
Modulation of Oxidative Stress-Induced Senescence during Non-Alcoholic Fatty Liver Disease

 , , and
, , and
Abstract
:1. Introduction
2. Molecular Mechanisms of NAFLD
3. Role of Senescence in the Development of NAFLD
3.1. Molecular Mechanisms of Cellular Senescence
3.2. Senescence Features in Liver Disease
3.3. Association of Cellular Senescence and NAFLD
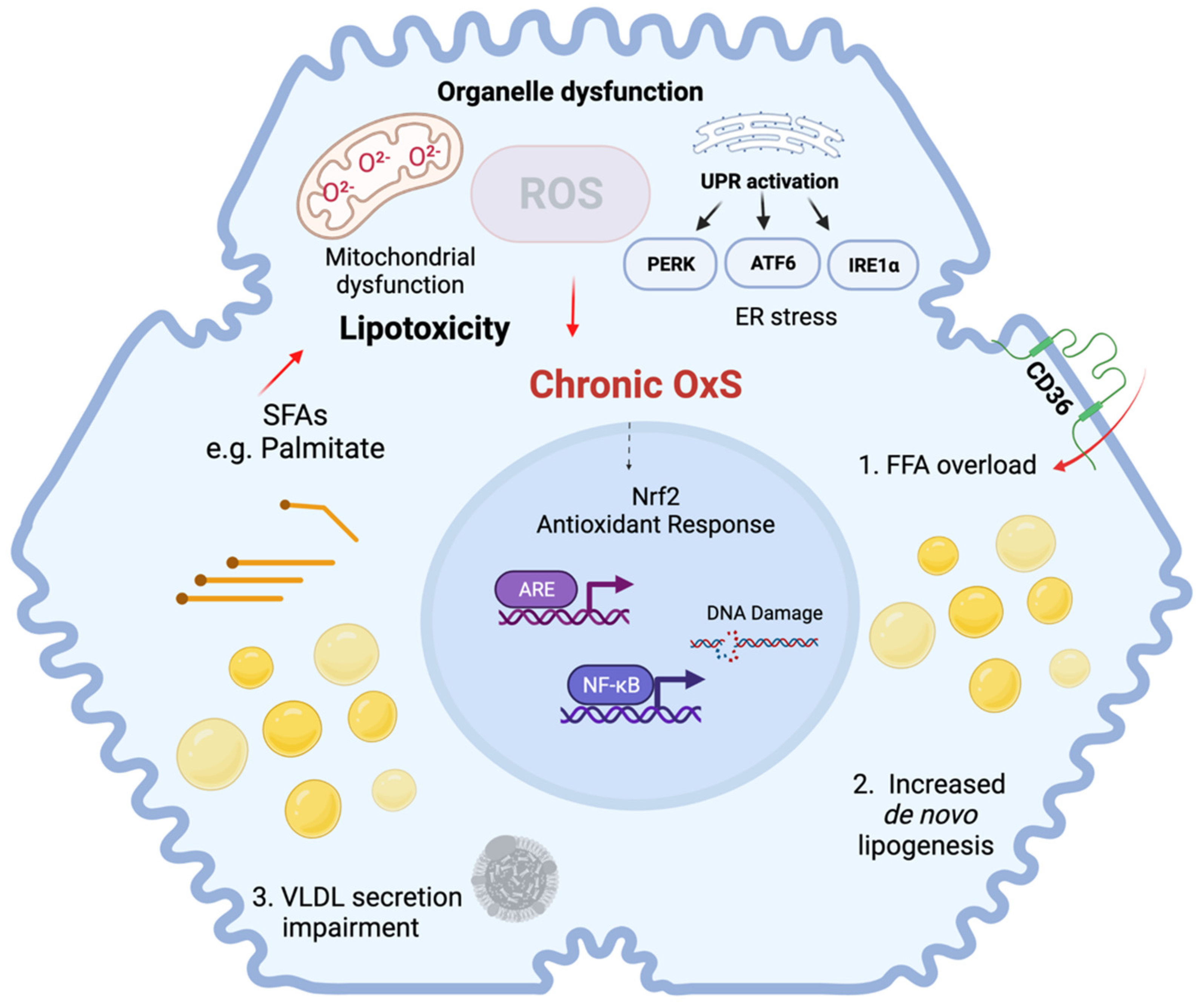
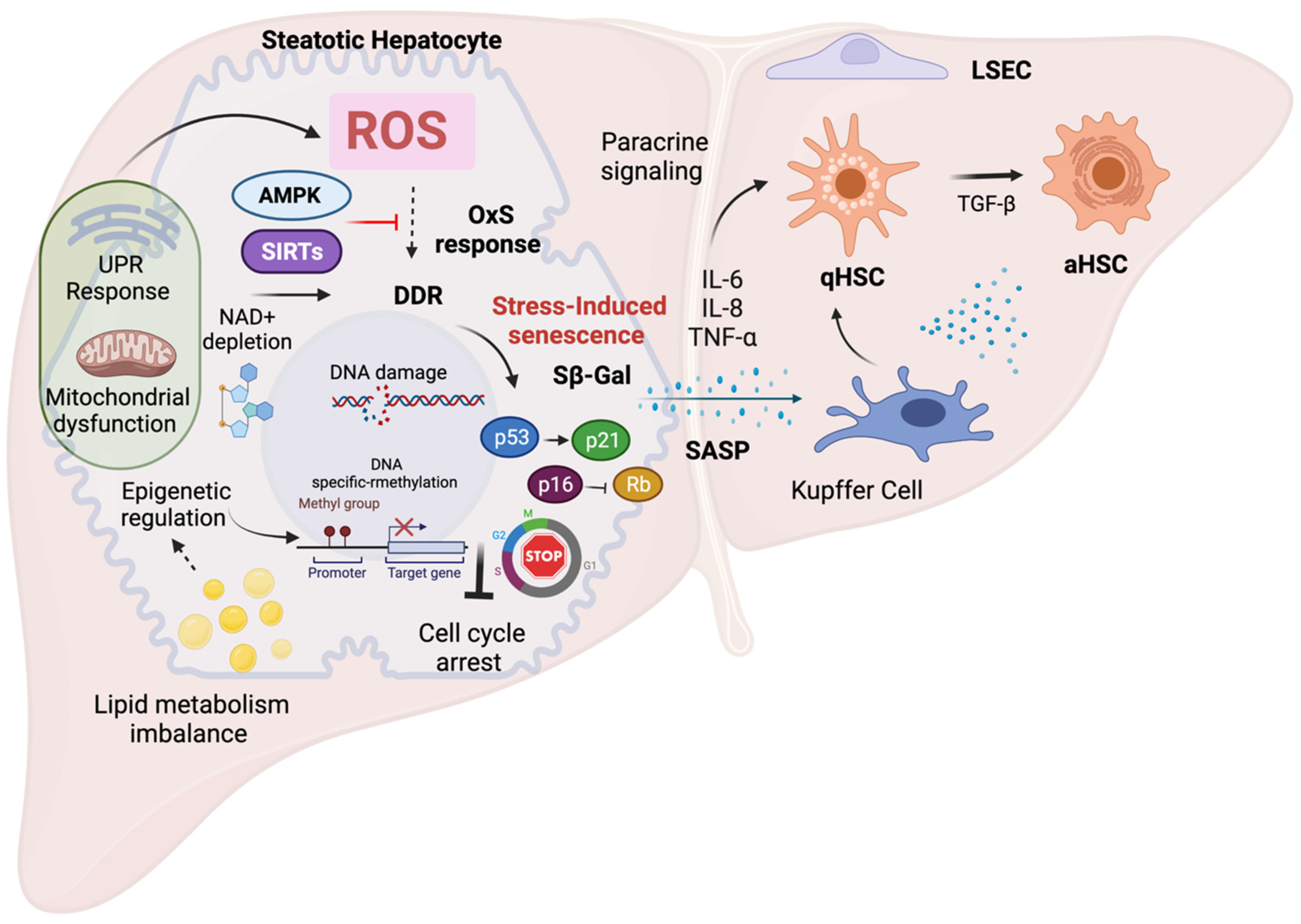
4. Molecular Mechanisms of Oxidative Stress Contributing to Senescence in NAFLD
4.1. Senescence and Oxidative Stress
4.2. Senescence and Hepatic Lipid Metabolism
4.3. Senescence and Mitochondrial Dysfunction
4.4. Senescence and ER Stress
4.5. Senescence and Epigenetic Modifications
5. Modulating OxS-Induced Senescence as a Potential Therapy in NAFLD
5.1. Targeting Oxidative Stress to Modulate Cellular Senescence

{kind=link}
{kind=link}
| Compound | Experimental Models | Molecular Mechanism | Experimental Findings | References |
|---|---|---|---|---|
| Vitamin D, Paricalcitol (vitD agonist) | In vivo Hamster model Sprague-Dawley rats on HFD 1 | ↑Nrf2 activators ↑SIRTs ROS inhibition |
| [93,180,210] |
| Resveratrol | In vitro Hepatic cell lines In vivo Rat steatosis models (HFD 1) | ↑AMPK activation ↑SIRTs ROS inhibition |
| [184,185,186,187] |
| Dietary Polyphenols (Flavononids) | In vivo mouse steatosis model | ↑Nrf2 activators ↑SIRTs ROS inhibition |
| [190,191,192,193,194,195,196] |
| Metformin | In vitro Primary rat hepatocytes Hepatic cell lines Human adipose stromal cells In vivo Mouse steatosis model | ↑AMPK activation, ↓NF-κB pathway |
| [200,201,203,204] |
| Rapamycin | In vivo Rat cirrhosis model Aging mice model Clinical randomized study | mTOR inhibition |
| [112,216,217,218] |
5.2. SASP Inhibition: Senostatic/Senomorphic Drugs
5.3. Targeting Senescent Cells and Survival Pathways: Senolytics
6. Conclusions and Perspectives
Funding
Conflicts of Interest
Abbreviations
| OxS | Oxidative stress |
| ROS | Reactive oxygen species |
| NAFLD | Non-alcoholic liver disease |
| NASH | Non-alcoholic steatohepatitis |
| MMP | Metalloproteinases |
| DDR | DNA damage response |
| SASP | Senescence-associated secretory phenotype |
| SIPS | Stress-induced premature senescence |
| DAMPs | Damage-associated molecular patterns |
| IL | Interleukin |
| TGF-β | Transforming growth factor-beta |
| ER | Endoplasmic Reticulum |
| OxPhos | Oxidative phosphorylation |
| Keap1-Nrf2 | Kelch-like ECH-associated protein 1-(Keap1-) nuclear factor-erythroid2-related factor 2 (Nrf2) |
| ARE | Antioxidant response element |
| UPR | Unfolded protein response |
| TG | Triglycerides |
| FFA | Free fatty acids |
| SFAs | Saturated fatty acids |
| MiDAS | Mitochondrial dysfunction associated senescence |
| MERCs | Mitochondrial ER contacts |
References
- Kleiner, D.E.; Makhlouf, H.R.; Program, D.; Investigation, P.; Branch, R.; Shams, A. Histology of NAFLD and NASH in Adults and Children. Clin. Liver Dis. 2017, 20, 293–312. [Google Scholar] [CrossRef] [Green Version]
- Yasui, K.; Hashimoto, E.; Komorizono, Y.; Koike, K.; Arii, S.; Imai, Y.; Shima, T.; Kanbara, Y.; Saibara, T.; Mori, T.; et al. Characteristics of Patients With Nonalcoholic Steatohepatitis Who Develop Hepatocellular Carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 428–433. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Spahis, S.; Delvin, E.; Borys, J.M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid. Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef]
- Pole, A.; Dimri, M.; Dimri, G.P. Oxidative stress, cellular senescence and ageing. AIMS Mol. Sci. 2016, 3, 300–324. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [Green Version]
- Ben-Porath, I.; Weinberg, R.A. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 2005, 37, 961–976. [Google Scholar] [CrossRef]
- Aravinthan, A.D.; Alexander, G.J.M. Senescence in chronic liver disease: Is the future in aging? J. Hepatol. 2016, 65, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Meijnikman, A.S.; Herrema, H.; Scheithauer, T.P.M.; Kroon, J.; Nieuwdorp, M.; Groen, A.K. Evaluating causality of cellular senescence in non-alcoholic fatty liver disease. JHEP Rep. 2021, 3, 100301. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Seebacher, F.; Zeigerer, A.; Kory, N.; Krahmer, N. Hepatic lipid droplet homeostasis and fatty liver disease. Semin. Cell Dev. Biol. 2020, 108, 72–81. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Scorletti, E.; Carr, R.M. A New Perspective on NAFLD: Focusing on Lipid Droplets; European Association for the Study of the Liver (EASL): Marburg, Germany, 2021; ISBN 2066858684. [Google Scholar]
- Geng, Y.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H.; Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2021, 15, 21–35. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: Implications for prevention and therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Gonzalez, A.; Huerta-Salgado, C.; Orozco-Aguilar, J.; Aguirre, F.; Tacchi, F.; Simon, F.; Cabello-Verrugio, C. Role of Oxidative Stress in Hepatic and Extrahepatic Dysfunctions during Nonalcoholic Fatty Liver Disease (NAFLD). Oxidative Med. Cell. Longev. 2020, 2020, 1617805. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [Green Version]
- Nassir, F.; Ibdah, J.A. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci 2014, 15, 8713–8742. [Google Scholar] [CrossRef] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [Green Version]
- Jana, B.A.; Chintamaneni, P.K.; Krishnamurthy, P.T.; Wadhwani, A.; Mohankumar, S.K. Cytosolic lipid excess-induced mitochondrial dysfunction is the cause or effect of high fat diet-induced skeletal muscle insulin resistance: A molecular insight. Mol. Biol. Rep. 2019, 46, 957–963. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial Oxidative Stress and Antioxidants Balance in Fatty Liver Disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [Green Version]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Wood, N.E.; Kositangool, P.; Hariri, H.; Marchand, A.J.; Henne, W.M. Nutrient Signaling, Stress Response, and Inter-organelle Communication Are Non-canonical Determinants of Cell Fate. Cell Rep. 2020, 33, 108446. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 485. [Google Scholar] [CrossRef]
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.A.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the Unique Role of the Retinoblastoma Tumor Suppressor during Cellular Senescence. Cancer Cell 2010, 17, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the regulation of cellular senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [Green Version]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [Green Version]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STI NG pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD + metabolism governs the proinflammatory senescence-associated secretome. Nat. Cell Biol. 2019, 21, 397–407. [Google Scholar] [CrossRef]
- Xiao, M.; Chen, W.; Wang, C.; Wu, Y.; Zhu, S.; Zeng, C.; Cai, Y.; Liu, C.; He, Z. Senescence and cell death in chronic liver injury: Roles and mechanisms underlying hepatocarcinogenesis. Oncotarget 2018, 9, 8772–8784. [Google Scholar] [CrossRef] [Green Version]
- Huda, N.; Liu, G.; Hong, H.; Yan, S.; Khambu, B.; Yin, X.M. Hepatic senescence, the good and the bad. World J. Gastroenterol. 2019, 25, 5069–5081. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Bezsonov, E.E.; Orekhov, A.N. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed. Pharmacother. 2021, 142, 112041. [Google Scholar] [CrossRef]
- Irvine, K.M.; Skoien, R.; Bokil, N.J.; Melino, M.; Thomas, G.P.; Loo, D.; Gabrielli, B.; Hill, M.M.; Sweet, M.J.; Clouston, A.D.; et al. Senescent human hepatocytes express a unique secretory phenotype and promote macrophage migration. World J. Gastroenterol. 2014, 20, 17851–17862. [Google Scholar] [CrossRef] [Green Version]
- Hunt, N.J.; Kang, S.W.S.; Lockwood, G.P.; Le Couteur, D.G.; Cogger, V.C. Hallmarks of Aging in the Liver. Comput. Struct. Biotechnol. J. 2019, 17, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Chen, F.; Li, J.X.; Liu, C.C.; Zhang, H.B.; Xia, Y.; Yu, B.; You, P.; Xiang, D.; Lu, L.; et al. Reversal of hepatocyte senescence after continuous in vivo cell proliferation. Hepatology 2014, 60, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; D’Adda Di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.G.; Mller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10, eaan1230. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Mehal, W.; Nagy, L.E.; Rotman, Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell. Mol. Immunol. 2021, 18, 73–91. [Google Scholar] [CrossRef]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer Cell Engulfment of Apoptotic Bodies Stimulates Death Ligand and Cytokine Expression. Hepatology 2003, 38, 1188–1198. [Google Scholar] [CrossRef]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Busch, C.J.; Hendrikx, T.; Weismann, D.; Jäckel, S.; Sofie, M.; Walenbergh, A.; Rendeiro, A.F.; Weißer, J.; Puhm, F.; Göderle, L.; et al. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology 2018, 65, 1181–1195. [Google Scholar] [CrossRef] [Green Version]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef]
- Gao, B.; Tsukamato, H. Inflammation in alcoholic and nonalcoholic fatty liver disease: Friend or foe? Gastroenterology 2016, 150, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, A.; Daily, K.P.; Criss, A.K. Assembly of NADPH oxidase in human neutrophils is modulated by the opacity-associated protein expression state of Neisseria gonorrhoeae. Infect. Immun. 2014, 82, 1036–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. IL-17 signaling in inflammatory cells, Kupffer cells and Hepatic Stellate cells exacerbates liver fibrosis. Gastroenterology 2012, 143, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, S.; Ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagnado, A.; Leslie, J.; Ruchaud-Sparagano, M.; Victorelli, S.; Hirsova, P.; Ogrodnik, M.; Collins, A.L.; Vizioli, M.G.; Habiballa, L.; Saretzki, G.; et al. Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. EMBO J. 2021, 40, e106048. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoridi, A.M.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Nonalcoholic Fatty Liver Disease and Progression to Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Panasiuk, A.; Dzieciol, J.; Panasiuk, B.; Prokopowicz, D. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J. Gastroenterol. 2006, 12, 6198–6202. [Google Scholar] [CrossRef]
- Akazawa, Y.; Nakashima, R.; Matsuda, K.; Okamaoto, K.; Hirano, R.; Kawasaki, H.; Miuma, S.; Miyaaki, H.; Malhi, H.; Abiru, S.; et al. Detection of DNA damage response in nonalcoholic fatty liver disease via p53-binding protein 1 nuclear expression. Mod. Pathol. 2019, 32, 997–1007. [Google Scholar] [CrossRef]
- Aravinthan, A.; Scarpini, C.; Tachtatzis, P.; Verma, S.; Penrhyn-Lowe, S.; Harvey, R.; Davies, S.E.; Allison, M.; Coleman, N.; Alexander, G. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J. Hepatol. 2013, 58, 549–556. [Google Scholar] [CrossRef]
- Laish, I.; Mannasse-Green, B.; Hadary, R.; Biron-Shental, T.; Konikoff, F.M.; Amiel, A.; Kitay-Cohen, Y. Telomere Dysfunction in Nonalcoholic Fatty Liver Disease and Cryptogenic Cirrhosis. Cytogenet. Genome Res. 2016, 150, 93–99. [Google Scholar] [CrossRef]
- Ping, F.; Li, Z.Y.; Lv, K.; Zhou, M.C.; Dong, Y.X.; Sun, Q.; Li, Y.X. Deoxyribonucleic acid telomere length shortening can predict the incidence of non-alcoholic fatty liver disease in patients with type 2 diabetes mellitus. J. Diabetes Investig. 2017, 8, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Aravinthan, A.; Mells, G.; Allison, M.; Leathart, J.; Kotronen, A.; Yki-Jarvinen, H.; Daly, A.K.; Day, C.P.; Anstee, Q.M.; Alexander, G. Gene polymorphisms of cellular senescence marker p21 and disease progression in non-alcohol-related fatty liver disease. Cell Cycle 2014, 13, 1489–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelmann, C.; Tacke, F. The Potential Role of Cellular Senescence in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 652. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Hasegawa, G.; Okada, H.; Senmaru, T.; Fukui, M.; Nakamura, N.; Sawada, M.; Kitawaki, J.; Okanoue, T.; Kishimoto, Y.; et al. Leprdb/db Mice with Senescence Marker Protein-30 Knockout (Leprdb/dbSmp30Y/-) Exhibit Increases in Small Dense-LDL and Severe Fatty Liver Despite Being Fed a Standard Diet. PLoS ONE 2013, 8, e65698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y.; Masutomi, H.; Noda, Y.; Ozawa, Y.; Takahashi, K.; Handa, S.; Maruyama, N.; Shimizu, T.; Ishigami, A. Senescence marker protein-30/superoxide dismutase 1 double knockout mice exhibit increased oxidative stress and hepatic steatosis. FEBS Open Bio 2014, 4, 522–532. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, I.I.; Katsarou, A.; Legaki, A.I.; Pyrina, I.; Ntostoglou, K.; Papatheodoridi, A.M.; Gercken, B.; Pateras, I.S.; Gorgoulis, V.G.; Koutsilieris, M.; et al. Hepatic senescence accompanies the development of NAFLD in non-aged mice independently of obesity. Int. J. Mol. Sci. 2021, 22, 3446. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z.; Hou, J.Y.; Zhang, R.H.; Yeh, M.M.; Williams, J.; Dela Peňa, A.; Francisco, R.; Osvath, S.R.; Brooling, J.; et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J. Gastroenterol. Hepatol. 2009, 24, 443–452. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Nishiyama, K.; Mataki, N.; Irie, R.; Minamino, T.; et al. P53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2012, 57, 837–843. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Jurk, D. Senescence explains age- and obesity-related liver steatosis. Cell Stress 2017, 8, 70–72. [Google Scholar] [CrossRef] [Green Version]
- Lohr, K.; Pachl, F.; Moghaddas Gholami, A.; Geillinger, K.E.; Daniel, H.; Kuster, B.; Klingenspor, M. Reduced mitochondrial mass and function add to age-related susceptibility toward diet-induced fatty liver in C57BL/6J mice. Physiol. Rep. 2016, 4, e12988. [Google Scholar] [CrossRef] [Green Version]
- Bhaskaran, S.; Pharaoh, G.; Ranjit, R.; Murphy, A.; Matsuzaki, S.; Nair, B.C.; Forbes, B.; Gispert, S.; Auburger, G.; Humphries, K.M.; et al. Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance. EMBO Rep. 2018, 19, e45009. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, Y.; Hu, S.; Xu, Y.; Stroup, D.; Pan, X.; Bawa, F.C.; Chen, S.; Gopoju, R.; Yin, L.; et al. Hepatocyte Nuclear Factor 4α Prevents the Steatosis-to-NASH Progression by Regulating p53 and Bile Acid Signaling (in mice). Hepatology 2021, 73, 2251–2265. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Wu, X.; Chen, H.; Xia, X.; Song, X.; Chen, S.; Lu, X.; Jin, J.; Su, Q.; Cai, D.; et al. Aging-induced aberrant RAGE/PPARα axis promotes hepatic steatosis via dysfunctional mitochondrial β oxidation. Aging Cell 2020, 19, e13238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Wang, B.; Luo, Y.; Shi, J.; Zhao, B. The p66shc-mediated regulation of hepatocyte senescence influences hepatic steatosis in nonalcoholic fatty liver disease. Med. Sci. Monit. 2020, 26, e921887-1. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.E.; Duan, L.; He, Y.; Yuan, C.; Wang, T.; Yuan, D.; Zhang, C.; Liu, C. Saturated Fatty Acids Promote Hepatocytic Senecence through Regulation of miR-34a/Cyclin-Dependent Kinase 6. Mol. Nutr. Food Res. 2020, 64, 2000383. [Google Scholar] [CrossRef]
- Daugherity, E.K.; Balmus, G.; Al Saei, A.; Moore, E.S.; Abi Abdallah, D.; Rogers, A.B.; Weiss, R.S.; Maurer, K.J. The DNA damage checkpoint protein ATM promotes hepatocellular apoptosis and fibrosis in a mouse model of non-alcoholic fatty liver disease. Cell Cycle 2012, 11, 1918–1928. [Google Scholar] [CrossRef] [Green Version]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 70–72. [Google Scholar] [CrossRef]
- Cindrova-Davies, T.; Fogarty, N.M.E.; Jones, C.J.P.; Kingdom, J.; Burton, G.J. Evidence of oxidative stress-induced senescence in mature, post-mature and pathological human placentas. Placenta 2018, 68, 15–22. [Google Scholar] [CrossRef]
- Saroyo, Y.B.; Wibowo, N.; Irwinda, R.; Prijanti, A.R.; Yunihastuti, E.; Bardosono, S.; Krisnadi, S.R.; Permata, P.I.; Wijaya, S.; Santawi, V.P.A. Oxidative Stress Induced Damage and Early Senescence in Preterm Placenta. J. Pregnancy 2021, 2021, 9923761. [Google Scholar] [CrossRef]
- Chen, Q.M.; Prowse, K.R.; Tu, V.C.; Purdom, S.; Linskens, M.H.K. Uncoupling the senescent phenotype from telomere shortening in hydrogen peroxide-treated fibroblasts. Exp. Cell Res. 2001, 265, 294–303. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Xu, M.; Yu, H.; Wang, L.; Li, X.; Rak, J.; Wang, S.; Zhao, R.C. Mesenchymal stem cell-derived small extracellular vesicles mitigate oxidative stress-induced senescence in endothelial cells via regulation of miR-146a/Src. Signal Transduct. Target. Ther. 2021, 6, 354. [Google Scholar] [CrossRef]
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 165. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.; Leu, J.I.; Basu, S.; Khaku, S.; Anokye-, F.; Liu, Q.; George, D.L.; Ahima, R.S.; Murphy, M.E. The P72R polymorphism of p53 predisposes to obesity and metabolic dysfunction. Cell Rep. 2016, 14, 2413–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldez, M.J.; Bjorklund, M.; Kaldis, P. Cell cycle regulation in NAFLD: When imbalanced metabolism limits cell division. Hepatol. Int. 2020, 14, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodenak-Kladniew, B.; Castro, A.; Stärkel, P.; Galle, M.; Crespo, R. 1,8-Cineole promotes G0/G1 cell cycle arrest and oxidative stress-induced senescence in HepG2 cells and sensitizes cells to anti-senescence drugs. Life Sci. 2020, 243, 117271. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Q.; Wang, H.; Qin, C.; Cui, X.; Li, L.; Liu, Y.; Chang, H. Induction of ROS and DNA damage-dependent senescence by icaritin contributes to its antitumor activity in hepatocellular carcinoma cells. Pharm. Biol. 2019, 57, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Song, A.; Ma, M.; Wang, P.; Zhang, X.; Lu, C.; Zhang, J.; Zheng, S.; Jin, H. Curcumol inhibits ferritinophagy to restrain hepatocyte senescence through YAP/NCOA4 in non-alcoholic fatty liver disease. Cell Prolif. 2021, 54, e13107. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The role of Nrf2 in liver disease: Novel molecular mechanisms and therapeutic approaches. Front. Pharmacol. 2019, 9, 1428. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Tarantini, S.; Nyúl-Tóth, Á.; Kiss, T.; Yabluchanskiy, A.; Csipo, T.; Balasubramanian, P.; Lipecz, A.; Benyo, Z.; Csiszar, A. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: From increased cellular senescence to the pathogenesis of age-related vascular diseases. GeroScience 2019, 41, 727–738. [Google Scholar] [CrossRef]
- Yu, C.; Xiao, J.H. The Keap1-Nrf2 System: A Mediator between Oxidative Stress and Aging. Oxid. Med. Cell. Longev. 2021, 2021, 6635460. [Google Scholar] [CrossRef]
- Lu, D.; Le, Y.; Ding, J.; Dou, X.; Mao, W.; Zhu, J.; Dou, X.; Mao, W.; Zhu, J.; Dou, X.; et al. CLIC1 Inhibition Protects against Cellular Senescence and Endothelial Dysfunction Via the Nrf2_HO-1 Pathway.pdf. Cell Biochem. Biophys. 2021, 79, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, R.; Qiao, W.; Zhang, W.; Chen, J.; Mao, L.; Goltzman, D.; Miao, D. 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling. Aging Cell 2019, 18, e12951. [Google Scholar] [CrossRef] [PubMed]
- Jie, Z.; Huan, Y.; Mengyun, W.; Yasha, L.; Huafeng, P.; Ke, Y. Nrf2 modulates immunosuppressive ability and cellular senescence of human umbilical cord mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2020, 526, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L.; Chang, S.; Millard, M.; Schreiber-Agus, N.; DePinho, R.A. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 2000, 287, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Tang, L.; Xin, G.; Li, S.; Ma, L.; Xu, Y.; Zhuang, M.; Xiong, Q.; Wei, Z.; Xing, Z.; et al. Oxidative stress mediated by lipid metabolism contributes to high glucose-induced senescence in retinal pigment epithelium. Free Radic. Biol. Med. 2019, 130, 48–58. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Krüger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1233. [Google Scholar] [CrossRef]
- Lizardo, D.Y.; Lin, Y.L.; Gokcumen, O.; Atilla-Gokcumen, G.E. Regulation of lipids is central to replicative senescence. Mol. Biosyst. 2017, 13, 498–509. [Google Scholar] [CrossRef]
- Saitou, M.; Lizardo, D.Y.; Taskent, R.O.; Millner, A.; Gokcumen, O.; Atilla-Gokcumen, G.E. An evolutionary transcriptomics approach links CD36 to membrane remodeling in replicative senescence. Mol. Omi. 2018, 14, 237–246. [Google Scholar] [CrossRef]
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov. 2017, 3, 17075. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, Y.; Huang, Y.; Zeng, H.; Hu, B.; Guan, L.; Zhang, H.; Yu, A.M.; Johnson, C.H.; J.Gonzalez, F.; et al. PPARα regulates tumor cell proliferation and senescence via a novel target gene carnitine palmitoyltransferase 1C. Carcinogenesis 2017, 38, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Seok, J.; Jung, H.S.; Park, S.; Lee, J.O.; Kim, C.J.; Kim, G.J. Alteration of fatty acid oxidation by increased CPT1A on replicative senescence of placenta-derived mesenchymal stem cells. Stem Cell Res. Ther. 2020, 11, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millner, A.; Ekin Atilla-Gokcumen, G. Lipid players of cellular senescence. Metabolites 2020, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolova-Karakashian, M. Sphingolipids at the Crossroads of NAFLD and Senescence. Adv. Cancer Res. 2018, 140, 155–190. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschênes-Simard, X.; Ferbeyre, G. Mitochondrial Dysfunction Contributes to Oncogene-Induced Senescence. Mol. Cell. Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.F.; Chen, T.; Wanagat, J.; Laflamme, M.; Marcinek, D.J.; Emond, M.J.; Ngo, C.P.; Prolla, T.A.; Rabinovitch, P.S. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Trimarchi, J.R.; Smith, P.J.S.; Keefe, D.L. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell 2002, 1, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-inducedsenescence via autophagic flux. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Jang, H.; Lee, S.M.; Lee, J.E.; Choi, J.; Kim, T.W.; Cho, E.J.; Youn, H.D. ATP-citrate lyase regulates cellular senescence via an AMPK- and p53-dependent pathway. FEBS J. 2015, 282, 361–371. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Choi, Y.S.; Kim, D.W.; Cheong, J.Y.; Song, K.Y.; Ryu, M.S.; Lim, I.K. Mitochondrial nucleoid remodeling and biogenesis are regulated by the p53-p21WAF1-PKCζ pathway in p16INK4a-silenced cells. Aging 2020, 12, 6700–6732. [Google Scholar] [CrossRef]
- Tsuchihashi, N.A.; Hayashi, K.; Dan, K.; Goto, F.; Nomura, Y.; Fujioka, M.; Kanzaki, S.; Komune, S.; Ogawa, K. Autophagy through 4EBP1 and AMPK regulates oxidative stress-induced premature senescence in auditory cells. Oncotarget 2015, 6, 3644–3655. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhang, W.; Zeng, L.Q.; Bai, H.; Li, J.; Zhou, J.; Zhou, G.Y.; Fang, C.W.; Wang, F.; Qin, X.J. Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol. 2020, 36, 101635. [Google Scholar] [CrossRef]
- Lew, L.C.; Hor, Y.Y.; Jaafar, M.H.; Lau, A.S.Y.; Lee, B.K.; Chuah, L.O.; Yap, K.P.; Azlan, A.; Azzam, G.; Choi, S.B.; et al. Lactobacillus strains alleviated hyperlipidemia and liver steatosis in aging rats via activation of ampk. Int. J. Mol. Sci. 2020, 21, 5872. [Google Scholar] [CrossRef]
- Lee, Y.H.; Yun, M.R.; Kim, H.M.; Jeon, B.H.; Park, B.C.; Lee, B.W.; Kang, E.S.; Lee, H.C.; Park, Y.W.; Cha, B.S. Exogenous administration of DLK1 ameliorates hepatic steatosis and regulates gluconeogenesis via activation of AMPK. Int. J. Obes. 2016, 40, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M.G.; Lin, T.; Shokhirev, M.N.; Pinto, A.F.M.; Ross, D.S.; et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019, 26, 192–208.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Zhou, C.C.; Yang, X.; Hua, X.; Liu, J.; Fan, M.B.; Li, G.Q.; Song, J.; Xu, T.Y.; Li, Z.Y.; Guan, Y.F.; et al. Hepatic NAD(+) deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br. J. Pharmacol. 2016, 173, 2352–2368. [Google Scholar] [CrossRef] [Green Version]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef]
- Lee, I.H. Mechanisms and disease implications of sirtuin-mediated autophagic regulation. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Liu, Y.H.; Fu, Y.C.; Liu, X.M.; Zhou, X.H. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar]
- Merksamer, P.I.; Liu, Y.; He, W.; Hirschey, M.D.; Chen, D.; Verdin, E. The sirtuins, oxidative stress and aging: An emerging link. Aging 2013, 5, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassir, F.; Ibdah, J.A. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 Is a Mitochondria-Localized Tumor Suppressor Required for Maintenance of Mitochondrial Integrity and Metabolism during Stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Jia, X.; Cui, Y.; Song, Y.; Wang, S.; Geng, Y.; Li, R.; Gao, W.; Fu, D. Sirt3-mediated mitophagy regulates AGEs-induced BMSCs senescence and senile osteoporosis. Redox Biol. 2021, 41, 101915. [Google Scholar] [CrossRef]
- Ma, C.; Sun, Y.; Pi, C.; Wang, H.; Sun, H.; Yu, X.; Shi, Y.; He, X. Sirt3 Attenuates Oxidative Stress Damage and Rescues Cellular Senescence in Rat Bone Marrow Mesenchymal Stem Cells by Targeting Superoxide Dismutase 2. Front. Cell Dev. Biol. 2020, 8, 599376. [Google Scholar] [CrossRef]
- Nassir, F.; Arndt, J.J.; Johnson, S.A.; Ibdah, J.A. Regulation of mitochondrial trifunctional protein modulates nonalcoholic fatty liver disease in mice. J. Lipid Res. 2018, 59, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.-H. SIRT3 as a Regulator of Non-alcoholic Fatty Liver Disease. J. Lifestyle Med. 2014, 4, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lu, X.; Zhou, Y.; Guo, X.; Chang, Y. Major royal jelly proteins prevents NAFLD by improving mitochondrial function and lipid accumulation through activating the AMPK / SIRT3 pathway in vitro. J. Food Sci. 2021, 86, 1105–1113. [Google Scholar] [CrossRef]
- Li, R.; Xin, T.; Li, D.; Wang, C.; Zhu, H.; Zhou, H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018, 18, 229–243. [Google Scholar] [CrossRef]
- Li, S.; Dou, X.; Ning, H.; Song, Q.; Wei, W.; Zhang, X.; Shen, C.; Li, J.; Sun, C.; Song, Z. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology 2017, 66, 936–952. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.H.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.Y. Mitophagy in the Pathogenesis of Liver Diseases. Cells 2020, 9, 831. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Chang, L.; Luo, Y.; Zhou, Y.; Zhang, J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019, 21, 101120. [Google Scholar] [CrossRef]
- Taylor, R.C. Aging and the UPR(ER). Brain Res. 2016, 1648, 588–593. [Google Scholar] [CrossRef]
- Matos, L.; Gouveia, A.M.; Almeida, H. ER stress response in human cellular models of senescence. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 924–935. [Google Scholar] [CrossRef] [Green Version]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, H.; Yan, X.; Gu, H.; Gu, Z.; Liu, F. Endoplasmic reticulum stress participates in the progress of senescence and apoptosis of osteoarthritis chondrocytes. Biochem. Biophys. Res. Commun. 2017, 491, 368–373. [Google Scholar] [CrossRef]
- Gómez-Santos, B.; Saenz de Urturi, D.; Nuñez-García, M.; Gonzalez-Romero, F.; Buque, X.; Aurrekoetxea, I.; Gutiérrez de Juan, V.; Gonzalez-Rellan, M.J.; García-Monzón, C.; González-Rodríguez, Á.; et al. Liver osteopontin is required to prevent the progression of age-related nonalcoholic fatty liver disease. Aging Cell 2020, 19, e13183. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Hua, B.; Cai, T.; Xu, L.; Li, E.; Huang, Y.; Sun, N.; Yan, Z.; Lu, C.; Qian, R. Clock mediates liver senescence by controlling ER stress. Aging 2017, 9, 2647–2665. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Kim, Y.; Lim, M.J.; Park, Y.G.; Park, S.I.; Sohn, J. The p38-activated ER stress-ATF6α axis mediates cellular senescence. FASEB J. 2019, 33, 2422–2434. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25. [Google Scholar] [CrossRef]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Sabath, N.; Levy-Adam, F.; Younis, A.; Rozales, K.; Meller, A.; Hadar, S.; Soueid-Baumgarten, S.; Shalgi, R. Cellular proteostasis decline in human senescence. Proc. Natl. Acad. Sci. USA 2020, 117, 31902–31913. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Martin, N.; Bernard, D. Cellular senescence links mitochondria-ER contacts and aging. Commun. Biol. 2021, 4, 1323. [Google Scholar] [CrossRef]
- Moltedo, O.; Remondelli, P.; Amodio, G. The mitochondria–endoplasmic reticulum contacts and their critical role in aging and age-associated diseases. Front. Cell Dev. Biol. 2019, 7, 172. [Google Scholar] [CrossRef]
- Janikiewicz, J.; Szymański, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszyński, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef]
- Ahumada-Castro, U.; Puebla-Huerta, A.; Cuevas-Espinoza, V.; Lovy, A.; Cardenas, J.C. Keeping zombies alive: The ER-mitochondria Ca2+ transfer in cellular senescence. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119099. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Vindrieux, D.; Goehrig, D.; Jaber, S.; Collin, G.; Griveau, A.; Wiel, C.; Bendridi, N.; Djebali, S.; Farfariello, V.; et al. Calcium channel ITPR2 and mitochondria–ER contacts promote cellular senescence and aging. Nat. Commun. 2021, 12, 720. [Google Scholar] [CrossRef]
- Ma, X.; Qian, H.; Chen, A.; Ni, H.M.; Ding, W.X. Perspectives on Mitochondria–ER and Mitochondria–Lipid Droplet Contact in Hepatocytes and Hepatic Lipid Metabolism. Cells 2021, 10, 2273. [Google Scholar] [CrossRef]
- Rieusset, J. Endoplasmic reticulum-mitochondria calcium signaling in hepatic metabolic diseases. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 865–876. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Friso, S.; Choi, S.W. Epigenetics in non-alcoholic fatty liver disease. Mol. Aspects Med. 2017, 54, 78–88. [Google Scholar] [CrossRef]
- Podrini, C.; Borghesan, M.; Greco, A.; Pazienza, V.; Mazzoccoli, G.; Vinciguerra, M. Redox homeostasis and epigenetics in non-alcoholic fatty liver disease (NAFLD). Curr. Pharm. Des. 2013, 19, 2737–2746. [Google Scholar] [CrossRef]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.D.; Wu, X.; Still, C.D.; Chu, X.; Petrick, A.T.; Gerhard, G.S.; Conneely, K.N.; DiStefano, J.K. Differential DNA methylation and changing cell-type proportions as fibrotic stage progresses in NAFLD. Clin. Epigenetics 2021, 13, 152. [Google Scholar] [CrossRef]
- Hotta, K.; Kitamoto, T.; Kitamoto, A.; Ogawa, Y.; Honda, Y.; Kessoku, T.; Yoneda, M.; Imajo, K.; Tomeno, W.; Saito, S.; et al. Identification of the genomic region under epigenetic regulation during non-alcoholic fatty liver disease progression. Hepatol. Res. 2018, 48, E320–E334. [Google Scholar] [CrossRef] [Green Version]
- Hotta, K.; Kitamoto, A.; Kitamoto, T.; Ogawa, Y.; Honda, Y.; Kessoku, T.; Yoneda, M.; Imajo, K.; Tomeno, W.; Saito, S.; et al. Identification of differentially methylated region (DMR) networks associated with progression of nonalcoholic fatty liver disease. Sci. Rep. 2018, 8, 13567. [Google Scholar] [CrossRef]
- Nishida, N.; Yada, N.; Hagiwara, S.; Sakurai, T.; Kitano, M.; Kudo, M. Unique features associated with hepatic oxidative DNA damage and DNA methylation in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 1646–1653. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Wang, L.; Feng, G.; Li, G.; Yu, M.; Li, Y.; Liu, C.; Yuan, X.; Zang, G.; et al. Impaired lipid metabolism by age-dependent DNA methylation alterations accelerates aging. Proc. Natl. Acad. Sci. USA 2020, 117, 4328–4336. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Z.; Jiang, Z.; Luo, P.; Liu, L.; Huang, Y.; Wang, H.; Wang, Y.; Long, L.; Tan, X.; et al. Cordycepin prevents radiation ulcer by inhibiting cell senescence via NRF2 and AMPK in rodents. Nat. Commun. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.Z.; Lin, Z.X.; Yuan, Q.J.; Li, Y.C.; Liang, J.L.; Zhan, J.Y.X.; Xie, Y.L.; Su, Z.R.; Liu, Y.H. Ameliorative effect of supercritical fluid extract of Chrysanthemum indicum Linnén against D-galactose induced brain and liver injury in senescent mice via suppression of oxidative stress, inflammation and apoptosis. J. Ethnopharmacol. 2019, 234, 44–56. [Google Scholar] [CrossRef]
- Zeng, L.; Lin, L.; Peng, Y.; Yuan, D.; Zhang, S.; Gong, Z.; Xiao, W. L-Theanine attenuates liver aging by inhibiting advanced glycation end products in D-galactose-induced rats and reversing an imbalance of oxidative stress and inflammation. Exp. Gerontol. 2020, 131, 110823. [Google Scholar] [CrossRef]
- Wu, J.L.; Wu, Q.P.; Yang, X.F.; Wei, M.K.; Zhang, J.M.; Huang, Q.; Zhou, X.Y. L-malate reverses oxidative stress and antioxidative defenses in liver and heart of aged rats. Physiol. Res. 2008, 57, 261–268. [Google Scholar] [CrossRef]
- Shin, J.H.; Jeon, H.J.; Park, J.; Chang, M.S. Epigallocatechin-3-gallate prevents oxidative stress-induced cellular senescence in human mesenchymal stem cells via Nrf2. Int. J. Mol. Med. 2016, 38, 1075–1082. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Yan, Y.; Teng, X.; Wen, X.; Li, N.; Peng, S.; Liu, W.; Donadeu, F.X.; Zhao, S.; Hua, J. Melatonin prevents senescence of canine adipose-derived mesenchymal stem cells through activating NRF2 and inhibiting ER stress. Aging 2018, 10, 2954–2972. [Google Scholar] [CrossRef]
- Gao, L.B.; Wang, Y.H.; Liu, Z.H.; Sun, Y.; Cai, P.; Jing, Q. Identification of a small molecule SR9009 that activates NRF2 to counteract cellular senescence. Aging Cell 2021, 20, e13483. [Google Scholar] [CrossRef]
- Al-ghamdi, H.A.; Al Fayez, F.F.; Bima, A.I.; Khawaji, T.M.; Elsamanoudy, A.Z. Study of Cellular Senescence and Vitamin D Deficiency in Nonalcoholic Fatty Liver Disease and The Potential Protective Effect of Vitamin D Supplementation. J. Clin. Exp. Hepatol. 2021, 11, 219–226. [Google Scholar] [CrossRef]
- Jia, R.; Yang, F.; Yan, P.; Ma, L.; Yang, L.; Li, L. Paricalcitol inhibits oxidative stress-induced cell senescence of the bile duct epithelium dependent on modulating Sirt1 pathway in cholestatic mice. Free Radic. Biol. Med. 2021, 169, 158–168. [Google Scholar] [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.Y.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ido, Y.; Duranton, A.; Lan, F.; Weikel, K.A.; Breton, L.; Ruderman, N.B. Resveratrol prevents oxidative stress-induced senescence and proliferative dysfunction by activating the AMPK-FOXO3 cascade in cultured primary human keratinocytes. PLoS ONE 2015, 10, e0115341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izdebska, M.; Piątkowska-Chmiel, I.; Korolczuk, A.; Herbet, M.; Gawrońska-Grzywacz, M.; Gieroba, R.; Sysa, M.; Czajkowska-Bania, K.; Cygal, M.; Korga, A.; et al. The beneficial effects of resveratrol on steatosis and mitochondrial oxidative stress in HepG2 cells. Can. J. Physiol. Pharmacol. 2017, 95, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Bujanda, L.; Hijona, E.; Larzabal, M.; Beraza, M.; Aldazabal, P.; García-Urkia, N.; Sarasqueta, C.; Cosme, A.; Irastorza, B.; González, A.; et al. Resveratrol inhibits nonalcoholic fatty liver disease in rats. BMC Gastroenterol. 2008, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Faghihzadeh, F.; Hekmatdoost, A.; Adibi, P. Resveratrol and liver: A systematic review. J. Res. Med. Sci. 2015, 20, 797–810. [Google Scholar] [CrossRef]
- Izzo, C.; Annunziata, M.; Melara, G.; Sciorio, R.; Dallio, M.; Masarone, M.; Federico, A.; Persico, M. The role of resveratrol in liver disease: A comprehensive review from in vitro to clinical trials. Nutrients 2021, 13, 933. [Google Scholar] [CrossRef]
- Colak, Y.; Ozturk, O.; Senates, E.; Tuncer, I.; Yorulmaz, E.; Adali, G.; Doganay, L.; Enc, F.Y. SIRT1 as a potential therapeutic target for treatment of nonalcoholic fatty liver disease. Med. Sci. Monit. 2011, 17, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Ding, R.B.; Bao, J.L.; Deng, C.X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef]
- Hua, Y.Q.; Zeng, Y.; Xu, J.; Xu, X. Le Naringenin alleviates nonalcoholic steatohepatitis in middle-aged Apoe−/−mice: Role of SIRT1. Phytomedicine 2021, 81, 153412. [Google Scholar] [CrossRef]
- Xu, X.; Zhu, X.P.; Bai, J.Y.; Xia, P.; Li, Y.; Lu, Y.; Li, X.Y.; Gao, X. Berberine alleviates nonalcoholic fatty liver induced by a high-fat diet in mice by activating SIRT3. FASEB J. 2019, 33, 7289–7300. [Google Scholar] [CrossRef]
- de Gregorio, E.; Colell, A.; Morales, A.; Marí, M. Relevance of SIRT1-NF-κB axis as therapeutic target to ameliorate inflammation in liver disease. Int. J. Mol. Sci. 2020, 21, 3858. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Jiang, J.; Zhang, G.; Bu, Y.; Zhang, G.; Zhao, X. Resveratrol and caloric restriction prevent hepatic steatosis by regulating SIRT1-autophagy pathway and alleviating endoplasmic reticulum stress in high-fat diet-fed rats. PLoS ONE 2017, 12, e0183541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.X.; Gao, J.G.; Wan, X.Y.; Chen, Y.; Xu, C.F.; Feng, Z.M.; Zeng, H.; Lin, Y.M.; Ma, H.; Xu, P.; et al. Allyl isothiocyanate ameliorates lipid accumulation and inflammation in nonalcoholic fatty liver disease via the Sirt1/AMPK and NF-κB signaling pathways. World J. Gastroenterol. 2019, 25, 5120–5133. [Google Scholar] [CrossRef]
- Rafiei, H.; Omidian, K.; Bandy, B. Dietary Polyphenols Protect Against Oleic Acid-Induced Steatosis in an in Vitro Model of NAFLD by Modulating Lipid Metabolism and Improving Mitochondrial Function. Nutrients 2019, 11, 541. [Google Scholar] [CrossRef] [Green Version]
- Stacchiotti, A.; Grossi, I.; García-Gómez, R.; Patel, G.A.; Salvi, A.; Lavazza, A.; De Petro, G.; Monsalve, M.; Rezzani, R. Melatonin Effects on Non-Alcoholic Fatty Liver Disease Are Related to MicroRNA-34a-5p/Sirt1 Axis and Autophagy. Cells 2019, 8, 1053. [Google Scholar] [CrossRef] [Green Version]
- Maharajan, N.; Ganesan, C.D.; Moon, C.; Jang, C.H.; Oh, W.K.; Cho, G.W. Licochalcone d ameliorates oxidative stress-induced senescence via ampk activation. Int. J. Mol. Sci. 2021, 22, 7324. [Google Scholar] [CrossRef]
- Foretz, M.; Even, P.C.; Viollet, B. AMPK activation reduces hepatic lipid content by increasing fat oxidation in vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- de la Rosa, L.C.; Vrenken, T.E.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Metformin protects primary rat hepatocytes against oxidative stress-induced apoptosis. Pharmacol. Res. Perspect. 2015, 3, e00125. [Google Scholar] [CrossRef]
- Geng, Y.; Hernández Villanueva, A.; Oun, A.; Buist-Homan, M.; Blokzijl, H.; Faber, K.N.; Dolga, A.; Moshage, H. Protective effect of metformin against palmitate-induced hepatic cell death. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165621. [Google Scholar] [CrossRef]
- Mora, F.A.A.; Musheshe, N.; Arroyave Ospina, J.C.; Geng, Y.; Soto, J.M.; Rodrigo, J.A.; Alieva, T.; Buist-Homan, M.; Lezoualc’h, F.; Cheng, X.; et al. Metformin protects against diclofenac-induced toxicity in primary rat hepatocytes by preserving mitochondrial integrity via a pathway involving EPAC. Biomed. Pharmacother. 2021, 143, 112072. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Le Pelletier, L.; Mantecon, M.; Gorwood, J.; Auclair, M.; Foresti, R.; Motterlini, R.; Laforge, M.; Atlan, M.; Fève, B.; Capeau, J.; et al. Metformin alleviates stress-induced cellular senescence of aging human adipose stromal cells and the ensuing adipocyte dysfunction. Elife 2021, 10, e62635. [Google Scholar] [CrossRef]
- Kakazu, E.; Mauer, A.S.; Yin, M.; Malhi, H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1 α -dependent manner. J. Lipid Res. 2016, 57, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Ekhlasi, G.; Shidfar, F.; Agah, S.; Merat, S.; Hosseini, A.F. Effects of Pomegranate and Orange Juice on Antioxidant Status in Non-Alcoholic Fatty Liver Disease Patients: A Randomized Clinical Trial. Int. J. Vitam. Nutr. Res. 2015, 85, 292–298. [Google Scholar] [CrossRef]
- Magosso, E.; Ansari, M.A.; Gopalan, Y.; Shuaib, I.L.; Wong, J.W.; Khan, N.A.K.; Abu Bakar, M.R.; Ng, B.H.; Yuen, K.H. Tocotrienols for normalisation of hepatic echogenic response in nonalcoholic fatty liver: A randomised placebo-controlled clinical trial. Nutr. J. 2013, 12, 166. [Google Scholar] [CrossRef] [Green Version]
- Pervez, M.A.; Khan, D.A.; Ijaz, A.; Khan, S. Effects of delta-tocotrienol supplementation on liver enzymes, inflammation, oxidative stress and hepatic steatosis in patients with nonalcoholic fatty liver disease. Turkish J. Gastroenterol. 2018, 29, 170–176. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, C.P.M.; Cotrim, H.P.; Stefano, J.T.; Siqueira, A.C.G.; Salgado, A.L.A.; Parise, E.R. N-acetylcysteine and/or ursodeoxycholic acid associated with metformin in non-alcoholic steatohepatitis: An open-label multicenter randomized controlled trial. Arq. Gastroenterol. 2019, 56, 184–190. [Google Scholar] [CrossRef]
- Sharifi, N.; Amani, R.; Hajiani, E.; Cheraghian, B. Does vitamin D improve liver enzymes, oxidative stress, and inflammatory biomarkers in adults with non-alcoholic fatty liver disease? A randomized clinical trial. Endocrine 2014, 47, 70–80. [Google Scholar] [CrossRef]
- Loffredo, L.; Baratta, F.; Ludovica, P.; Battaglia, S.; Carnevale, R.; Nocella, C.; Novo, M.; Pannitteri, G.; Ceci, F.; Angelico, F.; et al. Effects of dark chocolate on endothelial function in patients with non-alcoholic steatohepatitis. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 143–149. [Google Scholar] [CrossRef]
- Guo, H.; Zhong, R.; Liu, Y.; Jiang, X.; Tang, X.; Li, Z.; Xia, M.; Ling, W. Effects of bayberry juice on inflammatory and apoptotic markers in young adults with features of non-alcoholic fatty liver disease. Nutrition 2014, 30, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Asghari, S.; Rafraf, M.; Farzin, L.; Asghari-Jafarabadi, M.; Ghavami, S.-M.; Somi, M.-H. Effects of Pharmacologic Dose of Resveratrol Supplementation on Oxidative/Antioxidative Status Biomarkers in Nonalcoholic Fatty Liver Disease Patients: A Randomized, Double-Blind, Placebo-Controlled Trial. Adv. Pharm. Bull. 2018, 8, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Loffredo, L.; Del Ben, M.; Perri, L.; Carnevale, R.; Nocella, C.; Catasca, E.; Baratta, F.; Ceci, F.; Polimeni, L.; Gozzo, P.; et al. Effects of dark chocolate on NOX-2-generated oxidative stress in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2016, 44, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Dallio, M.; Masarone, M.; Gravina, A.G.; Di Sarno, R.; Tuccillo, C.; Cossiga, V.; Lama, S.; Stiuso, P.; Morisco, F.; et al. Evaluation of the Effect Derived from Silybin with Vitamin D and Vitamin E Administration on Clinical, Metabolic, Endothelial Dysfunction, Oxidative Stress Parameters, and Serological Worsening Markers in Nonalcoholic Fatty Liver Disease Patients. Oxid. Med. Cell. Longev. 2019, 2019, 8742075. [Google Scholar] [CrossRef]
- Fok, W.C.; Chen, Y.; Bokov, A.; Zhang, Y.; Salmon, A.B.; Diaz, V.; Javors, M.; Wood, W.H., 3rd; Zhang, Y.; Becker, K.G.; et al. Mice Fed Rapamycin Have an Increase in Lifespan Associated with Major Changes in the Liver Transcriptome. PLoS ONE 2014, 9, e83988. [Google Scholar] [CrossRef]
- Martínez-Cisuelo, V.; Gómez, J.; García-Junceda, I.; Naudí, A.; Cabré, R.; Mota-Martorell, N.; López-Torres, M.; González-Sánchez, M.; Pamplona, R.; Barja, G. Rapamycin reverses age-related increases in mitochondrial ROS production at complex I, oxidative stress, accumulation of mtDNA fragments inside nuclear DNA, and lipofuscin level, and increases autophagy, in the liver of middle-aged mice. Exp. Gerontol. 2016, 83, 130–138. [Google Scholar] [CrossRef]
- Cayo, A.; Segovia, R.; Venturini, W.; Moore-Carrasco, R.; Valenzuela, C.; Brown, N. Mtor activity and autophagy in senescent cells, a complex partnership. Int. J. Mol. Sci. 2021, 22, 8149. [Google Scholar] [CrossRef]
- Kang, C. Senolytics and senostatics: A two-pronged approach to target cellular senescence for delaying aging and age-related diseases. Mol. Cells 2019, 42, 821–827. [Google Scholar] [CrossRef]
- Mazzucco, A.E.; Smogorzewska, A.; Kang, C.; Luo, J.; Schlabach, M.R.; Xu, Q.; Patel, R.; Elledge, S.J. Genetic interrogation of replicative senescence uncovers a dual role for USP28 in coordinating the p53 and GATA4 branches of the senescence program. Genes Dev. 2017, 31, 1933–1938. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. LINE-1 derepression in senescent cells triggers interferon and inflammaging. Nature 2019, 566, 73. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Wang, S. The Role of Rapamycin in Healthspan Extension via the Delay of Organ Aging. Ageing Res. Rev. 2021, 70, 101376. [Google Scholar] [CrossRef] [PubMed]
- Neef, M.; Ledermann, M.; Saegesser, H.; Schneider, V.; Reichen, J. Low-dose oral rapamycin treatment reduces fibrogenesis, improves liver function, and prolongs survival in rats with established liver cirrhosis. J. Hepatol. 2006, 45, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Stavri, S.; Trusca, V.G.; Simionescu, M.; Gafencu, A.V. Metformin reduces the endotoxin-induced down-regulation of apolipoprotein e gene expression in macrophages. Biochem. Biophys. Res. Commun. 2015, 461, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Brandt, A.; Hernández-Arriaga, A.; Kehm, R.; Sánchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kita, Y.; Takamura, T.; Misu, H.; Ota, T.; Kurita, S.; Takeshita, Y.; Uno, M.; Matsuzawa-Nagata, N.; Kato, K.I.; Ando, H.; et al. Metformin Prevents and Reverses Inflammation in aNon-Diabetic Mouse Model of NonalcoholicSteatohepatitis.pdf. PLoS ONE 2012, 7, e43056. [Google Scholar] [CrossRef] [PubMed]
- Garinis, G.A.; Fruci, B.; Mazza, A.; De Siena, M.; Abenavoli, S.; Gulletta, E.; Ventura, V.; Greco, M.; Abenavoli, L.; Belfiore, A. Metformin versus dietary treatment in nonalcoholic hepatic steatosis: A randomized study. Int. J. Obes. 2010, 34, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Laberge, R.; Zhou, L.; Sarantos, M.R.; Rodier, F.; De Keizer, P.L.J.; Liu, S.; Demaria, M.; Cong, Y.; Kapahi, P.; Desprez, P.; et al. Glucocorticoids Suppress Selected Components of the Senescence-Associated Secretory Phenotype. Aging Cell 2013, 11, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.N.; Dasgupta, N.; Liu, T.; Adams, P.D.; Vizioli, M.G. Cytoplasmic chromatin fragments—from mechanisms to therapeutic potential. Elife 2021, 10, e63728. [Google Scholar] [CrossRef]
- Wiley, C.D.; Schaum, N.; Alimirah, F.; Lopez-Dominguez, J.A.; Orjalo, A.V.; Scott, G.; Desprez, P.Y.; Benz, C.; Davalos, A.R.; Campisi, J. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci. Rep. 2018, 8, 2–10. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA. 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Liu, X.; Wang, Y.; Chang, J.; Zhang, X.; Mackintosh, S.G.; Tackett, A.J.; He, Y.; Lv, D.; et al. Oxidation resistance 1 is a novel senolytic target. Aging Cell 2018, 17, e12780. [Google Scholar] [CrossRef]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.; Valanejad, L.; Cast, A.; Wright, M.; Garcia, J.M.; El-Serag, H.B.; Karns, R.; Timchenko, N.A. Elimination of Age-Associated Hepatic Steatosis and Correction of Aging Phenotype by Inhibition of cdk4-C/EBPα-p300 Axis. Cell Rep. 2018, 24, 1597–1609. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Valanejad, L.; Nguyen, T.P.; Lewis, K.; Wright, M.; Cast, A.; Stock, L.; Timchenko, L.; Timchenko, N.A. Activation of CDK4 Triggers Development of Non-alcoholic Fatty Liver Disease. Cell Rep. 2016, 16, 744–756. [Google Scholar] [CrossRef] [Green Version]
- Raffaele, M.; Kovacovicova, K.; Frohlich, J.; Lo Re, O.; Giallongo, S.; Oben, J.A.; Faldyna, M.; Leva, L.; Giannone, A.G.; Cabibi, D.; et al. Mild exacerbation of obesity- and age-dependent liver disease progression by senolytic cocktail dasatinib + quercetin. Cell Commun. Signal. 2021, 19, 44. [Google Scholar] [CrossRef]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic cocktail dasatinib+quercetin (D+Q) does not enhance the efficacy of senescence-inducing chemotherapy in liver cancer. Front. Oncol. 2018, 8, 459. [Google Scholar] [CrossRef] [PubMed]
| Senescence Markers | Patients and Samples | Findings | References |
|---|---|---|---|
| Expression of p53, Bax and Bcl-2. p53-binding protein 1 (53BP1)—positive foci formation | Hepatocytes with and without steatosis from patients at various stages of NAFLD. | Positive correlation between liver steatosis and p53 expression. Decreased level of anti-apoptotic protein Bcl-2 correlated with advancement of liver steatosis. | [57] |
| Liver tissue of NAFLD patients. | Number of abnormal 53BP1-positive foci in hepatocytes were significantly increased in NAFLD patients compared to controls, both in non-alcoholic fatty liver and non-alcoholic steatohepatitis. | [58] | |
| Telomere length/dysfunction, nuclear area, DNA damage and cell cycle phase markers. | Liver sections from patients with NAFLD and controls. | Hepatocyte telomeres were shorter in NAFLD patients than in controls. Hepatocytes in NAFLD patients demonstrated lack of cell cycle progression beyond G1/S phase and high-level expression of p21 and shortened telomere length. | [59] |
| Peripheral lymphocytes from patients with NAFLD, with cryptogenic cirrhosis (CC) and healthy, age-matched controls. | Shorter telomere length and increased cellular senescence were demonstrated in patients with NAFLD compared to the CC patients and healthy controls. | [60] | |
| Liver tissue from type 2 diabetes mellitus patients with NAFLD followed up for 6 years. | Type 2 diabetes mellitus patients who developed NAFLD showed shorter telomere length compared to T2DM patients who did not develop NAFLD. | [61] | |
| Variants of CDKN1A (p21) | Liver tissue from two cohorts of biopsy-proven NAFLD patients. | rs762623 SNP on CDKN1A was significantly associated with disease progression in NAFLD. CDKN1A variant rs762623 is associated with the development but not the progression of liver disease in NAFLD. | [62] |
| NAFLD Model | Experimental Conditions | Senescence Findings | References |
|---|---|---|---|
| Steatosis | Diabetic type 2-obese mice Leprdb/db, SMP30 knockout mice. | SMP30 knockout mice showed fatty liver accompanied by increased inflammation, oxidative stress and ER stress compared to controls mice. SPM30 loss also correlates with decreased expression of genes involved in fatty acid oxidation. | [64] |
| Steatosis | SMP30/SOD1 double knockout (SMP30/SOD1-DKO) mice: Superoxide dismutase 1 (SOD1) and SMP30. | High levels of oxidative stress due to concomitant deficiency of SMP30 and/or ascorbic acid and SOD1 cause abnormal lipid metabolism, hepatic lipid accumulation and premature death resulting from impaired VLDL secretion. | [65] |
| Steatosis | HFD 1 C57Bl/6 mice. | Liver fat accumulation and increased hepatic mRNA expression of steatosis-related genes is accompanied by hepatic senescence. | [66] |
| Steatohepatitis | Mice fed a MCD 2 diet. | MCD feeding enhanced hepatic p53 expression, corresponding to ~50% decrease in serum IGF-1, decreased Bcl-XL, enhanced cleavage of Bid into tBid and upregulation of p21. | [67] |
| Steatohepatitis | Male wild type and p53-deficient mice fed a MCD 2. | Hepatic p53 and p66Shc signaling was enhanced in a mouse NASH model. p53 deficiency suppressed the enhanced p66Shc signaling, decreased hepatic lipid peroxidation and the number of apoptotic hepatocytes and ameliorated progression of nutritional steatohepatitis. | [68] |
| Steatohepatitis | Obese mice (db/db) Ink-ATTAC mice on HFD 1. | Strong association between hepatic senescence and fat accumulation. Treatment with a senolytic significantly reduced liver fat accumulation in aged wild type mice and in obese mice (db/db). | [69] |
| Steatohepatitis | C57BL/6J mice fed a HFD 1. | Fat accumulation was negatively correlated with an age-related reduction in mitochondrial mass and aggravated by a reduced capacity of fatty acid oxidation in high fat-fed mice. | [70] |
| Steatohepatitis | ClpP knockout (ClpP−/−) mice fed ad libitum. | Caseinolytic peptidase P (ClpP) (protein initiation UPRmt). ClpP regulated mitochondrial function and its deficiency protects against hepatic steatosis. | [71] |
| Steatohepatitis | C57BL/6 mice with hepatocyte specific p53−/− fed a HFCH diet (high-fat/cholesterol/fructose). | Hepatocyte HNF4α protects against diet-induced development and progression of NAFLD, prevents hepatic triglyceride accumulation and promotes fatty acid oxidation but not in hepatocyte-specific p53−/− mice. | [72] |
| Steatohepatitis | Aged C57BL/6 mice fed with HFD or standard diet. | Upregulation of receptor for advanced glycation end products (RAGE) correlated with decreased PPARα levels and may play a critical role in aging-associated liver steatosis. | [73] |
| Steatohepatitis | HFD 1 rat model, HepG2 cell line, L02 cell line, NAFLD patients. | Steatosis and fat accumulation correlate with the induction of hepatic senescence and p66shc deficiency inhibits H2O2-induced senescence and lipid accumulation. p66shc and p21 expression correlate with the severity of NAFLD. | [74] |
| Steatohepatitis | Mice (C57BL/6) fed HFD 1 and BNL CL.2 cells with palmitate acid (PA). | Lipotoxicity-induced hepatocyte senescence is major risk factor for NAFLD. SA-β-gal positive staining was higher in hepatic tissues of HFD mice and in hepatocytes treated with PA. The expression level of senescence-associated genes, such as p21 and CDK6, were increased in fatty liver cells. These results revealed that fatty liver cells acquire a senescence phenotype. | [75] |
| Steatohepatitis | Mice fed high fat 1 or standard diets. | High intake of dietary fat induced ROS production and DNA damage in liver. Oxidative stress leads to fibrosis via activation of the ATM pathway. | [76] |
| Steatohepatitis | Diet-induced obese rat model: obesity prone (OP) and obesity-resistant (OR). | Hepatic cellular senescence pathway genes were induced via histone modifications in OP rats. Significant increase of expression of p16INK4a and p21 in OP rats. Increase of p16INK4a was associated with higher acetylation levels of histone H4 and lower methylation level of histone H3. | [77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedroza-Diaz, J.; Arroyave-Ospina, J.C.; Serna Salas, S.; Moshage, H. Modulation of Oxidative Stress-Induced Senescence during Non-Alcoholic Fatty Liver Disease. Antioxidants 2022, 11, 975. https://doi.org/10.3390/antiox11050975
Pedroza-Diaz J, Arroyave-Ospina JC, Serna Salas S, Moshage H. Modulation of Oxidative Stress-Induced Senescence during Non-Alcoholic Fatty Liver Disease. Antioxidants. 2022; 11(5):975. https://doi.org/10.3390/antiox11050975
Chicago/Turabian StylePedroza-Diaz, Johanna, Johanna C. Arroyave-Ospina, Sandra Serna Salas, and Han Moshage. 2022. "Modulation of Oxidative Stress-Induced Senescence during Non-Alcoholic Fatty Liver Disease" Antioxidants 11, no. 5: 975. https://doi.org/10.3390/antiox11050975