in colorectal cancer epithelial cells. Nat. Metab. 2021, 3, 969–982. [Google Scholar] [CrossRef]
- Wardman, P.; Candeias, L.P. Fenton Chemistry: An Introduction. Radiat. Res. 1996, 145, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.Y.; Buettner, G.R. Iron and dioxygen chemistry is an important route to initiation of biological free radical oxidations: An electron paramagnetic resonance spin trapping study. Free Radic. Biol. Med. 1999, 26, 1447–1456. [Google Scholar] [CrossRef]
- Breuer, W.; Shvartsman, M.; Cabantchik, Z.I. Intracellular labile iron. Int. J. Biochem. Cell Biol. 2008, 40, 350–354. [Google Scholar] [CrossRef]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res./Fundam. Mol. Mech. Mutagenes. 2003, 531, 81–92. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2− and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, 487–500.e8. [Google Scholar] [CrossRef] [Green Version]
- Dlouhy, A.C.; Outten, C.E. The iron metallome in eukaryotic organisms. Met. Ions Life Sci. 2013, 12, 241–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braymer, J.J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [Green Version]
- Crooks, D.R.; Ghosh, M.C.; Haller, R.G.; Tong, W.-H.; Rouault, T.A. Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood 2010, 115, 860–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider-Yin, X.; Gouya, L.; Dorsey, M.; Rüfenacht, U.; Deybach, J.-C.; Ferreira, G.C. Mutations in the iron-sulfur cluster ligands of the human ferrochelatase lead to erythropoietic protoporphyria. Blood 2000, 96, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.C.; Franco, R.; Lloyd, S.G.; Pereira, A.S.; Moura, I.; Moura, J.J.; Huynh, B.H. Mammalian ferrochelatase, a new addition to the metalloenzyme family. J. Biol. Chem. 1994, 269, 7062–7065. [Google Scholar] [CrossRef]
- Dailey, H.A.; Finnegan, M.G.; Johnson, M.K. Human ferrochelatase is an iron-sulfur protein. Biochemistry 1994, 33, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Fuss, J.O.; Tsai, C.-L.; Ishida, J.P.; Tainer, J.A. Emerging critical roles of Fe–S clusters in DNA replication and repair. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 1253–1271. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, J.; Makrantoni, V.; Ingledew, W.J.; Stark, M.J.R.; White, M.F. The DNA Repair Helicases XPD and FancJ Have Essential Iron-Sulfur Domains. Mol. Cell 2006, 23, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Klinge, S.; Hirst, J.; Maman, J.D.; Krude, T.; Pellegrini, L. An iron-sulfur domain of the eukaryotic primase is essential for RNA primer synthesis. Nat. Struct. Mol. Biol. 2007, 14, 875–877. [Google Scholar] [CrossRef] [Green Version]
- Netz, D.J.A.; Stith, C.M.; Stümpfig, M.; Köpf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.J.; Pierik, A.J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2011, 8, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Ter Beek, J.; Parkash, V.; Bylund, G.O.; Osterman, P.; Sauer-Eriksson, A.E.; Johansson, E. Structural evidence for an essential Fe–S cluster in the catalytic core domain of DNA polymerase ϵ. Nucleic Acids Res. 2019, 47, 5712–5722. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freibert, S.-A.; Goldberg, A.V.; Hacker, C.; Molik, S.; Dean, P.; Williams, T.A.; Nakjang, S.; Long, S.; Sendra, K.; Bill, E.; et al. Evolutionary conservation and in vitro reconstitution of microsporidian iron-sulfur cluster biosynthesis. Nat. Commun. 2017, 8, 13932. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.P.; Vishwanathan, V.; Bankapalli, K.; D’Silva, P. Iron-Sulfur Protein Assembly in Human Cells. In Reviews of Physiology, Biochemistry and Pharmacology Vol. 174; Nilius, B., de Tombe, P., Gudermann, T., Jahn, R., Lill, R., Petersen, O.H., Eds.; Springer International Publishing: Champaign, IL, USA, 2018; pp. 25–65. ISBN 978-3-319-78774-9. [Google Scholar]
- Yankovskaya, V.; Horsefield, R.; Törnroth, S.; Luna-Chavez, C.; Miyoshi, H.; Léger, C.; Byrne, B.; Cecchini, G.; Iwata, S. Architecture of Succinate Dehydrogenase and Reactive Oxygen Species Generation. Science 2003, 299, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, R.; Proteau, A.; Villarroya, M.; Moukadiri, I.; Zhang, L.; Trempe, J.-F.; Matte, A.; Armengod, M.E.; Cygler, M. Structural basis for Fe-S cluster assembly and tRNA thiolation mediated by IscS protein-protein interactions. PLoS Biol. 2010, 8, e1000354. [Google Scholar] [CrossRef]
- Urbina, H.D.; Silberg, J.J.; Hoff, K.G.; Vickery, L.E. Transfer of Sulfur from IscS to IscU during Fe/S Cluster Assembly. J. Biol. Chem. 2001, 276, 44521–44526. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Tonelli, M.; Markley, J.L. Disordered form of the scaffold protein IscU is the substrate for iron-sulfur cluster assembly on cysteine desulfurase. Proc. Natl. Acad. Sci. USA 2012, 109, 454–459. [Google Scholar] [CrossRef] [Green Version]
- Angerer, H. Eukaryotic LYR Proteins Interact with Mitochondrial Protein Complexes. Biology 2015, 4, 133–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.C.; Friemel, M.; Marum, J.E.; Tucker, E.J.; Bruno, D.L.; Riley, L.G.; Christodoulou, J.; Kirk, E.P.; Boneh, A.; DeGennaro, C.M.; et al. Mutations in LYRM4, encoding iron-sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum. Mol. Genet. 2013, 22, 4460–4473. [Google Scholar] [CrossRef]
- Wiedemann, N.; Urzica, E.; Guiard, B.; Müller, H.; Lohaus, C.; Meyer, H.E.; Ryan, M.T.; Meisinger, C.; Mühlenhoff, U.; Lill, R.; et al. Essential role of Isd11 in mitochondrial iron-sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 2006, 25, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Cai, K.; Frederick, R.O.; Tonelli, M.; Markley, J.L. Mitochondrial Cysteine Desulfurase and ISD11 Coexpressed in Escherichia coli Yield Complex Containing Acyl Carrier Protein. ACS Chem. Biol. 2017, 12, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Van Vranken, J.G.; Jeong, M.-Y.; Wei, P.; Chen, Y.-C.; Gygi, S.P.; Winge, D.R.; Rutter, J. The mitochondrial acyl carrier protein (ACP) coordinates mitochondrial fatty acid synthesis with iron sulfur cluster biogenesis. eLife 2016, 5, e17828. [Google Scholar] [CrossRef] [PubMed]
- Adam, A.C.; Bornhövd, C.; Prokisch, H.; Neupert, W.; Hell, K. The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO J. 2006, 25, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Boniecki, M.T.; Freibert, S.A.; Mühlenhoff, U.; Lill, R.; Cygler, M. Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat. Commun. 2017, 8, 1287. [Google Scholar] [CrossRef] [Green Version]
- Bridwell-Rabb, J.; Fox, N.G.; Tsai, C.-L.; Winn, A.M.; Barondeau, D.P. Human frataxin activates Fe-S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 2014, 53, 4904–4913. [Google Scholar] [CrossRef] [Green Version]
- Parent, A.; Elduque, X.; Cornu, D.; Belot, L.; Le Caer, J.-P.; Grandas, A.; Toledano, M.B.; D’Autréaux, B. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 2015, 6, 5686. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Gordon, D.M.; Pain, J.; Stemmler, T.L.; Dancis, A.; Pain, D. Frataxin directly stimulates mitochondrial cysteine desulfurase by exposing substrate-binding sites, and a mutant Fe-S cluster scaffold protein with frataxin-bypassing ability acts similarly. J. Biol. Chem. 2013, 288, 36773–36786. [Google Scholar] [CrossRef] [Green Version]
- Fox, N.G.; Yu, X.; Feng, X.; Bailey, H.J.; Martelli, A.; Nabhan, J.F.; Strain-Damerell, C.; Bulawa, C.; Yue, W.W.; Han, S. Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 2019, 10, 2210. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Napoli, E.; Cortopassi, G. Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum. Mol. Genet. 2007, 16, 929–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmucker, S.; Martelli, A.; Colin, F.; Page, A.; Wattenhofer-Donzé, M.; Reutenauer, L.; Puccio, H. Mammalian frataxin: An essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS ONE 2011, 6, e16199. [Google Scholar] [CrossRef] [PubMed]
- Marinoni, E.N.; de Oliveira, J.S.; Nicolet, Y.; Raulfs, E.C.; Amara, P.; Dean, D.R.; Fontecilla-Camps, J.C. (IscS-IscU)2 Complex Structures Provide Insights into Fe2S2 Biogenesis and Transfer. Angew. Chem. Int. Ed. 2012, 51, 5439–5442. [Google Scholar] [CrossRef]
- Colin, F.; Martelli, A.; Clémancey, M.; Latour, J.-M.; Gambarelli, S.; Zeppieri, L.; Birck, C.; Page, A.; Puccio, H.; Ollagnier de Choudens, S. Mammalian Frataxin Controls Sulfur Production and Iron Entry during de Novo Fe4S4 Cluster Assembly. J. Am. Chem. Soc. 2013, 135, 733–740. [Google Scholar] [CrossRef]
- Cai, K.; Frederick, R.O.; Tonelli, M.; Markley, J.L. Interactions of iron-bound frataxin with ISCU and ferredoxin on the cysteine desulfurase complex leading to Fe-S cluster assembly. J. Inorg. Biochem. 2018, 183, 107–116. [Google Scholar] [CrossRef]
- Yoon, T.; Cowan, J.A. Iron−Sulfur Cluster Biosynthesis. Characterization of Frataxin as an Iron Donor for Assembly of [2Fe-2S] Clusters in ISU-Type Proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dizin, E.; Cowan, J.A. Mapping iron binding sites on human frataxin: Implications for cluster assembly on the ISU Fe–S cluster scaffold protein. JBIC J. Biol. Inorg. Chem. 2008, 13, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Adinolfi, S.; Pastore, A. Ferredoxin, in conjunction with NADPH and ferredoxin-NADP reductase, transfers electrons to the IscS/IscU complex to promote iron-sulfur cluster assembly. Biochim. Biophys. Acta 2015, 1854, 1113–1117. [Google Scholar] [CrossRef] [Green Version]
- Webert, H.; Freibert, S.-A.; Gallo, A.; Heidenreich, T.; Linne, U.; Amlacher, S.; Hurt, E.; Mühlenhoff, U.; Banci, L.; Lill, R. Functional reconstitution of mitochondrial Fe/S cluster synthesis on Isu1 reveals the involvement of ferredoxin. Nat. Commun. 2014, 5, 5013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Müller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019, 10, 3566. [Google Scholar] [CrossRef] [Green Version]
- Puglisi, R.; Pastore, A. The role of chaperones in iron-sulfur cluster biogenesis. FEBS Lett. 2018, 592, 4011–4019. [Google Scholar] [CrossRef]
- Maio, N.; Rouault, T.A. Mammalian Fe-S proteins: Definition of a consensus motif recognized by the co-chaperone HSC20. Metallomics 2016, 8, 1032–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickery, L.E.; Cupp-Vickery, J.R. Molecular Chaperones HscA/Ssq1 and HscB/Jac1 and Their Roles in Iron-Sulfur Protein Maturation. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Cupp-Vickery, J.R.; Vickery, L.E. Crystal Structure of Hsc20, a J-type Co-chaperone from Escherichia coli. J. Mol. Biol. 2000, 304, 835–845. [Google Scholar] [CrossRef]
- Puglisi, R.; Yan, R.; Adinolfi, S.; Pastore, A. A New Tessera into the Interactome of the isc Operon: A Novel Interaction between HscB and IscS. Front. Mol. Biosci. 2016, 3, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silberg, J.J.; Vickery, L.E. Kinetic Characterization of the ATPase Cycle of the Molecular Chaperone Hsc66 from Escherichia coli. J. Biol. Chem. 2000, 275, 7779–7786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silberg, J.J.; Tapley, T.L.; Hoff, K.G.; Vickery, L.E. Regulation of the HscA ATPase Reaction Cycle by the Co-chaperone HscB and the Iron-Sulfur Cluster Assembly Protein IscU. J. Biol. Chem. 2004, 279, 53924–53931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzarska, M.A.; Dutkiewicz, R.; Freibert, S.-A.; Lill, R.; Mühlenhoff, U. The mitochondrial Hsp70 chaperone Ssq1 facilitates Fe/S cluster transfer from Isu1 to Grx5 by complex formation. Mol. Biol. Cell 2013, 24, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, F.; Iametti, S.; Morleo, A.; Ta, D.; Vickery, L.E. Studies on the mechanism of catalysis of iron-sulfur cluster transfer from IscU [2Fe2S] by HscA/HscB chaperones. Biochemistry 2008, 47, 12795–12801. [Google Scholar] [CrossRef] [PubMed]
- Mühlenhoff, U.; Gerber, J.; Richhardt, N.; Lill, R. Components involved in assembly and dislocation of iron-sulfur clusters on the scaffold protein Isu1p. EMBO J. 2003, 22, 4815–4825. [Google Scholar] [CrossRef] [Green Version]
- Weiler, B.D.; Brück, M.-C.; Kothe, I.; Bill, E.; Lill, R.; Mühlenhoff, U. Mitochondrial [4Fe-4S] protein assembly involves reductive [2Fe-2S] cluster fusion on ISCA1–ISCA2 by electron flow from ferredoxin FDX2. Proc. Natl. Acad. Sci. USA 2020, 117, 20555–20565. [Google Scholar] [CrossRef]
- Rodríguez-Manzaneque, M.T.; Tamarit, J.; Bellí, G.; Ros, J.; Herrero, E. Grx5 Is a Mitochondrial Glutaredoxin Required for the Activity of Iron/Sulfur Enzymes. Mol. Biol. Cell 2002, 13, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Sheftel, A.D.; Wilbrecht, C.; Stehling, O.; Niggemeyer, B.; Elsässer, H.-P.; Mühlenhoff, U.; Lill, R. The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fe-4S] protein maturation. Mol. Biol. Cell 2012, 23, 1157–1166. [Google Scholar] [CrossRef]
- Kim, K.-D.; Chung, W.-H.; Kim, H.-J.; Lee, K.-C.; Roe, J.-H. Monothiol glutaredoxin Grx5 interacts with Fe–S scaffold proteins Isa1 and Isa2 and supports Fe–S assembly and DNA integrity in mitochondria of fission yeast. Biochem. Biophys. Res. Commun. 2010, 392, 467–472. [Google Scholar] [CrossRef]
- Brancaccio, D.; Gallo, A.; Mikolajczyk, M.; Zovo, K.; Palumaa, P.; Novellino, E.; Piccioli, M.; Ciofi-Baffoni, S.; Banci, L. Formation of [4Fe-4S] Clusters in the Mitochondrial Iron–Sulfur Cluster Assembly Machinery. J. Am. Chem. Soc. 2014, 136, 16240–16250. [Google Scholar] [CrossRef]
- Azam, T.; Przybyla-Toscano, J.; Vignols, F.; Couturier, J.; Rouhier, N.; Johnson, M.K. [4Fe-4S] cluster trafficking mediated by Arabidopsis mitochondrial ISCA and NFU proteins. J. Biol. Chem. 2020, 295, 18367–18378. [Google Scholar] [CrossRef]
- Jain, A.; Singh, A.; Maio, N.; Rouault, T.A. Assembly of the [4Fe-4S] cluster of NFU1 requires the coordinated donation of two [2Fe-2S] clusters from the scaffold proteins, ISCU2 and ISCA1. Hum. Mol. Genet. 2020, 29, 3165–3182. [Google Scholar] [CrossRef] [PubMed]
- Cai, K.; Frederick, R.O.; Markley, J.L. ISCU interacts with NFU1, and ISCU[4Fe-4S] transfers its Fe-S cluster to NFU1 leading to the production of holo-NFU1. J. Struct. Biol. 2020, 210, 107491. [Google Scholar] [CrossRef] [PubMed]
- Talib, E.A.; Outten, C.E. Iron-sulfur cluster biogenesis, trafficking, and signaling: Roles for CGFS glutaredoxins and BolA proteins. Biochim. et Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 118847. [Google Scholar] [CrossRef] [PubMed]
- Nasta, V.; Suraci, D.; Gourdoupis, S.; Ciofi-Baffoni, S.; Banci, L. A pathway for assembling [4Fe-4S]2+ clusters in mitochondrial iron–sulfur protein biogenesis. FEBS J. 2020, 287, 2312–2327. [Google Scholar] [CrossRef] [PubMed]
- Stehling, O.; Lill, R. The role of mitochondria in cellular iron-sulfur protein biogenesis: Mechanisms, connected processes, and diseases. Cold Spring Harb. Perspect. Biol. 2013, 5, a011312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pondarré, C.; Antiochos, B.B.; Campagna, D.R.; Clarke, S.L.; Greer, E.L.; Deck, K.M.; McDonald, A.; Han, A.-P.; Medlock, A.; Kutok, J.L.; et al. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron–sulfur cluster biogenesis. Hum. Mol. Genet. 2006, 15, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Kispal, G.; Csere, P.; Prohl, C.; Lill, R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999, 18, 3981–3989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, R.; Kim, H.; Koppolu, U.M.K.; Ellis, E.A.; Scott, R.A.; Lindahl, P.A. Biophysical characterization of the iron in mitochondria from Atm1p-depleted Saccharomyces cerevisiae. Biochemistry 2009, 48, 9556–9568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, W.; Li, J.; Cowan, J.A. A structural model for glutathione-complexed iron-sulfur cluster as a substrate for ABCB7-type transporters. Chem. Commun. 2014, 50, 3795–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cowan, J.A. Glutathione-coordinated [2Fe-2S] cluster: A viable physiological substrate for mitochondrial ABCB7 transport. Chem. Commun. 2015, 51, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxid. Redox Signal 2019, 30, 1083–1095. [Google Scholar] [CrossRef]
- Lipper, C.H.; Paddock, M.L.; Onuchic, J.N.; Mittler, R.; Nechushtai, R.; Jennings, P.A. Cancer-Related NEET Proteins Transfer 2Fe-2S Clusters to Anamorsin, a Protein Required for Cytosolic Iron-Sulfur Cluster Biogenesis. PLoS ONE 2015, 10, e0139699. [Google Scholar] [CrossRef] [Green Version]
- Camponeschi, F.; Ciofi-Baffoni, S.; Banci, L. Anamorsin/Ndor1 Complex Reduces [2Fe–2S]-MitoNEET via a Transient Protein–Protein Interaction. J. Am. Chem. Soc. 2017, 139, 9479–9482. [Google Scholar] [CrossRef] [Green Version]
- Netz, D.J.A.; Stümpfig, M.; Doré, C.; Mühlenhoff, U.; Pierik, A.J.; Lill, R. Tah18 transfers electrons to Dre2 in cytosolic iron-sulfur protein biogenesis. Nat. Chem. Biol. 2010, 6, 758–765. [Google Scholar] [CrossRef]
- Pallesen, L.J.; Solodovnikova, N.; Sharma, A.K.; Walden, W.E. Interaction with Cfd1 increases the kinetic lability of FeS on the Nbp35 scaffold. J. Biol. Chem. 2013, 288, 23358–23367. [Google Scholar] [CrossRef] [Green Version]
- Netz, D.J.A.; Pierik, A.J.; Stümpfig, M.; Bill, E.; Sharma, A.K.; Pallesen, L.J.; Walden, W.E.; Lill, R. A bridging [4Fe-4S] cluster and nucleotide binding are essential for function of the Cfd1-Nbp35 complex as a scaffold in iron-sulfur protein maturation. J. Biol. Chem. 2012, 287, 12365–12378. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Solodovnikova, N.; Nicholson, T.; Antholine, W.; Walden, W.E. A novel eukaryotic factor for cytosolic Fe-S cluster assembly. EMBO J. 2003, 22, 4826–4835. [Google Scholar] [CrossRef] [Green Version]
- Hausmann, A.; Aguilar Netz, D.J.; Balk, J.; Pierik, A.J.; Mühlenhoff, U.; Lill, R. The eukaryotic P loop NTPase Nbp35: An essential component of the cytosolic and nuclear iron-sulfur protein assembly machinery. Proc. Natl. Acad. Sci. USA 2005, 102, 3266–3271. [Google Scholar] [CrossRef] [Green Version]
- Camponeschi, F.; Prusty, N.R.; Heider, S.A.E.; Ciofi-Baffoni, S.; Banci, L. GLRX3 Acts as a [2Fe-2S] Cluster Chaperone in the Cytosolic Iron–Sulfur Assembly Machinery Transferring [2Fe-2S] Clusters to NUBP1. J. Am. Chem. Soc. 2020, 142, 10794–10805. [Google Scholar] [CrossRef]
- Leipe, D.D.; Wolf, Y.I.; Koonin, E.V.; Aravind, L. Classification and evolution of P-loop GTPases and related ATPases11Edited by J. Thornton. J. Mol. Biol. 2002, 317, 41–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balk, J.; Pierik, A.J.; Aguilar Netz, D.J.; Mühlenhoff, U.; Lill, R. Nar1p, a conserved eukaryotic protein with similarity to Fe-only hydrogenases, functions in cytosolic iron-sulphur protein biogenesis. Biochem. Soc. Trans. 2005, 33, 86–89. [Google Scholar] [CrossRef] [Green Version]
- Urzica, E.; Pierik, A.J.; Mühlenhoff, U.; Lill, R. Crucial Role of Conserved Cysteine Residues in the Assembly of Two Iron−Sulfur Clusters on the CIA Protein Nar1. Biochemistry 2009, 48, 4946–4958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.; Lee, F.S. A role for IOP1 in mammalian cytosolic iron-sulfur protein biogenesis. J. Biol. Chem. 2008, 283, 9231–9238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gari, K.; León Ortiz, A.M.; Borel, V.; Flynn, H.; Skehel, J.M.; Boulton, S.J. MMS19 Links Cytoplasmic Iron-Sulfur Cluster Assembly to DNA Metabolism. Science 2012, 337, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Kassube, S.A.; Thomä, N.H. Structural insights into Fe–S protein biogenesis by the CIA targeting complex. Nat. Struct. Mol. Biol. 2020, 27, 735–742. [Google Scholar] [CrossRef]
- Odermatt, D.C.; Gari, K. The CIA Targeting Complex Is Highly Regulated and Provides Two Distinct Binding Sites for Client Iron-Sulfur Proteins. Cell Rep. 2017, 18, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Seki, M.; Takeda, Y.; Iwai, K.; Tanaka, K. IOP1 Protein Is an External Component of the Human Cytosolic Iron-Sulfur Cluster Assembly (CIA) Machinery and Functions in the MMS19 Protein-dependent CIA Pathway. J. Biol. Chem. 2013, 288, 16680–16689. [Google Scholar] [CrossRef] [Green Version]
- Stehling, O.; Vashisht, A.A.; Mascarenhas, J.; Jonsson, Z.O.; Sharma, T.; Netz, D.J.A.; Pierik, A.J.; Wohlschlegel, J.A.; Lill, R. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 2012, 337, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Puig, S.; Ramos-Alonso, L.; Romero, A.M.; Martínez-Pastor, M.T. The elemental role of iron in DNA synthesis and repair. Metallomics 2017, 9, 1483–1500. [Google Scholar] [CrossRef] [Green Version]
- Bartha, Á.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef] [PubMed]
- Funauchi, Y.; Tanikawa, C.; Yi Lo, P.H.; Mori, J.; Daigo, Y.; Takano, A.; Miyagi, Y.; Okawa, A.; Nakamura, Y.; Matsuda, K. Regulation of iron homeostasis by the p53-ISCU pathway. Sci. Rep. 2015, 5, 16497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, E.; Ramachandran, A.; McCormick, R.; Gee, H.; Blancher, C.; Crosby, M.; Devlin, C.; Blick, C.; Buffa, F.; Li, J.-L.; et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS ONE 2010, 5, e10345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanova, A.; Magrané, J. Mitochondrial dysfunction in neurons in Friedreich’s ataxia. Mol. Cell. Neurosci. 2020, 102, 103419. [Google Scholar] [CrossRef]
- Ackroyd, R.; Shorthouse, A.J.; Stephenson, T.J. Gastric carcinoma in siblings with Friedreich’s ataxia. Eur. J. Surg. Oncol. (EJSO) 1996, 22, 301–303. [Google Scholar] [CrossRef]
- Kidd, A.; Coleman, R.; Whiteford, M.; Barron, L.H.; Simpson, S.A.; Haites, N.E. Breast cancer in two sisters with Friedreich’s ataxia. Eur. J. Surg. Oncol. (EJSO) 2001, 27, 512–514. [Google Scholar] [CrossRef]
- Tsou, A.Y.; Paulsen, E.K.; Lagedrost, S.J.; Perlman, S.L.; Mathews, K.D.; Wilmot, G.R.; Ravina, B.; Koeppen, A.H.; Lynch, D.R. Mortality in Friedreich ataxia. J. Neurol. Sci. 2011, 307, 46–49. [Google Scholar] [CrossRef]
- White, M.C.; Holman, D.M.; Boehm, J.E.; Peipins, L.A.; Grossman, M.; Henley, S.J. Age and cancer risk: A potentially modifiable relationship. Am. J. Prev. Med. 2014, 46, S7–S15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karthikeyan, G.; Lewis, L.K.; Resnick, M.A. The mitochondrial protein frataxin prevents nuclear damage. Hum. Mol. Genet. 2002, 11, 1351–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thierbach, R.; Drewes, G.; Fusser, M.; Voigt, A.; Kuhlow, D.; Blume, U.; Schulz, T.J.; Reiche, C.; Glatt, H.; Epe, B.; et al. The Friedreich’s ataxia protein frataxin modulates DNA base excision repair in prokaryotes and mammals. Biochem. J. 2010, 432, 165–172. [Google Scholar] [CrossRef]
- Khonsari, H.; Schneider, M.; Al-Mahdawi, S.; Chianea, Y.G.; Themis, M.; Parris, C.; Pook, M.A.; Themis, M. Lentivirus-meditated frataxin gene delivery reverses genome instability in Friedreich ataxia patient and mouse model fibroblasts. Gene Ther. 2016, 23, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guccini, I.; Serio, D.; Condò, I.; Rufini, A.; Tomassini, B.; Mangiola, A.; Maira, G.; Anile, C.; Fina, D.; Pallone, F.; et al. Frataxin participates to the hypoxia-induced response in tumors. Cell Death Dis. 2011, 2, e123. [Google Scholar] [CrossRef] [PubMed]
- Thierbach, R.; Schulz, T.J.; Isken, F.; Voigt, A.; Mietzner, B.; Drewes, G.; von Kleist-Retzow, J.-C.; Wiesner, R.J.; Magnuson, M.A.; Puccio, H.; et al. Targeted disruption of hepatic frataxin expression causes impaired mitochondrial function, decreased life span and tumor growth in mice. Hum. Mol. Genet. 2005, 14, 3857–3864. [Google Scholar] [CrossRef]
- Schulz, T.J.; Thierbach, R.; Voigt, A.; Drewes, G.; Mietzner, B.; Steinberg, P.; Pfeiffer, A.F.H.; Ristow, M. Induction of Oxidative Metabolism by Mitochondrial Frataxin Inhibits Cancer Growth: OTTO WARBURG REVISITED. J. Biol. Chem. 2006, 281, 977–981. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef]
- Lee, J.; You, J.H.; Shin, D.; Roh, J.-L. Inhibition of Glutaredoxin 5 predisposes Cisplatin-resistant Head and Neck Cancer Cells to Ferroptosis. Theranostics 2020, 10, 7775–7786. [Google Scholar] [CrossRef]
- Wu, P.-K.; Hong, S.-K.; Veeranki, S.; Karkhanis, M.; Starenki, D.; Plaza, J.A.; Park, J.-I. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol. Cell Biol. 2013, 33, 4051–4067. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.-K.; Hong, S.-K.; Starenki, D.; Oshima, K.; Shao, H.; Gestwicki, J.E.; Tsai, S.; Park, J.-I. Mortalin/HSPA9 targeting selectively induces KRAS tumor cell death by perturbing mitochondrial membrane permeability. Oncogene 2020, 39, 4257–4270. [Google Scholar] [CrossRef]
- Bai, F.; Morcos, F.; Sohn, Y.-S.; Darash-Yahana, M.; Rezende, C.O.; Lipper, C.H.; Paddock, M.L.; Song, L.; Luo, Y.; Holt, S.H.; et al. The Fe-S cluster-containing NEET proteins mitoNEET and NAF-1 as chemotherapeutic targets in breast cancer. Proc. Natl Acad. Sci. USA 2015, 112, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darash-Yahana, M.; Pozniak, Y.; Lu, M.; Sohn, Y.-S.; Karmi, O.; Tamir, S.; Bai, F.; Song, L.; Jennings, P.A.; Pikarsky, E.; et al. Breast cancer tumorigenicity is dependent on high expression levels of NAF-1 and the lability of its Fe-S clusters. Proc. Natl. Acad. Sci. USA 2016, 113, 10890–10895. [Google Scholar] [CrossRef] [Green Version]
- Weon, J.L.; Yang, S.W.; Potts, P.R. Cytosolic Iron-Sulfur Assembly Is Evolutionarily Tuned by a Cancer-Amplified Ubiquitin Ligase. Mol. Cell 2018, 69, 113–125.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelièvre, P.; Sancey, L.; Coll, J.-L.; Deniaud, A.; Busser, B. Iron Dysregulation in Human Cancer: Altered Metabolism, Biomarkers for Diagnosis, Prognosis, Monitoring and Rationale for Therapy. Cancers 2020, 12, 3524. [Google Scholar] [CrossRef] [PubMed]
- Kunos, C.A.; Radivoyevitch, T.; Kresak, A.; Dawson, D.; Jacobberger, J.; Yang, B.; Abdul-Karim, F.W. Elevated ribonucleotide reductase levels associate with suppressed radiochemotherapy response in human cervical cancers. Int. J. Gynecol. Cancer 2012, 22, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Gao, L.; Yan, X. Human ferritin for tumor detection and therapy. WIREs Nanomed. Nanobiotechnol. 2013, 5, 287–298. [Google Scholar] [CrossRef] [PubMed]
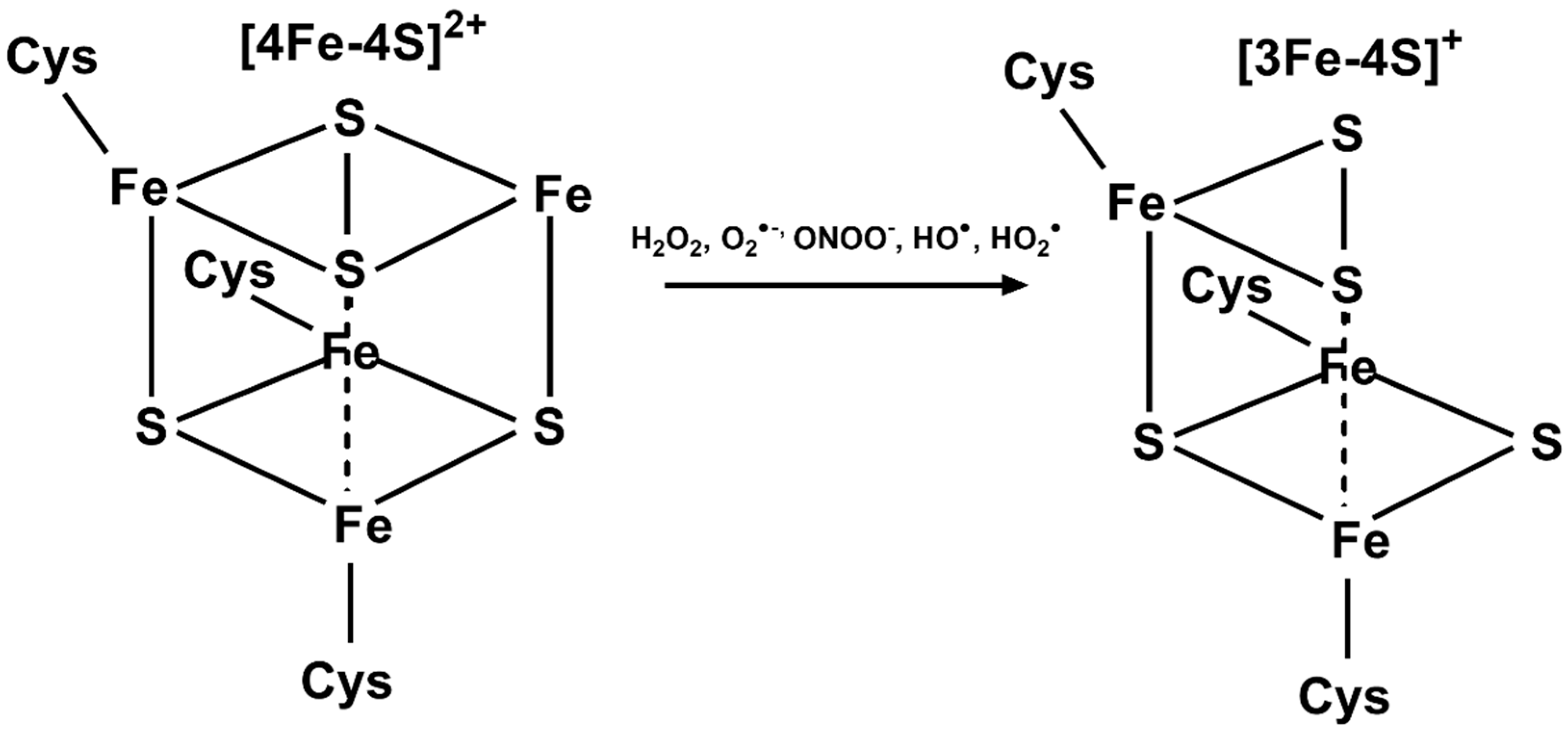
- Bulteau, A.-L.; Ikeda-Saito, M.; Szweda, L.I. Redox-Dependent Modulation of Aconitase Activity in Intact Mitochondria. Biochemistry 2003, 42, 14846–14855. [Google Scholar] [CrossRef]
- Gardner, P.R. Superoxide-Driven Aconitase FE–S Center Cycling. Biosci. Rep. 1997, 17, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Cantu, D.; Schaack, J.; Patel, M. Oxidative Inactivation of Mitochondrial Aconitase Results in Iron and H2O2-Mediated Neurotoxicity in Rat Primary Mesencephalic Cultures. PLoS ONE 2009, 4, e7095. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Bradley, M.D.; Wagner, B.A.; Buettner, G.R.; Monga, V.; Milhem, M.; Spitz, D.R.; Allen, B.G. Redox active metals and H2O2 mediate the increased efficacy of pharmacological ascorbate in combination with gemcitabine or radiation in pre-clinical sarcoma models. Redox Biol. 2018, 14, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.; Jurkiewicz, B.A. Catalytic Metals, Ascorbate and Free Radicals: Combinations to Avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-Dose Parenteral Ascorbate Enhanced Chemosensitivity of Ovarian Cancer and Reduced Toxicity of Chemotherapy. Sci. Transl. Med. 2014, 6, 222ra18. [Google Scholar] [CrossRef]
- Polireddy, K.; Dong, R.; Reed, G.; Yu, J.; Chen, P.; Williamson, S.; Violet, P.-C.; Pessetto, Z.; Godwin, A.K.; Fan, F.; et al. High Dose Parenteral Ascorbate Inhibited Pancreatic Cancer Growth and Metastasis: Mechanisms and a Phase I/IIa study. Sci. Rep. 2017, 7, 17188. [Google Scholar] [CrossRef] [Green Version]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Roberts, L.J.; et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, D.A.; Mitchell, E.; Bazzan, A.J.; Littman, S.; Zabrecky, G.; Yeo, C.J.; Pillai, M.V.; Newberg, A.B.; Deshmukh, S.; Levine, M. Phase I Evaluation of Intravenous Ascorbic Acid in Combination with Gemcitabine and Erlotinib in Patients with Metastatic Pancreatic Cancer. PLoS ONE 2012, 7, e29794. [Google Scholar] [CrossRef]
- Allen, B.G.; Bodeker, K.L.; Smith, M.C.; Monga, V.; Sandhu, S.; Hohl, R.; Carlisle, T.; Brown, H.; Hollenbeck, N.; Vollstedt, S.; et al. First-in-Human Phase I Clinical Trial of Pharmacologic Ascorbate Combined with Radiation and Temozolomide for Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2019, 25, 6590–6597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and natural iron chelators: Therapeutic potential and clinical use. Future Med. Chem. 2009, 1, 1643–1670. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, O.; O’Sullivan, J. Iron chelators in cancer therapy. BioMetals 2020, 33, 201–215. [Google Scholar] [CrossRef]
- Keberle, H. The Biochemistry of Desferrioxamine and its Relation to Iron Metabolism. Ann. N. Y. Acad. Sci. 1964, 119, 758–768. [Google Scholar] [CrossRef]
- Propper, R.D.; Cooper, B.; Rufo, R.R.; Nienhuis, A.W.; Anderson, W.F.; Bunn, H.F.; Rosenthal, A.; Nathan, D.G. Continuous Subcutaneous Administration of Deferoxamine in Patients with Iron Overload. N. Engl. J. Med. 1977, 297, 418–423. [Google Scholar] [CrossRef]
- Estrov, Z.; Tawa, A.; Wang, X.-H.; Dubé, I.D.; Sulh, H.; Cohen, A.; Gelfand, E.W.; Freedman, M.H. In Vitro and In Vivo Effects of Deferoxamine in Neonatal Acute Leukemia. Blood 1987, 69, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Brard, L.; Granai, C.O.; Swamy, N. Iron chelators deferoxamine and diethylenetriamine pentaacetic acid induce apoptosis in ovarian carcinoma. Gynecol. Oncol. 2006, 100, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Simonart, T.; Boelaert, J.R.; Mosselmans, R.; Andrei, G.; Noel, J.-C.; De Clercq, E.; Snoeck, R. Antiproliferative and Apoptotic Effects of Iron Chelators on Human Cervical Carcinoma Cells. Gynecol. Oncol. 2002, 85, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, S.; Liu, P. Deferoxamine Enhanced Mitochondrial Iron Accumulation and Promoted Cell Migration in Triple-Negative MDA-MB-231 Breast Cancer Cells Via a ROS-Dependent Mechanism. Int. J. Mol. Sci. 2019, 20, 4952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval-Acuña, C.; Torrealba, N.; Tomkova, V.; Jadhav, S.B.; Blazkova, K.; Merta, L.; Lettlova, S.; Adamcová, M.K.; Rosel, D.; Brábek, J.; et al. Targeting Mitochondrial Iron Metabolism Suppresses Tumor Growth and Metastasis by Inducing Mitochondrial Dysfunction and Mitophagy. Cancer Res. 2021, 81, 2289. [Google Scholar] [CrossRef] [PubMed]
- Dreicer, R.; Kemp, J.D.; Stegink, L.D.; Cardillo, T.; Davis, C.S.; Forest, P.K.; See, W.A. A Phase II Trial of Deferoxamine in Patients with Hormone-Refractory Metastatic Prostate Cancer. Cancer Investig. 1997, 15, 311–317. [Google Scholar] [CrossRef]
- Donfrancesco, A.; Deb, G.; Dominici, C.; Pileggi, D.; Castello, M.A.; Helson, L. Effects of a Single Course of Deferoxamine in Neuroblastoma Patients. Cancer Res. 1990, 50, 4929. [Google Scholar] [PubMed]
- Brittenham, G.M.; Griffith, P.M.; Nienhuis, A.W.; McLaren, C.E.; Young, N.S.; Tucker, E.E.; Allen, C.J.; Farrell, D.E.; Harris, J.W. Efficacy of Deferoxamine in Preventing Complications of Iron Overload in Patients with Thalassemia Major. N. Engl. J. Med. 1994, 331, 567–573. [Google Scholar] [CrossRef]
- Bedford, M.R.; Ford, S.J.; Horniblow, R.D.; Iqbal, T.H.; Tselepis, C. Iron Chelation in the Treatment of Cancer: A New Role for Deferasirox? J. Clin. Pharmacol. 2013, 53, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Chantrel-Groussard, K.; Gaboriau, F.; Pasdeloup, N.; Havouis, R.; Nick, H.; Pierre, J.-L.; Brissot, P.; Lescoat, G. The new orally active iron chelator ICL670A exhibits a higher antiproliferative effect in human hepatocyte cultures than O-trensox. Eur. J. Pharmacol. 2006, 541, 129–137. [Google Scholar] [CrossRef]
- Lee, D.-H.; Jang, P.S.; Chung, N.G.; Cho, B.; Jeong, D.C.; Kim, H.K. Deferasirox shows in vitro and in vivo antileukemic effects on murine leukemic cell lines regardless of iron status. Exp. Hematol. 2013, 41, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Harima, H.; Kaino, S.; Takami, T.; Shinoda, S.; Matsumoto, T.; Fujisawa, K.; Yamamoto, N.; Yamasaki, T.; Sakaida, I. Deferasirox, a novel oral iron chelator, shows antiproliferative activity against pancreatic cancer in vitro and in vivo. BMC Cancer 2016, 16, 702. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.G.; Kim, J.-L.; Park, J.; Lee, S.; Park, S.J.; Kim, J.S.; Choi, C.W. Effects of oral iron chelator deferasirox on human malignant lymphoma cells. Korean J. Hematol. 2012, 47, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Lui, G.Y.L.; Obeidy, P.; Ford, S.J.; Tselepis, C.; Sharp, D.M.; Jansson, P.J.; Kalinowski, D.S.; Kovacevic, Z.; Lovejoy, D.B.; Richardson, D.R. The Iron Chelator, Deferasirox, as a Novel Strategy for Cancer Treatment: Oral Activity Against Human Lung Tumor Xenografts and Molecular Mechanism of Action. Mol. Pharm. 2013, 83, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-L.; Kang, H.-N.; Kang, M.H.; Yoo, Y.A.; Kim, J.S.; Choi, C.W. The Oral Iron Chelator Deferasirox Induces Apoptosis in Myeloid Leukemia Cells by Targeting Caspase. Acta. Haematol. 2011, 126, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.L.; Lee, D.-H.; Na, Y.J.; Kim, B.R.; Jeong, Y.A.; Lee, S.I.; Kang, S.; Joung, S.Y.; Lee, S.-Y.; Oh, S.C.; et al. Iron chelator-induced apoptosis via the ER stress pathway in gastric cancer cells. Tumor Biol. 2016, 37, 9709–9719. [Google Scholar] [CrossRef]
- Ford, S.J.; Obeidy, P.; Lovejoy, D.B.; Bedford, M.; Nichols, L.; Chadwick, C.; Tucker, O.; Lui, G.Y.L.; Kalinowski, D.S.; Jansson, P.J.; et al. Deferasirox (ICL670A) effectively inhibits oesophageal cancer growth in vitro and in vivo. Br. J. Pharm. 2013, 168, 1316–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorillo, M.; Tóth, F.; Brindisi, M.; Sotgia, F.; Lisanti, M.P. Deferiprone (DFP) Targets Cancer Stem Cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production. Cells 2020, 9, 1529. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.; Cappellini, M.D. Update on the use of deferasirox in the management of iron overload. Ther. Clin. Risk Manag. 2009, 5, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Green, M.A.; Welch, M.J. Gallium radiopharmaceutical chemistry. Int. J. Radiat. Appl. Instrum. Part B Nucl. Med. Biol. 1989, 16, 435–448. [Google Scholar] [CrossRef]
- Chitambar, C.R. The therapeutic potential of iron-targeting gallium compounds in human disease: From basic research to clinical application. Pharmacol. Res. 2017, 115, 56–64. [Google Scholar] [CrossRef]
- Chen, D.C.P.; Newman, B.; Turkall, R.M.; Tsan, M.-F. Transferrin receptors and gallium-67 uptake in vitro. Eur. J. Nucl. Med. 1982, 7, 536–540. [Google Scholar] [CrossRef]
- Collery, P.; Keppler, B.; Madoulet, C.; Desoize, B. Gallium in cancer treatment. Crit. Rev. Oncol./Hematol. 2002, 42, 283–296. [Google Scholar] [CrossRef]
- Hayes, R.; Rafter, J.; Byrd, B.; Carlton, J. Studies of the in vivo entry of Ga-67 into normal and malignant tissue. J. Nucl. Med. 1981, 22, 325–332. [Google Scholar]
- Larson, S.M.; Grunbaumi, Z.; Raseyz, J.S. The role of transferrins in gallium uptake. Int. J. Nucl. Med. Biol. 1981, 8, 257–266. [Google Scholar] [CrossRef]
- Chitambar, C.R.; Narasimhan, J.; Guy, J.; Sem, D.S.; O’Brien, W.J. Inhibition of Ribonucleotide Reductase by Gallium in Murine Leukemic L1210 Cells. Cancer Res. 1991, 51, 6199–6201. [Google Scholar]
- Chitambar, C.R.; Wereley, J.P.; Matsuyama, S. Gallium-induced cell death in lymphoma: Role of transferrin receptor cycling, involvement of Bax and the mitochondria, and effects of proteasome inhibition. Mol. Cancer Ther. 2006, 5, 2834–2843. [Google Scholar] [CrossRef] [Green Version]
- Chitambar, C.R.; Zivkovic-Gilgenbach, Z.; Narasimhan, J.; Antholine, W.E. Development of Drug Resistance to Gallium Nitrate through Modulation of Cellular Iron Uptake. Cancer Res. 1990, 50, 4468–4472. [Google Scholar]
- Chitambar, C.R. Gallium nitrate for the treatment of non-Hodgkin’s lymphoma. Expert Opin. Investig. Drugs 2004, 13, 531–541. [Google Scholar] [CrossRef]
- Straus, D.J. Gallium nitrate in the treatment of lymphoma. Semin. Oncol. 2003, 30, 25–33. [Google Scholar] [CrossRef]
- Einhorn, L. Gallium nitrate in the treatment of bladder cancer. Semin. Oncol. 2003, 30, 34–41. [Google Scholar] [CrossRef]
- Pro, B.; Bociek, R.G.; Chitambar, C.R.; Gregory, S.A.; Leonard, J.P.; Smith, S.; Novick, S. Phase 2 Multicenter Trial of Gallium Nitrate in Patients with Advanced Non-Hodgkin’s Lymphoma (NHL). Blood 2004, 104, 2487. [Google Scholar] [CrossRef]
- Webster, L.K.; Olver, I.N.; Stokes, K.H.; Sephton, R.G.; Hillcoat, B.L.; Bishop, J.F. A pharmacokinetic and phase II study of gallium nitrate in patients with non-small cell lung cancer. Cancer Chemother. Pharmacol. 2000, 45, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, L.R.; Tanner, T.; Godfrey, C.; Noll, B. Chemistry and pharmacokinetics of gallium maltolate, a compound with high oral gallium bioavailability. Met. Based Drugs 2000, 7, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Chitambar, C.R.; Purpi, D.P.; Woodliff, J.; Yang, M.; Wereley, J.P. Development of Gallium Compounds for Treatment of Lymphoma: Gallium Maltolate, a Novel Hydroxypyrone Gallium Compound, Induces Apoptosis and Circumvents Lymphoma Cell Resistance to Gallium Nitrate. J. Pharm. Exp. Ther. 2007, 322, 1228–1236. [Google Scholar] [CrossRef] [Green Version]
- Chitambar, C.R.; Al-Gizawiy, M.M.; Alhajala, H.S.; Pechman, K.R.; Wereley, J.P.; Wujek, R.; Clark, P.A.; Kuo, J.S.; Antholine, W.E.; Schmainda, K.M. Gallium Maltolate Disrupts Tumor Iron Metabolism and Retards the Growth of Glioblastoma by Inhibiting Mitochondrial Function and Ribonucleotide Reductase. Mol. Cancer Ther. 2018, 17, 1240–1250. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Stümpfig, M.; Zhang, C.; An, X.; Stubbe, J.; Lill, R.; Huang, M. The diferric-tyrosyl radical cluster of ribonucleotide reductase and cytosolic iron-sulfur clusters have distinct and similar biogenesis requirements. J. Biol. Chem. 2017, 292, 11445–11451. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Role in Fe–S Formation | Cancer Types | Implication in Tumors b |
|---|---|---|---|
| ISCU | [2Fe-2S] scaffold | AML, bladder, breast, colon, lung, ovary, prostate, rectum, renal (clear cell), stomach, testis, uterine | ↓ |
| Adrenal, liver, pancreas, renal (chromophobe), thyroid | ↑ | ||
| NFS1 | S donation in [2Fe-2S] synthesis | Adrenal, AML, bladder, colon, esophageal, lung, pancreas, rectum, renal (chromophobe), uterine | ↑ |
| Renal (clear cell), testis, thyroid | ↓ | ||
| FXN | IscU scaffold stability Fe2+ donation | AML, breast, colon, esophageal, lung, prostate, rectum, renal (chromophobe), skin, stomach, testis, thyroid, uterine | ↑ |
| Liver, renal (clear cell) | ↓ | ||
| LYRM4 | IscU scaffold stability | AML, breast, colon, esophageal, liver, lung, ovary, pancreas, prostate, rectum, renal (papillary), skin, stomach, testis, thyroid, uterine | ↑ |
| Renal (chromophobe) | ↓ | ||
| ACP1 | IscU scaffold stability | AML, bladder, breast, colon, esophageal, liver, lung, ovary, prostate, pancreas, rectum, skin, testis, thyroid, uterine | ↑ |
| Renal (chromophobe) | ↓ | ||
| GLRX5 | [2Fe-2S] trafficking | Adrenal, breast, bladder, esophageal, liver, lung, ovary, pancreas, prostate, rectum, renal (chromophobe), skin, stomach, thyroid, uterine | ↑ |
| AML, renal (clear cell), testis | ↓ | ||
| HSC20 | [2Fe-2S] trafficking | AML, liver, lung, pancreas, prostate, renal (clear cell), skin, testis, thyroid, uterine | ↑ |
| Adrenal, ovary, rectum, renal (chromophobe), stomach | ↓ | ||
| HSPA9 | [2Fe-2S] trafficking | Adrenal, AML, breast, colon, liver, lung, pancreas, prostate, rectum, renal (chromophobe), skin, testis, uterine | ↑ |
| Gene | Role in Fe–S Formation | Cancer Types | Implication in Tumors b |
|---|---|---|---|
| ABCB7 | Fe–S trafficking across inner mitochondrial membrane | AML, colon, esophageal, liver, lung (adenocarcinoma), pancreas, rectum, renal (clear cell and chromophobe), testis | ↓ |
| Adrenal, breast, ovary, skin, uterine | ↑ | ||
| CISD1 | Fe–S donation for extramitochondrial trafficking | Adrenal, AML, breast, colon, esophageal, liver, lung, ovary, pancreas, prostate, renal (chromophobe), skin, stomach, uterine | ↑ |
| testis | ↓ | ||
| CISD2 | Fe–S donation for extramitochondrial trafficking | Adrenal, AML, bladder, breast, colon, esophageal, liver, lung, ovary, pancreas, prostate, rectum, renal (clear cell, chromophobe, papillary), skin, stomach, testis, thyroid, uterine | ↑ |
| CIAPIN1 | Cytosolic Fe–S transfer | Adrenal, AML, bladder, breast, colon, liver, lung, ovary, pancreas, prostate, rectum, renal (papillary), skin, stomach, uterine | ↑ |
| Renal (clear cell), testis, thyroid | ↓ | ||
| NDOR1 | Electron transfer to CIAPIN1 for de novo cluster transfer | AML, bladder, esophageal, ovary, stomach | ↑ |
| Colon, lung, prostate, renal (clear cell), testis, thyroid, uterine | ↓ | ||
| NUBP1 | Cytosolic [4Fe-4S] formation/transfer | Adrenal, AML, breast, colon, esophageal, liver, lung, ovary, pancreas, prostate, rectum, renal (clear cell, chromophobe, papillary), skin, stomach, testis, thyroid, uterine | ↑ |
| NUBP2 | Cytosolic [4Fe-4S] formation/transfer | Adrenal, bladder, breast, colon, liver, lung, prostate, rectum, renal (clear cell, chromophobe, papillary), skin, testis, uterine | ↑ |
| Stomach | ↓ | ||
| CIAO3 | Cytosolic [4Fe-4S] formation/transfer | Adrenal, AML, bladder, breast, liver, lung, pancreas, rectum, renal (chromophobe, papillary), uterine | ↑ |
| Esophageal, stomach, testis, thyroid | ↓ | ||
| CIAO1 | Cytosolic [4Fe-4S] transfer | Adrenal, AML, bladder, breast, colon, esophageal, liver, lung, ovary, pancreas, prostate, rectum, renal (clear cell, chromophobe, papillary), skin, stomach, testis, thyroid, uterine | ↑ |
| CIAO2B | Cytosolic [4Fe-4S] transfer | AML, bladder, breast, colon, esophageal, lung, ovary, pancreas, prostate, rectum, renal (chromophobe), renal (papillary), skin, stomach, testis, thyroid, uterine | ↑ |
| MMS19 | Insertion of [4Fe-4S] cluster into target apo-proteins | AML, liver, pancreas, renal (papillary) | ↑ |
| Breast, lung, ovary, prostate, renal (clear cell and chromophobe), skin, testis, thyroid, uterine | ↓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petronek, M.S.; Spitz, D.R.; Allen, B.G. Iron–Sulfur Cluster Biogenesis as a Critical Target in Cancer. Antioxidants 2021, 10, 1458. https://doi.org/10.3390/antiox10091458
Petronek MS, Spitz DR, Allen BG. Iron–Sulfur Cluster Biogenesis as a Critical Target in Cancer. Antioxidants. 2021; 10(9):1458. https://doi.org/10.3390/antiox10091458
Chicago/Turabian StylePetronek, Michael S., Douglas R. Spitz, and Bryan G. Allen. 2021. "Iron–Sulfur Cluster Biogenesis as a Critical Target in Cancer" Antioxidants 10, no. 9: 1458. https://doi.org/10.3390/antiox10091458
APA StylePetronek, M. S., Spitz, D. R., & Allen, B. G. (2021). Iron–Sulfur Cluster Biogenesis as a Critical Target in Cancer. Antioxidants, 10(9), 1458. https://doi.org/10.3390/antiox10091458