
Cigarette Smoke Impairs Airway Epithelial Wound Repair: Role of Modulation of Epithelial-Mesenchymal Transition Processes and Notch-1 Signaling

 , and
, and
Abstract
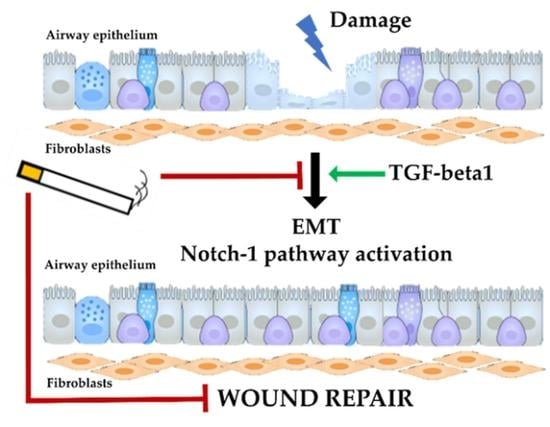
1. Introduction
2. Materials and Methods
2.1. Human Primary Bronchial Epithelial Cells (PBECs) Culture
2.2. Fibroblast and PBEC Co-Culture
2.3. Cigarette Smoke Extract (CSE) Stimulation
2.4. Whole Cigarette Smoke (WCS) Stimulation
2.5. Wound Healing Assay
2.6. Immunofluorescence by Confocal Microscopy
2.7. Gene Expression Evaluation
2.8. Statistics
3. Results
3.1. Cigarette Smoke Reduces Epithelial Wound Repair: Impact of EMT Mechanism in Submerged-PBECs
3.2. Cigarette Smoke Reduces Epithelial Wound Repair: Impact of Inhibition of Notch Signaling in Submerged-PBECs
3.3. Cigarette Smoke Reduces E-Cadherin Gene and Protein Expression in ALI-PBECs
3.4. Cigarette Smoke Reduces Vimentin Gene and Protein Expression in ALI-PBECs
3.5. Cigarette Smoke Regulates the Gene Expression of EMT Transcription Factors in ALI-PBECs and in an ALI-PBECs/Fibroblasts Co-Culture Model
3.6. Cigarette Smoke Decreases Gene Expression of MMP9 in ALI-PBECs and in the ALI-PBECs/Fibroblasts Co-Culture Model
3.7. Cigarette Smoke Decreases Gene Expression of TP63 and SCGB1A1 in ALI-PBECs and in the ALI-PBECs/Fibroblasts Co-Culture Model
3.8. Cigarette Smoke Decreases the Gene Expression of NOTCH1, JAG1 and HES1 in ALI-PBECs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelsen, S.G. Respiratory epithelial cell responses to cigarette smoke: The unfolded protein response. Pulm. Pharmacol. Ther. 2012, 25, 447–452. [Google Scholar] [CrossRef]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and antioxidant balance in the airways and airway diseases. Eur. J. Pharmacol. 2006, 533, 222–239. [Google Scholar] [CrossRef]
- Drost, E.M.; Skwarski, K.M.; Sauleda, J.; Soler, N.; Roca, J.; Agusti, A.; MacNee, W. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax 2005, 60, 293–300. [Google Scholar] [CrossRef]
- Pace, E.; Ferraro, M.; Di Vincenzo, S.; Cipollina, C.; Gerbino, S.; Cigna, D.; Caputo, V.; Balsamo, R.; Lanata, L.; Gjomarkaj, M. Comparative cytoprotective effects of carbocysteine and fluticasone propionate in cigarette smoke extract-stimulated bronchial epithelial cells. Cell Stress Chaperones 2013, 18, 733–743. [Google Scholar] [CrossRef]
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef]
- Bishop, A.E. Pulmonary epithelial stem cells. Cell Prolif. 2004, 37, 89–96. [Google Scholar] [CrossRef]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587. [Google Scholar] [CrossRef]
- Cano Sanchez, M.; Lancel, S.; Boulanger, E.; Neviere, R. Targeting Oxidative Stress and Mitochondrial Dysfunction in the Treatment of Impaired Wound Healing: A Systematic Review. Antioxidants 2018, 7, 98. [Google Scholar] [CrossRef]
- Willis, B.C.; duBois, R.M.; Borok, Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc. Am. Thorac. Soc. 2006, 3, 377–382. [Google Scholar] [CrossRef]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef]
- Koch, U.; Lehal, R.; Radtke, F. Stem cells living with a Notch. Development 2013, 140, 689–704. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertran, E.; Perez-Pomares, J.M.; Diez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisua-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Kong, D.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett. 2011, 307, 26–36. [Google Scholar] [CrossRef]
- Yuan, X.; Wu, H.; Han, N.; Xu, H.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Notch signaling and EMT in non-small cell lung cancer: Biological significance and therapeutic application. J. Hematol. Oncol. 2014, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhang, L.; He, C.S.; Xu, F.; Liu, J.L.; Hu, Z.H.; Zhao, L.P.; Tian, Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell Biochem. 2012, 113, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Saad, S.; Stanners, S.R.; Yong, R.; Tang, O.; Pollock, C.A. Notch mediated epithelial to mesenchymal transformation is associated with increased expression of the Snail transcription factor. Int. J. Biochem. Cell Biol. 2010, 42, 1115–1122. [Google Scholar] [CrossRef]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial–mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Tilley, A.E.; Harvey, B.G.; Heguy, A.; Hackett, N.R.; Wang, R.; O’Connor, T.P.; Crystal, R.G. Down-regulation of the notch pathway in human airway epithelium in association with smoking and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2009, 179, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Di Sano, C.; D’Anna, C.; Ferraro, M.; Chiappara, G.; Sangiorgi, C.; Di Vincenzo, S.; Bertani, A.; Vitulo, P.; Bruno, A.; Dino, P.; et al. Impaired activation of Notch-1 signaling hinders repair processes of bronchial epithelial cells exposed to cigarette smoke. Toxicol. Lett. 2020, 326, 61–69. [Google Scholar] [CrossRef]
- Faux, S.P.; Tai, T.; Thorne, D.; Xu, Y.; Breheny, D.; Gaca, M. The role of oxidative stress in the biological responses of lung epithelial cells to cigarette smoke. Biomarkers 2009, 14 (Suppl. S1), 90–96. [Google Scholar] [CrossRef]
- Pace, E.; Ferraro, M.; Siena, L.; Melis, M.; Montalbano, A.M.; Johnson, M.; Bonsignore, M.R.; Bonsignore, G.; Gjomarkaj, M. Cigarette smoke increases Toll-like receptor 4 and modifies lipopolysaccharide-mediated responses in airway epithelial cells. Immunology 2008, 124, 401–411. [Google Scholar] [CrossRef]
- Amatngalim, G.D.; Schrumpf, J.A.; Dishchekenian, F.; Mertens, T.C.J.; Ninaber, D.K.; van der Linden, A.C.; Pilette, C.; Taube, C.; Hiemstra, P.S.; van der Does, A.M. Aberrant epithelial differentiation by cigarette smoke dysregulates respiratory host defence. Eur. Respir. J. 2018, 51, 1701009. [Google Scholar] [CrossRef]
- Schamberger, A.C.; Staab-Weijnitz, C.A.; Mise-Racek, N.; Eickelberg, O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci. Rep. 2015, 5, 8163. [Google Scholar] [CrossRef] [PubMed]
- Brekman, A.; Walters, M.S.; Tilley, A.E.; Crystal, R.G. FOXJ1 prevents cilia growth inhibition by cigarette smoke in human airway epithelium in vitro. Am. J. Respir. Cell Mol. Biol. 2014, 51, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Amatngalim, G.D.; Broekman, W.; Daniel, N.M.; van der Vlugt, L.E.; van Schadewijk, A.; Taube, C.; Hiemstra, P.S. Cigarette Smoke Modulates Repair and Innate Immunity following Injury to Airway Epithelial Cells. PLoS ONE 2016, 11, e0166255. [Google Scholar] [CrossRef] [PubMed]
- Luppi, F.; Aarbiou, J.; van Wetering, S.; Rahman, I.; de Boer, W.I.; Rabe, K.F.; Hiemstra, P.S. Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: Role of glutathione. Respir. Res. 2005, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Avila, G.; Sommer, B.; Garcia-Hernandez, A.A.; Ramos, C. Matrix Metalloproteinases’ Role in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1245, 97–131. [Google Scholar] [CrossRef]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef]
- Puchelle, E.; Zahm, J.-M.; Tournier, J.-M.; Coraux, C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 726–733. [Google Scholar] [CrossRef]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 1838–1851. [Google Scholar] [CrossRef]
- Toivola, D.M.; Tao, G.Z.; Habtezion, A.; Liao, J.; Omary, M.B. Cellular integrity plus: Organelle-related and protein-targeting functions of intermediate filaments. Trends Cell Biol. 2005, 15, 608–617. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Miyazawa, K. Transcriptional and Post-transcriptional Regulation in TGF-β-mediated epithelial-mesenchymal transition. J. Biochem. 2012, 151, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Wang, W.L.; Chang, Y.L.; Wu, C.T.; Chao, Y.C.; Kao, S.H.; Yuan, A.; Lin, C.W.; Yang, S.C.; Chan, W.K.; et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 2009, 11, 694–704. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Lazaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef] [PubMed]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, A.; Beltran, M.; Peiro, S.; de Herreros, A.G. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Ma, L.; Jiang, M.; Zhao, X.; Sun, J.; Pan, Q.; Chu, S. Cigarette and IL-17A synergistically induce bronchial epithelial-mesenchymal transition via activating IL-17R/NF-κB signaling. BMC Pulm. Med. 2020, 20, 26. [Google Scholar] [CrossRef]
- Xia, H.; Xue, J.; Xu, H.; Lin, M.; Shi, M.; Sun, Q.; Xiao, T.; Dai, X.; Wu, L.; Li, J. Andrographolide antagonizes the cigarette smoke-induced epithelial-mesenchymal transition and pulmonary dysfunction through anti-inflammatory inhibiting HOTAIR. Toxicology 2019, 422, 84–94. [Google Scholar] [CrossRef]
- Wang, Z.; Fang, K.; Wang, G.; Guan, X.; Pang, Z.; Guo, Y.; Yuan, Y.; Ran, N.; Liu, Y.; Wang, F. Protective effect of amygdalin on epithelial–mesenchymal transformation in experimental chronic obstructive pulmonary disease mice. Phytother. Res. 2019, 33, 808–817. [Google Scholar] [CrossRef]
- Jiang, B.; Guan, Y.; Shen, H.-J.; Zhang, L.-H.; Jiang, J.-X.; Dong, X.-W.; Shen, H.-H.; Xie, Q.-M. Akt/PKB signaling regulates cigarette smoke-induced pulmonary epithelial-mesenchymal transition. Lung Cancer 2018, 122, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Brune, K.A.; Putcha, N.; Mandke, P.; O’Neal, W.K.; Shade, D.; Srivastava, V.; Wang, M.; Lam, H.; An, S.S. Cigarette smoke disrupts monolayer integrity by altering epithelial cell-cell adhesion and cortical tension. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 313, L581–L591. [Google Scholar] [CrossRef]
- Milara, J.; Peiró, T.; Serrano, A.; Guijarro, R.; Zaragozá, C.; Tenor, H.; Cortijo, J. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm. Pharmacol. Ther. 2014, 28, 138–148. [Google Scholar] [CrossRef]
- Milara, J.; Peiró, T.; Serrano, A.; Artigues, E.; Aparicio, J.; Tenor, H.; Sanz, C.; Cortijo, J. Simvastatin increases the ability of roflumilast N-oxide to inhibit cigarette smoke-induced epithelial to mesenchymal transition in well-differentiated human bronchial epithelial cells in vitro. COPD: J. Chronic Obstr. Pulm. Dis. 2015, 12, 327–338. [Google Scholar] [CrossRef]
- Mahmood, M.Q.; Sohal, S.S.; Shukla, S.D.; Ward, C.; Hardikar, A.; Noor, W.D.; Muller, H.K.; Knight, D.A.; Walters, E.H. Epithelial mesenchymal transition in smokers: Large versus small airways and relation to airflow obstruction. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 1515. [Google Scholar] [CrossRef] [PubMed]
- Sohal, S.S.; Reid, D.; Soltani, A.; Ward, C.; Weston, S.; Muller, H.K.; Wood-Baker, R.; Walters, E.H. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology 2010, 15, 930–938. [Google Scholar] [CrossRef]
- Osei, E.T.; Hackett, T.L. Epithelial-mesenchymal crosstalk in COPD: An update from in vitro model studies. Int. J. Biochem. Cell Biol. 2020, 125, 105775. [Google Scholar] [CrossRef]
- Evans, M.J.; Van Winkle, L.S.; Fanucchi, M.V.; Plopper, C.G. The attenuated fibroblast sheath of the respiratory tract epithelial-mesenchymal trophic unit. Am. J. Respir. Cell Mol. Biol. 1999, 21, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Mechanisms of TGFbeta-Induced Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 63. [Google Scholar] [CrossRef]
- Espinoza, I.; Pochampally, R.; Xing, F.; Watabe, K.; Miele, L. Notch signaling: Targeting cancer stem cells and epithelial-to-mesenchymal transition. OncoTargets Ther. 2013, 6, 1249–1259. [Google Scholar] [CrossRef]
- Aaron, S.D.; Donaldson, G.C.; Whitmore, G.A.; Hurst, J.R.; Ramsay, T.; Wedzicha, J.A. Time course and pattern of COPD exacerbation onset. Thorax 2012, 67, 238–243. [Google Scholar] [CrossRef]
- Hong, K.U.; Reynolds, S.D.; Watkins, S.; Fuchs, E.; Stripp, B.R. Basal cells are a multipotent progenitor capable of renewing the bronchial epithelium. Am. J. Pathol. 2004, 164, 577–588. [Google Scholar] [CrossRef]
- Tsao, P.N.; Vasconcelos, M.; Izvolsky, K.I.; Qian, J.; Lu, J.; Cardoso, W.V. Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development 2009, 136, 2297–2307. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name | Forward Sequence (5’-->3’) | Reverse Sequence (5’-->3’) | Accession Number (GenBank) |
|---|---|---|---|---|
| CDH1 | E-cadherin | TTCCCAACTCCTCTCCTG | AAACCTTGCCTTCTTTGTC | NM_001317185.2; NM_001317186.2; NM_001317184.2; NM_004360.5 |
| VIMENTIN | Vimentin | TTGAACGCAAAGTGGAATC | AGGTCAGGCTTGGAAACA | NM_003380.5 |
| ZEB-1 | ZEB-1 | TTACACCTTTGCATACAGAACCC | GATGATGAATGCGAGTCAGATGC | NM_001128128.3 |
| SNAIL1 | Snail1 | TCGGAAGCCTAACTACAGCGA | AGATGAGCATTGGCAGCGAG | NM_005985.4 |
| SNAIL2 | Slug | TGTGACAAGGAATATGTGAGCC | TGAGCCCTCAGATTTGACCTG | NM_003068.5 |
| TP63 | TP63 | CCACCTGGACGTATTCCACTG | TCGAATCAAATGACTAGGAGGGG | NM_001329964.2 |
| SCGB1A1 | SCGB1A1 | ACATGAGGGAGGCAGGGGCTC | ACTCAAAGCATGGCAGCGGCA | NM_003357.5 |
| RPL13A | RPL13A | AAGGTGGTGGTCGTACGCTGTG | CGGGAAGGGTTGGTGTTCATCC | NM_012423.4 |
| ATP5B | ATP5B | TCACCCAGGCTGGTTCAGA | AGTGGCCAGGGTAGGCTGAT | NM_001686.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Vincenzo, S.; Ninaber, D.K.; Cipollina, C.; Ferraro, M.; Hiemstra, P.S.; Pace, E. Cigarette Smoke Impairs Airway Epithelial Wound Repair: Role of Modulation of Epithelial-Mesenchymal Transition Processes and Notch-1 Signaling. Antioxidants 2022, 11, 2018. https://doi.org/10.3390/antiox11102018
Di Vincenzo S, Ninaber DK, Cipollina C, Ferraro M, Hiemstra PS, Pace E. Cigarette Smoke Impairs Airway Epithelial Wound Repair: Role of Modulation of Epithelial-Mesenchymal Transition Processes and Notch-1 Signaling. Antioxidants. 2022; 11(10):2018. https://doi.org/10.3390/antiox11102018
Chicago/Turabian StyleDi Vincenzo, Serena, Dennis K. Ninaber, Chiara Cipollina, Maria Ferraro, Pieter S. Hiemstra, and Elisabetta Pace. 2022. "Cigarette Smoke Impairs Airway Epithelial Wound Repair: Role of Modulation of Epithelial-Mesenchymal Transition Processes and Notch-1 Signaling" Antioxidants 11, no. 10: 2018. https://doi.org/10.3390/antiox11102018
APA StyleDi Vincenzo, S., Ninaber, D. K., Cipollina, C., Ferraro, M., Hiemstra, P. S., & Pace, E. (2022). Cigarette Smoke Impairs Airway Epithelial Wound Repair: Role of Modulation of Epithelial-Mesenchymal Transition Processes and Notch-1 Signaling. Antioxidants, 11(10), 2018. https://doi.org/10.3390/antiox11102018