
CDDO-Me Attenuates CA1 Neuronal Death by Facilitating RalBP1-Mediated Mitochondrial Fission and 4-HNE Efflux in the Rat Hippocampus Following Status Epilepticus

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Chemicals
2.2. Surgery, CDDO-Me Infusion, and RalBP1 Knockdown
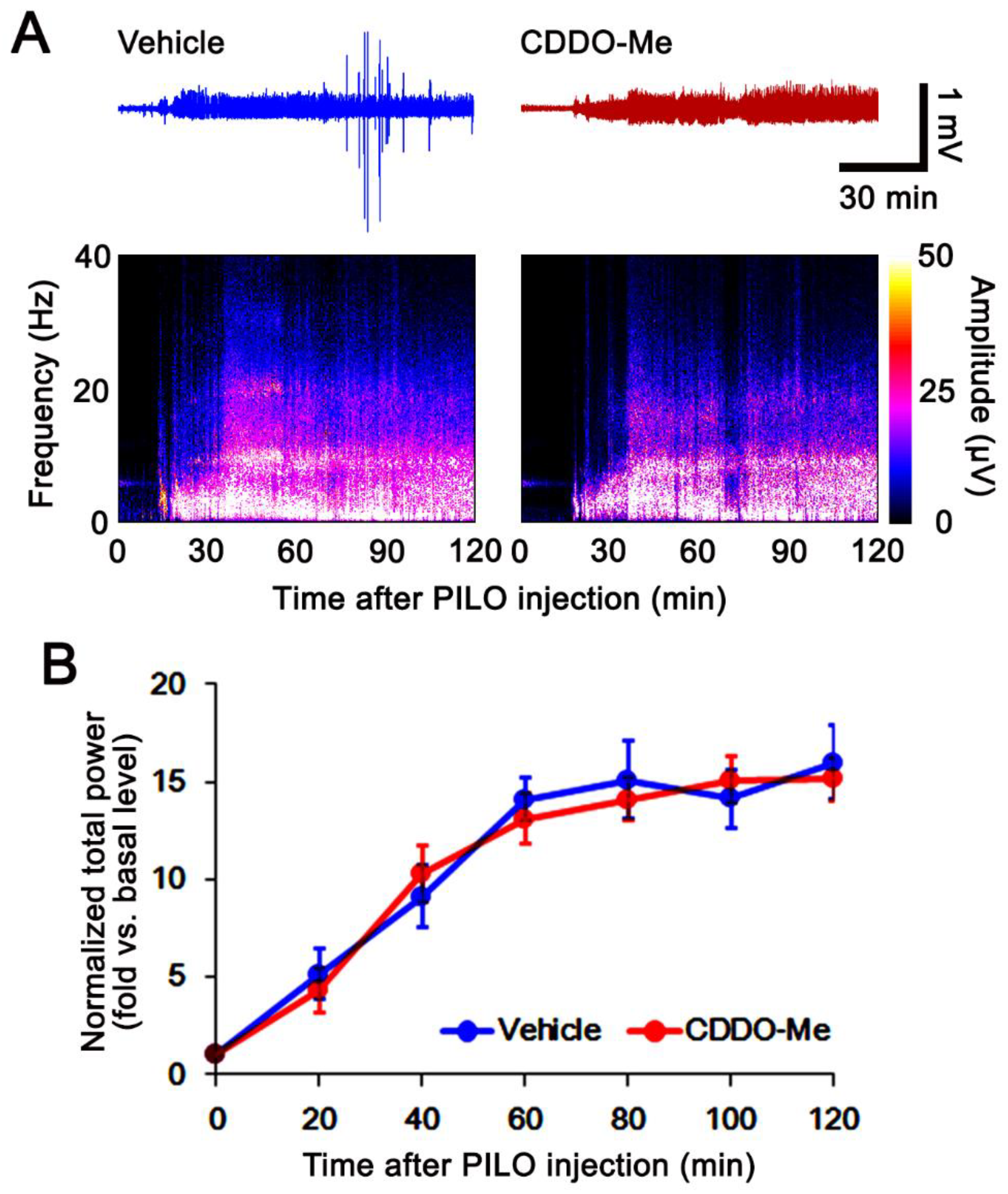
2.3. SE Induction and Electroencephalogram (EEG) Analysis
2.4. Tissue Processing
2.5. Immunohistochemistry, Measurements of Mitochondrial Length, and Neuronal Damage
2.6. Western Blot
2.7. Quantification of Data and Statistical Analysis
3. Results
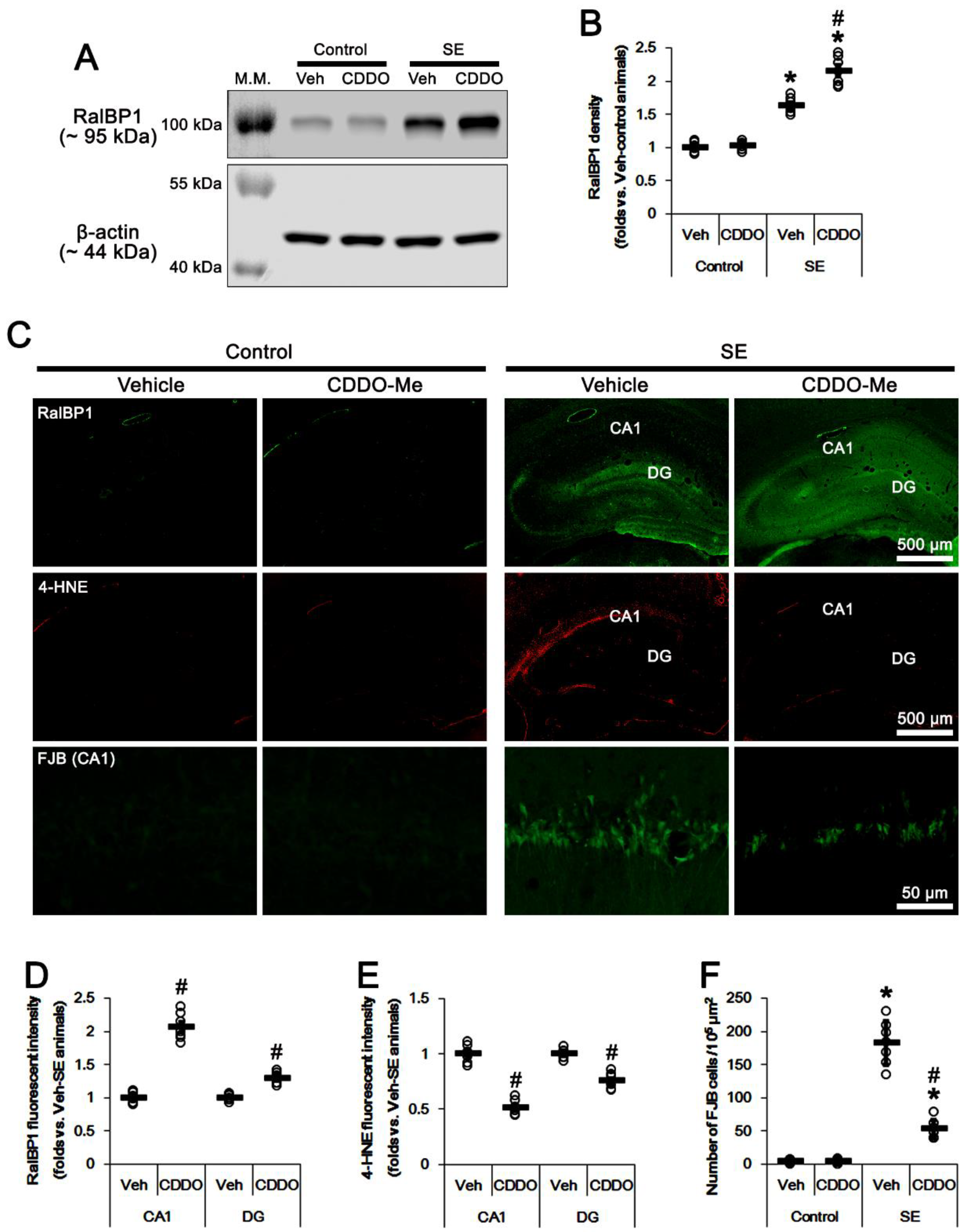
3.1. CDDO-Me Upregulates RalBP1 Protein Levels but Reduces 4-HNE Signals in the CA1 Region Following SE
3.2. CDDO-Me Facilitates Mitochondrial Fission in CA1 Neurons under Physiological and Post-SE Conditions
3.3. RalBP1 siRNA Does Not Affect Mitochondrial Dynamics and CA1 Neuronal Death Induced by SE
3.4. RalBP1 Knockdown Diminishes the Effects of CDDO-Me on Aberrant Mitochondrial Dynamics and CA1 Neuronal Death Following SE
4. Discussion
5. Limitation of the Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell. Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Kang, T.C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef]
- Kashatus, D.F.; Lim, K.H.; Brady, D.C.; Pershing, N.L.; Cox, A.D.; Counter, C.M. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 2011, 13, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Shorvon, S.; Sen, A. What is status epilepticus and what do we know about its epidemiology? Seizure 2020, 75, 131–136. [Google Scholar] [CrossRef]
- Bauman, K.; Devinsky, O. Seizure Clusters: Morbidity and Mortality. Front. Neurol. 2021, 12, 636045. [Google Scholar] [CrossRef]
- Sloviter, R.S.; Zappone, C.A.; Harvey, B.D.; Bumanglag, A.V.; Bender, R.A.; Frotscher, M. “Dormant basket cell” hypothesis revisited: Relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J. Comp. Neurol. 2003, 459, 44–76. [Google Scholar] [CrossRef]
- Walker, M.C. Pathophysiology of status epilepticus. Neurosci. Lett. 2018, 667, 84–91. [Google Scholar] [CrossRef]
- Kim, J.E.; Ryu, H.J.; Kim, M.J.; Kang, T.C. LIM kinase-2 induces programmed necrotic neuronal death via dysfunction of DRP1-mediated mitochondrial fission. Cell Death Differ. 2014, 21, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Park, H.; Kim, T.H.; Kang, T.C. LONP1 Regulates Mitochondrial Accumulations of HMGB1 and Caspase-3 in CA1 and PV Neurons following Status Epilepticus. Int. J. Mol. Sci. 2021, 22, 2275. [Google Scholar] [CrossRef]
- Singhal, S.S.; Yadav, S.; Roth, C.; Singhal, J. RLIP76: A novel glutathione-conjugate and multi-drug transporter. Biochem. Pharmacol. 2009, 77, 761–769. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, Y.C.; Sharma, R.; Cheng, J.Z.; Yang, Y.; Sharma, A.; Singhal, S.S.; Awasthi, S. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Mol. Asp. Med. 2003, 24, 219–230. [Google Scholar] [CrossRef]
- Feig, L.A. Ral-GTPases: Approaching their 15 minutes of fame. Trends Cell Biol. 2003, 13, 419–425. [Google Scholar] [CrossRef]
- Camonis, J.H.; White, M.A. Ral-GTPases: Corrupting the exocyst in cancer cells. Trends Cell Biol. 2005, 15, 327–332. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell. Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.C. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef] [Green Version]
- Takagi, T.; Kitashoji, A.; Iwawaki, T.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Temporal activation of Nrf2 in the penumbra and Nrf2 activator-mediated neuroprotection in ischemia-reperfusion injury. Free Radic. Biol. Med. 2014, 72, 124–133. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, H.; Kang, T.C. CDDO-Me Distinctly Regulates Regional Specific Astroglial Responses to Status Epilepticus via ERK1/2-Nrf2, PTEN-PI3K-AKT and NFκB Signaling Pathways. Antioxidants 2020, 9, 1026. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. CDDO-Me Selectively Attenuates CA1 Neuronal Death Induced by Status Epilepticus via Facilitating Mitochondrial Fission Independent of LONP. Cells 2019, 8, 833. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, J.T.; Strack, S. Functional characterization of phosphorylation sites in dynamin-related protein. Methods Enzymol. 2009, 457, 231–253. [Google Scholar]
- Merrill, R.A.; Dagda, R.K.; Dickey, A.S.; Cribbs, J.T.; Green, S.H.; Usachev, Y.M.; Strack, S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Salgia, R.; Singhal, S.; Horne, D.; Awasthi, S. RLIP: An existential requirement for breast carcinogenesis. Biochim. Biophys. Acta Rev. Cancer. 2019, 1871, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.R.; Kang, T.C. TRPC6-mediated ERK1/2 phosphorylation prevents dentate granule cell degeneration via inhibiting mitochondrial elongation. Neuropharmacology 2017, 121, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. TRPC6-Mediated ERK1/2 Activation Increases Dentate Granule Cell Resistance to Status Epilepticus Via Regulating Lon Protease-1 Expression and Mitochondrial Dynamics. Cells 2019, 8, 1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, J.; Nagaprashantha, L.; Vatsyayan, R.; Awasthi, S.; Singhal, S.S. RLIP76, a glutathione-conjugate transporter, plays a major role in the pathogenesis of metabolic syndrome. PLoS ONE 2011, 6, e24688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Huang, L.; Zheng, W.; An, J.; Zhan, Z.; Wang, L.; Chen, Z.; Liu, L. Recurrent nonsevere hypoglycemia exacerbates imbalance of mitochondrial homeostasis leading to synapse injury and cognitive deficit in diabetes. Am. J. Physiol. Endocrinol. Metab. 2018, 315, 973–986. [Google Scholar] [CrossRef]
- Sehrawat, A.; Yadav, S.; Awasthi, Y.C.; Basu, A.; Warden, C.; Awasthi, S. P300 regulates the human RLIP76 promoter activity and gene expression. Biochem. Pharmacol. 2013, 85, 1203–1211. [Google Scholar] [CrossRef] [Green Version]
- Bennani-Baiti, B.; Toegel, S.; Viernstein, H.; Urban, E.; Noe, C.R.; Bennani-Baiti, I.M. Inflammation Modulates RLIP76/RALBP1 Electrophile-Glutathione Conjugate Transporter and Housekeeping Genes in Human Blood-Brain Barrier Endothelial Cells. PLoS ONE 2015, 10, e0139101. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Raina, D.; Meyer, C.; Kharbanda, S.; Kufe, D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J. Biol. Chem. 2006, 281, 35764–35769. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Park, H.; Choi, S.H.; Kong, M.J.; Kim, J.E.; Kang, T.C. CDDO-Me Attenuates Vasogenic Edema and Astroglial Death by Regulating NF-κB p65 Phosphorylations and Nrf2 Expression following Status Epilepticus. Int. J. Mol. Sci. 2019, 20, 4862. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Park, H.; Lee, J.E.; Kang, T.C. CDDO-Me Inhibits Microglial Activation and Monocyte Infiltration by Abrogating NFκB- and p38 MAPK-Mediated Signaling Pathways following Status Epilepticus. Cells 2020, 9, 1123. [Google Scholar] [CrossRef]
- Tian, C.; Gao, L.; Zhang, A.; Hackfort, B.T.; Zucker, I.H. Therapeutic Effects of Nrf2 Activation by Bardoxolone Methyl in Chronic Heart Failure. J. Pharmacol. Exp. Ther. 2019, 371, 642–651. [Google Scholar] [CrossRef]
- Ganner, A.; Pfeiffer, Z.C.; Wingendorf, L.; Kreis, S.; Klein, M.; Walz, G.; Neumann-Haefelin, E. The acetyltransferase p300 regulates NRF2 stability and localization. Biochem. Biophys. Res. Commun. 2020, 524, 895–902. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Dev. Ther. 2014, 8, 2075–2088. [Google Scholar]
- Kovac, S.; Dinkova Kostova, A.T.; Herrmann, A.M.; Melzer, N.; Meuth, S.G.; Gorji, A. Metabolic and homeostatic changes in seizures and acquired epilepsy-mitochondria, calcium dynamics and reactive oxygen species. Int. J. Mol. Sci. 2017, 18, 1935. [Google Scholar] [CrossRef] [Green Version]
- Barel, O.; Christine, V.; Malicdan, M.; Ben-Zeev, B.; Kandel, J.; Pri-Chen, H.; Stephen, J.; Castro, I.G.; Metz, J.; Atawa, O.; et al. Deleterious variants in TRAK1 disrupt mitochondrial movement and cause fatal encephalopathy. Brain 2017, 140, 568–581. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Vanstone, J.R.; Smith, A.M.; McBride, S.; Naas, T.; Holcik, M.; Antoun, G.; Harper, M.E.; Michaud, J.; Sell, E.; Chakraborty, P.; et al. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur. J. Hum. Genet. 2016, 24, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Kang, T.C. Differential Roles of Mitochondrial Translocation of Active Caspase-3 and HMGB1 in Neuronal Death Induced by Status Epilepticus. Front. Cell. Neurosci. 2018, 12, 301. [Google Scholar] [CrossRef] [Green Version]
- Lucchi, C.; Curia, G.; Vinet, J.; Gualtieri, F.; Bresciani, E.; Locatelli, V.; Torsello, A.; Biagini, G. Protective but not anticonvulsant effects of ghrelin and JMV-1843 in the pilocarpine model of Status epilepticus. PLoS ONE 2013, 8, e72716. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, J.E.; Kwak, S.E.; Choi, K.C.; Kim, D.W.; Kwon, O.S.; Choi, S.Y.; Kang, T.C. Spatiotemporal characteristics of astroglial death in the rat hippocampo-entorhinal complex following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2008, 511, 581–598. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, Y.J.; Kim, J.Y.; Kang, T.C. PARP1 activation/expression modulates regional-specific neuronal and glial responses to seizure in a hemodynamic-independent manner. Cell Death Dis. 2014, 5, e1362. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Yeo, S.I.; Ryu, H.J.; Kim, M.J.; Kim, D.S.; Jo, S.M.; Kang, T.C. Astroglial loss and edema formation in the rat piriform cortex and hippocampus following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2010, 518, 4612–4628. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kang, T.C. p47Phox/CDK5/DRP1-Mediated Mitochondrial Fission Evokes PV Cell Degeneration in the Rat Dentate Gyrus following Status Epilepticus. Front. Cell. Neurosci. 2017, 11, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.M.; Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kim, M.J.; Choi, H.C.; Song, H.K.; Kang, T.C. Up-regulation of endothelial endothelin-1 expression prior to vasogenic edema formation in the rat piriform cortex following status epilepticus. Neurosci. Lett. 2011, 501, 25–30. [Google Scholar] [CrossRef]
- Ryu, H.J.; Kim, J.E.; Kim, Y.J.; Kim, J.Y.; Kim, W.I.; Choi, S.Y.; Kim, M.J.; Kang, T.C. Endothelial transient receptor potential conical channel (TRPC)-3 activation induces vasogenic edema formation in the rat piriform cortex following status epilepticus. Cell. Mol. Neurobiol. 2013, 33, 575–585. [Google Scholar] [CrossRef]
- Kim, J.E.; Ryu, H.J.; Kang, T.C. Status epilepticus induces vasogenic edema via tumor necrosis factor-α/ endothelin-1-mediated two different pathways. PLoS ONE 2013, 8, e74458. [Google Scholar] [CrossRef] [Green Version]
- Gualtieri, F.; Curia, G.; Marinelli, C.; Biagini, G. Increased perivascular laminin predicts damage to astrocytes in CA3 and piriform cortex following chemoconvulsive treatments. Neuroscience 2012, 218, 278–294. [Google Scholar] [CrossRef]
- Jeon, A.R.; Kim, J.E. PDI Knockdown Inhibits Seizure Activity in Acute Seizure and Chronic Epilepsy Rat Models via S-Nitrosylation-Independent Thiolation on NMDA Receptor. Front. Cell. Neurosci. 2018, 12, 438. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Kim, J.E. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. 2018, 9, 869. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| 4-HNE | Rabbit | Alpha Diagnostic (#HNE11-S) | 1:500 (IH) |
| Mitochondrial marker (Mitochondrial complex IV subunit 1, MTCO1) | Mouse | Abcam (#ab14705) | 1:500 (IH) |
| RalBP1 | Rabbit | Abcam (#ab133549) | 1:500 (IH) 1:10,000 (WB) |
| β-actin | Mouse | Sigma (#A5316) | 1:6000 (WB) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-E.; Lee, D.-S.; Kim, T.-H.; Kang, T.-C. CDDO-Me Attenuates CA1 Neuronal Death by Facilitating RalBP1-Mediated Mitochondrial Fission and 4-HNE Efflux in the Rat Hippocampus Following Status Epilepticus. Antioxidants 2022, 11, 985. https://doi.org/10.3390/antiox11050985
Kim J-E, Lee D-S, Kim T-H, Kang T-C. CDDO-Me Attenuates CA1 Neuronal Death by Facilitating RalBP1-Mediated Mitochondrial Fission and 4-HNE Efflux in the Rat Hippocampus Following Status Epilepticus. Antioxidants. 2022; 11(5):985. https://doi.org/10.3390/antiox11050985
Chicago/Turabian StyleKim, Ji-Eun, Duk-Shin Lee, Tae-Hyun Kim, and Tae-Cheon Kang. 2022. "CDDO-Me Attenuates CA1 Neuronal Death by Facilitating RalBP1-Mediated Mitochondrial Fission and 4-HNE Efflux in the Rat Hippocampus Following Status Epilepticus" Antioxidants 11, no. 5: 985. https://doi.org/10.3390/antiox11050985