
Extracellular Signal-Regulated Kinase 1 Alone Is Dispensable for Hyperoxia-Mediated Alveolar and Pulmonary Vascular Simplification in Neonatal Mice


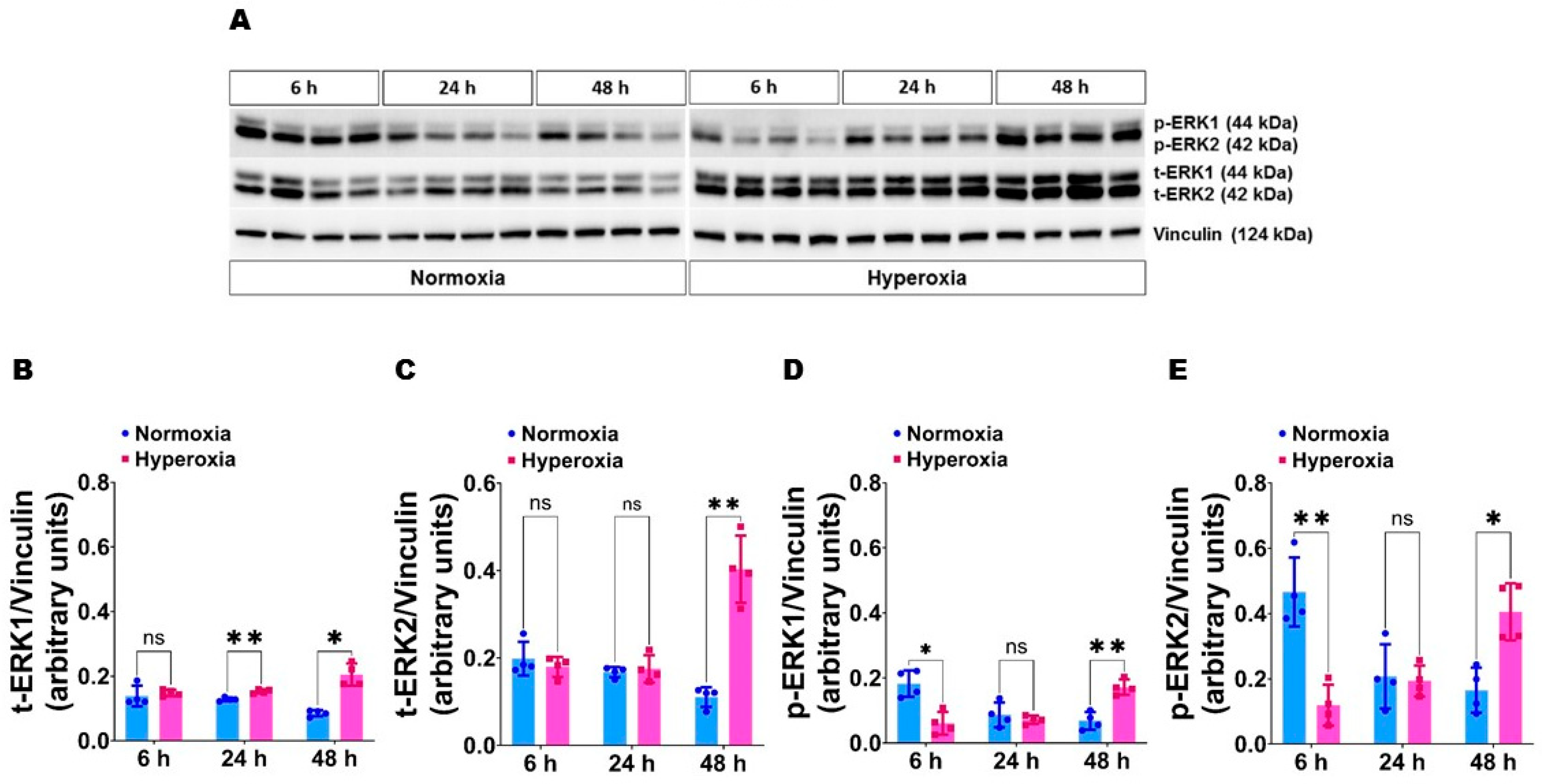{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
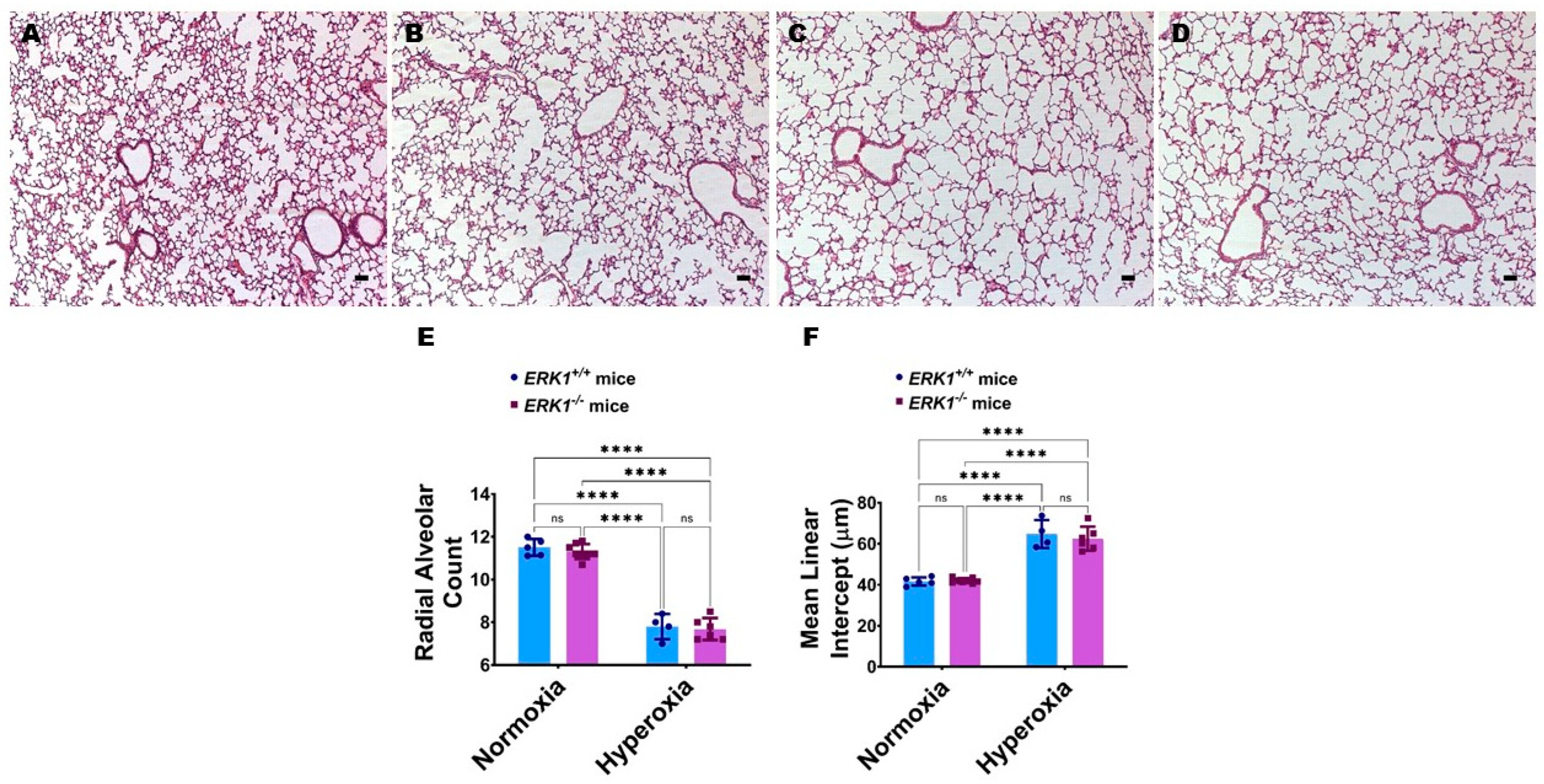{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Vitro Experiments
2.1.1. Cell Culture
2.1.2. Transfection Experiments
2.1.3. Exposure of Cells to Hyperoxia
2.1.4. Western Blot Assays
2.1.5. Tubule Formation Assay
2.1.6. Measurement of H2O2 Generation
2.1.7. Statistical Analyses
2.2. In Vivo Experiments
2.2.1. Animals
2.2.2. Hyperoxia Experiments
2.2.3. Analyses of Lung Alveolarization and Vascularization
2.2.4. Lung Tissue Extraction for Genomic and Proteomic Studies
2.2.5. Real-Time RT- PCR Assays
2.2.6. Western Blot Assays
2.2.7. Statistical Analyses
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thébaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Lapcharoensap, W.; Bennett, M.V.; Xu, X.; Lee, H.C.; Dukhovny, D. Hospitalization costs associated with bronchopulmonary dysplasia in the first year of life. J. Perinatol. 2020, 40, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, J.L.; Pakrashi, T.; Jones, D.E.; South, A.P.; Shah, T.A. Echocardiographic detection of pulmonary hypertension in extremely low birth weight infants with bronchopulmonary dysplasia requiring prolonged positive pressure ventilation. J. Perinatol. 2011, 31, 635–640. [Google Scholar] [CrossRef] [Green Version]
- Arjaans, S.; Zwart, E.A.H.; Ploegstra, M.J.; Bos, A.F.; Kooi, E.M.W.; Hillege, H.L.; Berger, R.M.F. Identification of gaps in the current knowledge on pulmonary hypertension in extremely preterm infants: A systematic review and meta-analysis. Paediatr. Perinat. Epidemiol. 2018, 32, 258–267. [Google Scholar] [CrossRef]
- Nakanishi, H.; Uchiyama, A.; Kusuda, S. Impact of pulmonary hypertension on neurodevelopmental outcome in preterm infants with bronchopulmonary dysplasia: A cohort study. J. Perinatol. 2016, 36, 890–896. [Google Scholar] [CrossRef] [Green Version]
- Berkelhamer, S.K.; Mestan, K.K.; Steinhorn, R. An update on the diagnosis and management of bronchopulmonary dysplasia (BPD)-associated pulmonary hypertension. Semin. Perinatol. 2018, 42, 432–443. [Google Scholar] [CrossRef]
- Critser, P.J.; Higano, N.S.; Tkach, J.A.; Olson, E.S.; Spielberg, D.R.; Kingma, P.S.; Fleck, R.J.; Lang, S.M.; Moore, R.A.; Taylor, M.D.; et al. Cardiac MRI Evaluation of Neonatal Bronchopulmonary Dysplasia Associated Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, 73–82. [Google Scholar] [CrossRef]
- Husain, A.N.; Siddiqui, N.H.; Stocker, J.T. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum. Pathol. 1998, 29, 710–717. [Google Scholar] [CrossRef]
- Coalson, J.J. Pathology of new bronchopulmonary dysplasia. Semin. Neonatol. 2003, 8, 73–81. [Google Scholar] [CrossRef]
- Thebaud, B.; Abman, S.H. Bronchopulmonary dysplasia: Where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am. J. Respir. Crit. Care Med. 2007, 175, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Appuhn, S.V.; Siebert, S.; Myti, D.; Wrede, C.; Solaligue, D.E.S.; Pérez-Bravo, D.; Brandenberger, C.; Schipke, J.; Morty, R.E.; Grothausmann, R.; et al. Capillary Changes Precede Disordered Alveolarization in a Mouse Model of Bronchopulmonary Dysplasia. Am. J. Respir. Cell Mol. Biol. 2021, 65, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Gwini, S.M.; Menahem, S.; Allison, B.J.; Miller, S.L.; Polglase, G.R. Preterm growth restriction and bronchopulmonary dysplasia: The vascular hypothesis and related physiology. J. Physiol. 2019, 597, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Wallace, B.; Peisl, A.; Seedorf, G.; Nowlin, T.; Kim, C.; Bosco, J.; Kenniston, J.; Keefe, D.; Abman, S.H. Anti-sFlt-1 Therapy Preserves Lung Alveolar and Vascular Growth in Antenatal Models of Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2018, 197, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Markham, N.E.; Balasubramaniam, V.; Tang, J.R.; Maxey, A.; Kinsella, J.P.; Abman, S.H. Inhaled nitric oxide enhances distal lung growth after exposure to hyperoxia in neonatal rats. Pediatric Res. 2005, 58, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.R.; Markham, N.E.; Lin, Y.J.; McMurtry, I.F.; Maxey, A.; Kinsella, J.P.; Abman, S.H. Inhaled nitric oxide attenuates pulmonary hypertension and improves lung growth in infant rats after neonatal treatment with a VEGF receptor inhibitor. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L344–L351. [Google Scholar] [CrossRef]
- Kunig, A.M.; Balasubramaniam, V.; Markham, N.E.; Seedorf, G.; Gien, J.; Abman, S.H. Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1068–L1078. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.C.; Donohue, P.; Gilmore, M.; Cristofalo, E.; Wilson, R.F.; Weiner, J.Z.; Robinson, K. Inhaled nitric oxide in preterm infants. Evid. Rep. Technol. Assess. 2010, 195, 1–315. [Google Scholar]
- Donohue, P.K.; Gilmore, M.M.; Cristofalo, E.; Wilson, R.F.; Weiner, J.Z.; Lau, B.D.; Robinson, K.A.; Allen, M.C. Inhaled nitric oxide in preterm infants: A systematic review. Pediatrics 2011, 127, e414–e422. [Google Scholar] [CrossRef]
- Gadhia, M.M.; Cutter, G.R.; Abman, S.H.; Kinsella, J.P. Effects of early inhaled nitric oxide therapy and vitamin A supplementation on the risk for bronchopulmonary dysplasia in premature newborns with respiratory failure. J. Pediatrics 2014, 164, 744–748. [Google Scholar] [CrossRef] [Green Version]
- Madurga, A.; Mizikova, I.; Ruiz-Camp, J.; Morty, R.E. Recent advances in late lung development and the pathogenesis of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L893–L905. [Google Scholar] [CrossRef]
- English, J.; Pearson, G.; Wilsbacher, J.; Swantek, J.; Karandikar, M.; Xu, S.; Cobb, M.H. New insights into the control of MAP kinase pathways. Exp. Cell Res. 1999, 253, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Gabay, L.; Seger, R.; Shilo, B.Z. MAP kinase in situ activation atlas during Drosophila embryogenesis. Development 1997, 124, 3535–3541. [Google Scholar] [CrossRef] [PubMed]
- Kashimata, M.; Sayeed, S.; Ka, A.; Onetti-Muda, A.; Sakagami, H.; Faraggiana, T.; Gresik, E.W. The ERK-1/2 signaling pathway is involved in the stimulation of branching morphogenesis of fetal mouse submandibular glands by EGF. Dev. Biol. 2000, 220, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Karihaloo, A.; O’Rourke, D.A.; Nickel, C.; Spokes, K.; Cantley, L.G. Differential MAPK pathways utilized for HGF- and EGF-dependent renal epithelial morphogenesis. J. Biol. Chem. 2001, 276, 9166–9173. [Google Scholar] [CrossRef] [Green Version]
- Niemann, C.; Brinkmann, V.; Birchmeier, W. Hepatocyte growth factor and neuregulin in mammary gland cell morphogenesis. Adv. Exp. Med. Biol. 2000, 480, 9–18. [Google Scholar] [CrossRef]
- Kling, D.E.; Lorenzo, H.K.; Trbovich, A.M.; Kinane, T.B.; Donahoe, P.K.; Schnitzer, J.J. MEK-1/2 inhibition reduces branching morphogenesis and causes mesenchymal cell apoptosis in fetal rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L370–L378. [Google Scholar] [CrossRef]
- Menon, R.T.; Shrestha, A.K.; Barrios, R.; Shivanna, B. Hyperoxia Disrupts Extracellular Signal-Regulated Kinases 1/2-Induced Angiogenesis in the Developing Lungs. Int. J. Mol. Sci. 2018, 19, 1525. [Google Scholar] [CrossRef] [Green Version]
- Menon, R.T.; Shrestha, A.K.; Barrios, R.; Reynolds, C.; Shivanna, B. Tie-2 Cre-Mediated Deficiency of Extracellular Signal-Regulated Kinase 2 Potentiates Experimental Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension in Neonatal Mice. Int. J. Mol. Sci. 2020, 21, 2408. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.; Baveja, R.; Liang, O.D.; Fernandez-Gonzalez, A.; Lee, C.; Mitsialis, S.A.; Kourembanas, S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.J.; Berkelhamer, S.K.; Kim, G.A.; Taylor, J.M.; O’Shea, K.M.; Steinhorn, R.H.; Farrow, K.N. Disrupted Pulmonary Artery cGMP Signaling in Mice with Hyperoxia-Induced Pulmonary Hypertension. Am J Respir Cell Mol Biol 2014, 50, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Menon, R.T.; Shrestha, A.K.; Reynolds, C.L.; Barrios, R.; Shivanna, B. Long-term pulmonary and cardiovascular morbidities of neonatal hyperoxia exposure in mice. Int. J. Biochem. Cell Biol. 2018, 94, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.L.; Zhang, S.; Shrestha, A.K.; Barrios, R.; Shivanna, B. Phenotypic assessment of pulmonary hypertension using high-resolution echocardiography is feasible in neonatal mice with experimental bronchopulmonary dysplasia and pulmonary hypertension: A step toward preventing chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 1597–1605. [Google Scholar] [CrossRef] [Green Version]
- Elsaie, A.; Menon, R.T.; Shrestha, A.K.; Gowda, S.H.; Varghese, N.P.; Barrios, R.J.; Blanco, C.L.; Konduri, G.G.; Shivanna, B. Endothelial Adenosine Monophosphate-Activated Protein Kinase-Alpha1 Deficiency Potentiates Hyperoxia-Induced Experimental Bronchopulmonary Dysplasia and Pulmonary Hypertension. Antioxidants 2021, 10, 1913. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.K.; Bettini, M.L.; Menon, R.T.; Gopal, V.Y.N.; Huang, S.; Edwards, D.P.; Pammi, M.; Barrios, R.; Shivanna, B. Consequences of Early Postnatal Lipopolysaccharide Exposure on Developing Lungs in Mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L229–L244. [Google Scholar] [CrossRef]
- Arnaoutova, I.; Kleinman, H.K. In vitro angiogenesis: Endothelial cell tube formation on gelled basement membrane extract. Nat. Protoc. 2010, 5, 628–635. [Google Scholar] [CrossRef]
- Ribatti, D.; Guidolin, D.; Conconi, M.T.; Nico, B.; Baiguera, S.; Parnigotto, P.P.; Vacca, A.; Nussdorfer, G.G. Vinblastine inhibits the angiogenic response induced by adrenomedullin in vitro and in vivo. Oncogene 2003, 22, 6458–6461. [Google Scholar] [CrossRef] [Green Version]
- Shivanna, B.; Zhang, W.; Jiang, W.; Welty, S.E.; Couroucli, X.I.; Wang, L.; Moorthy, B. Functional deficiency of aryl hydrocarbon receptor augments oxygen toxicity-induced alveolar simplification in newborn mice. Toxicol. Appl. Pharmacol. 2013, 267, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Shivanna, B.; Chu, C.; Welty, S.E.; Jiang, W.; Wang, L.; Couroucli, X.I.; Moorthy, B. Omeprazole attenuates hyperoxic injury in H441 cells via the aryl hydrocarbon receptor. Free. Radic. Biol. Med. 2011, 51, 1910–1917. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.J.; Pryhuber, G.S.; Huyck, H.; Watkins, R.H.; Metlay, L.A.; Maniscalco, W.M. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 164, 1971–1980. [Google Scholar] [CrossRef]
- Menon, R.T.; Shrestha, A.K.; Reynolds, C.L.; Barrios, R.; Caron, K.M.; Shivanna, B. Adrenomedullin Is Necessary to Resolve Hyperoxia-Induced Experimental Bronchopulmonary Dysplasia and Pulmonary Hypertension in Mice. Am. J. Pathol. 2020, 190, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Frémin, C.; Saba-El-Leil, M.K.; Lévesque, K.; Ang, S.L.; Meloche, S. Functional Redundancy of ERK1 and ERK2 MAP Kinases during Development. Cell Rep. 2015, 12, 913–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscà, R.; Pouysségur, J.; Lenormand, P. ERK1 and ERK2 Map Kinases: Specific Roles or Functional Redundancy? Front. Cell Dev. Biol. 2016, 4, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavria, G.; Vercoulen, Y.; Yeo, M.; Paterson, H.; Karasarides, M.; Marais, R.; Bird, D.; Marshall, C.J. ERK-MAPK signaling opposes Rho-kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell 2006, 9, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, R.; Zabuawala, T.; Huang, H.; Zhang, J.; Gulati, P.; Fernandez, S.; Karlo, J.C.; Landreth, G.E.; Leone, G.; Ostrowski, M.C. Erk1 and Erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS ONE 2009, 4, e8283. [Google Scholar] [CrossRef]
- Pagès, G.; Guérin, S.; Grall, D.; Bonino, F.; Smith, A.; Anjuere, F.; Auberger, P.; Pouysségur, J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 1999, 286, 1374–1377. [Google Scholar] [CrossRef]
- Ricard, N.; Zhang, J.; Zhuang, Z.W.; Simons, M. Isoform-Specific Roles of ERK1 and ERK2 in Arteriogenesis. Cells 2019, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Silva, D.M.; Nardiello, C.; Pozarska, A.; Morty, R.E. Recent advances in the mechanisms of lung alveolarization and the pathogenesis of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1239–L1272. [Google Scholar] [CrossRef]
- Morty, R.E. Recent advances in the pathogenesis of BPD. Semin. Perinatol. 2018, 42, 404–412. [Google Scholar] [CrossRef]
- Kim, Y.E.; Park, W.S.; Ahn, S.Y.; Sung, D.K.; Sung, S.I.; Kim, J.H.; Chang, Y.S. WKYMVm hexapeptide, a strong formyl peptide receptor 2 agonist, attenuates hyperoxia-induced lung injuries in newborn mice. Sci. Rep. 2019, 9, 6815. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Sasidhar, M.; Chupp, G.L.; Flavell, R.A.; Choi, A.M.; Lee, P.J. Reactive oxygen species and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase mediate hyperoxia-induced cell death in lung epithelium. Am. J. Respir. Cell Mol. Biol. 2003, 28, 305–315. [Google Scholar] [CrossRef]
- Carnesecchi, S.; Deffert, C.; Pagano, A.; Garrido-Urbani, S.; Metrailler-Ruchonnet, I.; Schappi, M.; Donati, Y.; Matthay, M.A.; Krause, K.H.; Argiroffo, C.B. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2009, 180, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, R.; Villarreal, P.; Husain, S.; Liu, J.; Sakurai, T.; Tou, E.; Torday, J.S.; Rehan, V.K. Curcumin protects the developing lung against long-term hyperoxic injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L301–L311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Fu, J.; Xue, X. Association of the proliferation of lung fibroblasts with the ERK1/2 signaling pathway in neonatal rats with hyperoxia-induced lung fibrosis. Exp. Ther. Med. 2019, 17, 701–708. [Google Scholar] [CrossRef] [PubMed]
- McGowan, S.E.; Torday, J.S. The pulmonary lipofibroblast (lipid interstitial cell) and its contributions to alveolar development. Annu. Rev. Physiol. 1997, 59, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Torday, J.S.; Torres, E.; Rehan, V.K. The role of fibroblast transdifferentiation in lung epithelial cell proliferation, differentiation, and repair in vitro. Pediatric Pathol. Mol. Med. 2003, 22, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Saugstad, O.D. Oxygen and oxidative stress in bronchopulmonary dysplasia. J. Perinat. Med. 2010, 38, 571–577. [Google Scholar] [CrossRef]
- Bhandari, V. Hyperoxia-derived lung damage in preterm infants. Semin. Fetal Neonatal Med. 2010, 15, 223–229. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menon, R.T.; Thapa, S.; Shrestha, A.K.; Barrios, R.; Shivanna, B. Extracellular Signal-Regulated Kinase 1 Alone Is Dispensable for Hyperoxia-Mediated Alveolar and Pulmonary Vascular Simplification in Neonatal Mice. Antioxidants 2022, 11, 1130. https://doi.org/10.3390/antiox11061130
Menon RT, Thapa S, Shrestha AK, Barrios R, Shivanna B. Extracellular Signal-Regulated Kinase 1 Alone Is Dispensable for Hyperoxia-Mediated Alveolar and Pulmonary Vascular Simplification in Neonatal Mice. Antioxidants. 2022; 11(6):1130. https://doi.org/10.3390/antiox11061130
Chicago/Turabian StyleMenon, Renuka T., Shyam Thapa, Amrit Kumar Shrestha, Roberto Barrios, and Binoy Shivanna. 2022. "Extracellular Signal-Regulated Kinase 1 Alone Is Dispensable for Hyperoxia-Mediated Alveolar and Pulmonary Vascular Simplification in Neonatal Mice" Antioxidants 11, no. 6: 1130. https://doi.org/10.3390/antiox11061130
APA StyleMenon, R. T., Thapa, S., Shrestha, A. K., Barrios, R., & Shivanna, B. (2022). Extracellular Signal-Regulated Kinase 1 Alone Is Dispensable for Hyperoxia-Mediated Alveolar and Pulmonary Vascular Simplification in Neonatal Mice. Antioxidants, 11(6), 1130. https://doi.org/10.3390/antiox11061130