
Sphingolipids and Atherosclerosis: The Dual Role of Ceramide and Sphingosine-1-Phosphate
 , , , , , , ,
, , , , , , ,
Abstract
:
1. Sphingolipids and Atherosclerosis
2. Ceramide and Sphingosine-1-Phosphate Metabolism
3. Ceramide
3.1. Ceramide and Endothelial Dysfunction
3.2. Ceramide and Lipoproteins
3.3. Ceramide and Monocytes Recruitment
3.4. Ceramide, VSMCs and Effects on Atherosclerosis Plaques Formation

3.5. Ceramide Plasma Levels and CAD
4. Sphingosine-1-Phosphate
4.1. S1P and Endothelial Dysfunction
4.2. S1P and Lipoproteins
4.3. S1P and Monocytes Recruitment
4.4. S1P and VSMCs
4.5. S1P Analogs and Effects on Atherosclerosis Plaques Formation
4.6. S1P Plasma Levels and CAD
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Park, T.S.; Devi, S.; Sharma, A.; Kim, G.T.; Cho, K.H. De Novo Sphingolipid Biosynthesis in Atherosclerosis. Adv. Exp. Med. Biol. 2022, 1372, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Piccoli, M.; Ghiroldi, A.; Monasky, M.M.; Rota, P.; La Rocca, P.; Tarantino, A.; D’Imperio, S.; Signorelli, P.; Pappone, C.; et al. The antithetic role of ceramide and sphingosine-1-phosphate in cardiac dysfunction. J. Cell. Physiol. 2021, 236, 4857–4873. [Google Scholar] [CrossRef] [PubMed]
- Engin, A.B. What Is Lipotoxicity? Adv. Exp. Med. Biol. 2017, 960, 197–220. [Google Scholar] [CrossRef] [PubMed]
- Saddoughi, S.A.; Ogretmen, B. Diverse functions of ceramide in cancer cell death and proliferation. Adv. Cancer Res. 2013, 117, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A.; Chaurasia, B.; Holland, W.L. Metabolic Messengers: Ceramides. Nat. Metab. 2019, 1, 1051–1058. [Google Scholar] [CrossRef]
- Garic, D.; De Sanctis, J.B.; Shah, J.; Dumut, D.C.; Radzioch, D. Biochemistry of very-long-chain and long-chain ceramides in cystic fibrosis and other diseases: The importance of side chain. Prog. Lipid Res. 2019, 74, 130–144. [Google Scholar] [CrossRef]
- Borodzicz-Jazdzyk, S.; Jazdzyk, P.; Lysik, W.; Cudnoch-Jedrzejewska, A.; Czarzasta, K. Sphingolipid metabolism and signaling in cardiovascular diseases. Front. Cardiovasc. Med. 2022, 9, 915961. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, H.; Qu, P.; Qiao, X.; Guo, L.; Liu, L. Novel insight on the role of Macrophages in atherosclerosis: Focus on polarization, apoptosis and efferocytosis. Int. Immunopharmacol. 2022, 113, 109260. [Google Scholar] [CrossRef]
- Smith, E.B. Intimal and medial lipids in human aortas. Lancet 1960, 1, 799–803. [Google Scholar] [CrossRef]
- Edsfeldt, A.; Duner, P.; Stahlman, M.; Mollet, I.G.; Asciutto, G.; Grufman, H.; Nitulescu, M.; Persson, A.F.; Fisher, R.M.; Melander, O.; et al. Sphingolipids Contribute to Human Atherosclerotic Plaque Inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1132–1140. [Google Scholar] [CrossRef]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb. Vasc. Biol. 1995, 15, 1512–1531. [Google Scholar] [CrossRef]
- Sun, H.J.; Wu, Z.Y.; Nie, X.W.; Bian, J.S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2019, 10, 1568. [Google Scholar] [CrossRef] [Green Version]
- Berenji Ardestani, S.; Eftedal, I.; Pedersen, M.; Jeppesen, P.B.; Norregaard, R.; Matchkov, V.V. Endothelial dysfunction in small arteries and early signs of atherosclerosis in ApoE knockout rats. Sci. Rep. 2020, 10, 15296. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Breton-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Haigh, S.; Barman, S.; Fulton, D.J. From form to function: The role of Nox4 in the cardiovascular system. Front. Physiol. 2012, 3, 412. [Google Scholar] [CrossRef] [Green Version]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Chun, J.N.; Jung, H.Y.; Choi, C.; Bae, Y.S. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc. Res. 2006, 72, 447–455. [Google Scholar] [CrossRef]
- Muller, G.; Goettsch, C.; Morawietz, H. Oxidative stress and endothelial dysfunction. Hamostaseologie 2007, 27, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Cohen, R.; Ullrich, V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium 2004, 11, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Stahl, W.; Sevanian, A. Nutritional, dietary and postprandial oxidative stress. J. Nutr. 2005, 135, 969–972. [Google Scholar] [CrossRef] [Green Version]
- Di Pietro, N.; Formoso, G.; Pandolfi, A. Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis. Vasc. Pharmacol. 2016, 84, 1–7. [Google Scholar] [CrossRef]
- Malekmohammad, K.; Sewell, R.D.E.; Rafieian-Kopaei, M. Antioxidants and Atherosclerosis: Mechanistic Aspects. Biomolecules 2019, 9, 301. [Google Scholar] [CrossRef] [Green Version]
- Quehenberger, O. Thematic review series: The immune system and atherogenesis. Molecular mechanisms regulating monocyte recruitment in atherosclerosis. J. Lipid Res. 2005, 46, 1582–1590. [Google Scholar] [CrossRef] [Green Version]
- Krieglstein, C.F.; Granger, D.N. Adhesion molecules and their role in vascular disease. Am. J. Hypertens. 2001, 14, 44S–54S. [Google Scholar] [CrossRef] [Green Version]
- Navab, M.; Imes, S.S.; Hama, S.Y.; Hough, G.P.; Ross, L.A.; Bork, R.W.; Valente, A.J.; Berliner, J.A.; Drinkwater, D.C.; Laks, H.; et al. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J. Clin. Investig. 1991, 88, 2039–2046. [Google Scholar] [CrossRef]
- Drechsler, M.; Duchene, J.; Soehnlein, O. Chemokines control mobilization, recruitment, and fate of monocytes in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1050–1055. [Google Scholar] [CrossRef] [Green Version]
- Van Gils, J.M.; Zwaginga, J.J.; Hordijk, P.L. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J. Leukoc. Biol. 2009, 85, 195–204. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Weber, C. Platelets as immune cells: Bridging inflammation and cardiovascular disease. Circ. Res. 2007, 100, 27–40. [Google Scholar] [CrossRef]
- Tabas, I.; Garcia-Cardena, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Mietus-Snyder, M.; Gowri, M.S.; Pitas, R.E. Class A scavenger receptor up-regulation in smooth muscle cells by oxidized low density lipoprotein. Enhancement by calcium flux and concurrent cyclooxygenase-2 up-regulation. J. Biol. Chem. 2000, 275, 17661–17670. [Google Scholar] [CrossRef] [Green Version]
- Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation 2000, 102, 82–87. [Google Scholar] [CrossRef]
- Hegyi, L.; Skepper, J.N.; Cary, N.R.; Mitchinson, M.J. Foam cell apoptosis and the development of the lipid core of human atherosclerosis. J. Pathol. 1996, 180, 423–429. [Google Scholar] [CrossRef]
- Thondapu, V.; Bourantas, C.V.; Foin, N.; Jang, I.K.; Serruys, P.W.; Barlis, P. Biomechanical stress in coronary atherosclerosis: Emerging insights from computational modelling. Eur. Heart J. 2017, 38, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Y.; Taviani, V.; Tang, T.; Sadat, U.; Young, V.; Patterson, A.; Graves, M.; Gillard, J.H. The mechanical triggers of plaque rupture: Shear stress vs pressure gradient. Br. J. Radiol. 2009, 82, S39–S45. [Google Scholar] [CrossRef]
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zietzer, A.; Dusing, P.; Reese, L.; Nickenig, G.; Jansen, F. Ceramide Metabolism in Cardiovascular Disease: A Network With High Therapeutic Potential. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Futerman, A.H. Mammalian ceramide synthases. IUBMB Life 2010, 62, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Lachkar, F.; Ferre, P.; Foufelle, F.; Papaioannou, A. Dihydroceramides: Their emerging physiological roles and functions in cancer and metabolic diseases. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E122–E130. [Google Scholar] [CrossRef] [PubMed]
- Alka, K.; Mohammad, G.; Kowluru, R.A. Regulation of serine palmitoyl-transferase and Rac1-Nox2 signaling in diabetic retinopathy. Sci. Rep. 2022, 12, 16740. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, J.; Yang, M.; Guo, J.; Li, D.; Li, Y. Lipidomics Analysis Reveals a Protective Effect of Myriocin on Cerebral Ischemia/Reperfusion Model Rats. J. Mol. NeuroSci. 2022, 72, 1846–1858. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Talbot, C.L.; Chandravanshi, B.; Ksiazek, A.; Sood, A.; Chowdhury, K.H.; Maschek, J.A.; Cox, J.; Babu, A.K.S.; Paz, H.A.; et al. Cordyceps inhibits ceramide biosynthesis and improves insulin resistance and hepatic steatosis. Sci. Rep. 2022, 12, 7273. [Google Scholar] [CrossRef]
- Mingione, A.; Dei Cas, M.; Bonezzi, F.; Caretti, A.; Piccoli, M.; Anastasia, L.; Ghidoni, R.; Paroni, R.; Signorelli, P. Inhibition of Sphingolipid Synthesis as a Phenotype-Modifying Therapy in Cystic Fibrosis. Cell. Physiol. Biochem. 2020, 54, 110–125. [Google Scholar] [CrossRef]
- Bonezzi, F.; Piccoli, M.; Dei Cas, M.; Paroni, R.; Mingione, A.; Monasky, M.M.; Caretti, A.; Riganti, C.; Ghidoni, R.; Pappone, C.; et al. Sphingolipid Synthesis Inhibition by Myriocin Administration Enhances Lipid Consumption and Ameliorates Lipid Response to Myocardial Ischemia Reperfusion Injury. Front. Physiol. 2019, 10, 986. [Google Scholar] [CrossRef] [Green Version]
- Goni, F.M.; Alonso, A. Sphingomyelinases: Enzymology and membrane activity. FEBS Lett. 2002, 531, 38–46. [Google Scholar] [CrossRef]
- Marchesini, N.; Osta, W.; Bielawski, J.; Luberto, C.; Obeid, L.M.; Hannun, Y.A. Role for mammalian neutral sphingomyelinase 2 in confluence-induced growth arrest of MCF7 cells. J. Biol. Chem. 2004, 279, 25101–25111. [Google Scholar] [CrossRef] [Green Version]
- Breiden, B.; Sandhoff, K. Acid Sphingomyelinase, a Lysosomal and Secretory Phospholipase C, Is Key for Cellular Phospholipid Catabolism. Int. J. Mol. Sci. 2021, 22, 9001. [Google Scholar] [CrossRef]
- Marathe, S.; Schissel, S.L.; Yellin, M.J.; Beatini, N.; Mintzer, R.; Williams, K.J.; Tabas, I. Human vascular endothelial cells are a rich and regulatable source of secretory sphingomyelinase. Implications for early atherogenesis and ceramide-mediated cell signaling. J. Biol. Chem. 1998, 273, 4081–4088. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, C.A.; He, Q.; Ponnusamy, S.; Ogretmen, B.; Huang, Y.; Sheikh, M.S. Neutral sphingomyelinase-3 is a DNA damage and nongenotoxic stress-regulated gene that is deregulated in human malignancies. Mol. Cancer Res. 2008, 6, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, V.; Canals, D.; Luberto, C.; Snider, J.; Voelkel-Johnson, C.; Obeid, L.M.; Hannun, Y.A. Critical determinants of mitochondria-associated neutral sphingomyelinase (MA-nSMase) for mitochondrial localization. Biochim. Biophys. Acta 2015, 1850, 628–639. [Google Scholar] [CrossRef] [Green Version]
- Zietzer, A.; Jahnel, A.L.; Bulic, M.; Gutbrod, K.; Dusing, P.; Hosen, M.R.; Dormann, P.; Werner, N.; Nickenig, G.; Jansen, F. Activation of neutral sphingomyelinase 2 through hyperglycemia contributes to endothelial apoptosis via vesicle-bound intercellular transfer of ceramides. Cell Mol. Life Sci. 2021, 79, 48. [Google Scholar] [CrossRef]
- Auge, N.; Maupas-Schwalm, F.; Elbaz, M.; Thiers, J.C.; Waysbort, A.; Itohara, S.; Krell, H.W.; Salvayre, R.; Negre-Salvayre, A. Role for matrix metalloproteinase-2 in oxidized low-density lipoprotein-induced activation of the sphingomyelin/ceramide pathway and smooth muscle cell proliferation. Circulation 2004, 110, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Wollny, T.; Watek, M.; Durnas, B.; Niemirowicz, K.; Piktel, E.; Zendzian-Piotrowska, M.; Gozdz, S.; Bucki, R. Sphingosine-1-Phosphate Metabolism and Its Role in the Development of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2017, 18, 741. [Google Scholar] [CrossRef] [Green Version]
- Le Stunff, H.; Milstien, S.; Spiegel, S. Generation and metabolism of bioactive sphingosine-1-phosphate. J. Cell. Biochem. 2004, 92, 882–899. [Google Scholar] [CrossRef]
- Yanagida, K.; Hla, T. Vascular and Immunobiology of the Circulatory Sphingosine 1-Phosphate Gradient. Annu. Rev. Physiol. 2017, 79, 67–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keul, P.; Sattler, K.; Levkau, B. HDL and its sphingosine-1-phosphate content in cardioprotection. Heart Fail. Rev. 2007, 12, 301–306. [Google Scholar] [CrossRef]
- Alonso, A.; Goni, F.M. The Physical Properties of Ceramides in Membranes. Annu. Rev. Biophys. 2018, 47, 633–654. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H., Jr. De novo sphingolipid biosynthesis: A necessary, but dangerous, pathway. J. Biol. Chem. 2002, 277, 25843–25846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim. Biophys. Acta 2003, 1632, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Pewzner-Jung, Y.; Ben-Dor, S.; Futerman, A.H. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J. Biol. Chem. 2006, 281, 25001–25005. [Google Scholar] [CrossRef] [Green Version]
- Pavoine, C.; Pecker, F. Sphingomyelinases: Their regulation and roles in cardiovascular pathophysiology. Cardiovasc. Res. 2009, 82, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.C.; Goldberg, I.J.; Park, T.S. Sphingolipids and cardiovascular diseases: Lipoprotein metabolism, atherosclerosis and cardiomyopathy. Adv. Exp. Med. Biol. 2011, 721, 19–39. [Google Scholar] [CrossRef]
- Price, D.T.; Vita, J.A.; Keaney, J.F., Jr. Redox control of vascular nitric oxide bioavailability. Antioxid. Redox Signal. 2000, 2, 919–935. [Google Scholar] [CrossRef]
- Rubanyi, G.M.; Vanhoutte, P.M. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am. J. Physiol. 1986, 250, H822–H827. [Google Scholar] [CrossRef]
- Li, P.L.; Zhang, Y. Cross talk between ceramide and redox signaling: Implications for endothelial dysfunction and renal disease. In Sphingolipids in Disease; Handbook of Experimental Pharmacology; Springer: Vienna, Austria, 2013; pp. 171–197. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Becker, K.A.; Zhang, Y. Ceramide in redox signaling and cardiovascular diseases. Cell. Physiol. Biochem. 2010, 26, 41–48. [Google Scholar] [CrossRef]
- Frazziano, G.; Moreno, L.; Moral-Sanz, J.; Menendez, C.; Escolano, L.; Gonzalez, C.; Villamor, E.; Alvarez-Sala, J.L.; Cogolludo, A.L.; Perez-Vizcaino, F. Neutral sphingomyelinase, NADPH oxidase and reactive oxygen species. Role in acute hypoxic pulmonary vasoconstriction. J. Cell. Physiol. 2011, 226, 2633–2640. [Google Scholar] [CrossRef]
- Jesko, H.; Stepien, A.; Lukiw, W.J.; Strosznajder, R.P. The Cross-Talk Between Sphingolipids and Insulin-Like Growth Factor Signaling: Significance for Aging and Neurodegeneration. Mol. NeuroBiol. 2019, 56, 3501–3521. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Schuchman, E.H. Ceramide and Ischemia/Reperfusion Injury. J. Lipids 2018, 2018, 3646725. [Google Scholar] [CrossRef] [Green Version]
- Cinq-Frais, C.; Coatrieux, C.; Grazide, M.H.; Hannun, Y.A.; Negre-Salvayre, A.; Salvayre, R.; Auge, N. A signaling cascade mediated by ceramide, src and PDGFRbeta coordinates the activation of the redox-sensitive neutral sphingomyelinase-2 and sphingosine kinase-1. Biochim. Biophys. Acta 2013, 1831, 1344–1356. [Google Scholar] [CrossRef]
- Levy, M.; Castillo, S.S.; Goldkorn, T. nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem. Biophys. Res. Commun. 2006, 344, 900–905. [Google Scholar] [CrossRef] [Green Version]
- Dumitru, C.A.; Gulbins, E. TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene 2006, 25, 5612–5625. [Google Scholar] [CrossRef] [Green Version]
- Malaplate-Armand, C.; Florent-Bechard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.L.; Oster, T.; Pillot, T. Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. NeuroBiol. Dis. 2006, 23, 178–189. [Google Scholar] [CrossRef]
- Martin, S.F.; Sawai, H.; Villalba, J.M.; Hannun, Y.A. Redox regulation of neutral sphingomyelinase-1 activity in HEK293 cells through a GSH-dependent mechanism. Arch. Biochem. Biophys. 2007, 459, 295–300. [Google Scholar] [CrossRef]
- Siskind, L.J.; Colombini, M. The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J. Biol. Chem. 2000, 275, 38640–38644. [Google Scholar] [CrossRef] [Green Version]
- Funai, K.; Summers, S.A.; Rutter, J. Reign in the membrane: How common lipids govern mitochondrial function. Curr. Opin. Cell Biol. 2020, 63, 162–173. [Google Scholar] [CrossRef]
- Kogot-Levin, A.; Saada, A. Ceramide and the mitochondrial respiratory chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.; Dulovic-Mahlow, M.; Mandik, F.; Frese, L.; Kanana, Y.; Haissatou Diaw, S.; Depperschmidt, J.; Bohm, C.; Rohr, J.; Lohnau, T.; et al. Ceramide accumulation induces mitophagy and impairs beta-oxidation in PINK1 deficiency. Proc. Natl. Acad. Sci. USA 2021, 118, e2025347118. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Junk, P.; Huwiler, A.; Burkhardt, C.; Wallerath, T.; Pfeilschifter, J.; Forstermann, U. Dual effect of ceramide on human endothelial cells: Induction of oxidative stress and transcriptional upregulation of endothelial nitric oxide synthase. Circulation 2002, 106, 2250–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.X.; Zou, A.P.; Li, P.L. Ceramide reduces endothelium-dependent vasodilation by increasing superoxide production in small bovine coronary arteries. Circ. Res. 2001, 88, 824–831. [Google Scholar] [CrossRef] [Green Version]
- Bourbon, N.A.; Yun, J.; Kester, M. Ceramide directly activates protein kinase C zeta to regulate a stress-activated protein kinase signaling complex. J. Biol. Chem. 2000, 275, 35617–35623. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yao, B.; Delikat, S.; Bayoumy, S.; Lin, X.H.; Basu, S.; McGinley, M.; Chan-Hui, P.Y.; Lichenstein, H.; Kolesnick, R. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell 1997, 89, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Corda, S.; Laplace, C.; Vicaut, E.; Duranteau, J. Rapid reactive oxygen species production by mitochondria in endothelial cells exposed to tumor necrosis factor-alpha is mediated by ceramide. Am. J. Respir. Cell Mol. Biol. 2001, 24, 762–768. [Google Scholar] [CrossRef]
- Ji, Y.; Chen, J.; Pang, L.; Chen, C.; Ye, J.; Liu, H.; Chen, H.; Zhang, S.; Liu, S.; Liu, B.; et al. The Acid Sphingomyelinase Inhibitor Amitriptyline Ameliorates TNF-alpha-Induced Endothelial Dysfunction. Cardiovasc. Drugs Ther. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Xu, J.; Yeh, C.H.; Chen, S.; He, L.; Sensi, S.L.; Canzoniero, L.M.; Choi, D.W.; Hsu, C.Y. Involvement of de novo ceramide biosynthesis in tumor necrosis factor-alpha/cycloheximide-induced cerebral endothelial cell death. J. Biol. Chem. 1998, 273, 16521–16526. [Google Scholar] [CrossRef] [Green Version]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef]
- Birbes, H.; El Bawab, S.; Hannun, Y.A.; Obeid, L.M. Selective hydrolysis of a mitochondrial pool of sphingomyelin induces apoptosis. FASEB J. 2001, 15, 2669–2679. [Google Scholar] [CrossRef]
- Lin, X.; Fuks, Z.; Kolesnick, R. Ceramide mediates radiation-induced death of endothelium. Crit. Care Med. 2000, 28, N87–N93. [Google Scholar] [CrossRef]
- Schweitzer, K.S.; Hatoum, H.; Brown, M.B.; Gupta, M.; Justice, M.J.; Beteck, B.; Van Demark, M.; Gu, Y.; Presson, R.G., Jr.; Hubbard, W.C.; et al. Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: Role of oxidative stress and ceramides. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L836–L846. [Google Scholar] [CrossRef]
- Zhang, A.Y.; Yi, F.; Zhang, G.; Gulbins, E.; Li, P.L. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 2006, 47, 74–80. [Google Scholar] [CrossRef]
- Zhang, A.Y.; Yi, F.; Jin, S.; Xia, M.; Chen, Q.Z.; Gulbins, E.; Li, P.L. Acid sphingomyelinase and its redox amplification in formation of lipid raft redox signaling platforms in endothelial cells. Antioxid. Redox Signal. 2007, 9, 817–828. [Google Scholar] [CrossRef]
- Niaudet, C.; Bonnaud, S.; Guillonneau, M.; Gouard, S.; Gaugler, M.H.; Dutoit, S.; Ripoche, N.; Dubois, N.; Trichet, V.; Corre, I.; et al. Plasma membrane reorganization links acid sphingomyelinase/ceramide to p38 MAPK pathways in endothelial cells apoptosis. Cell. Signal. 2017, 33, 10–21. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ. Res. 2016, 119, 375–396. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A. The sphingomyelin cycle and the second messenger function of ceramide. J. Biol. Chem. 1994, 269, 3125–3128. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Holland, W.L.; Wilson, L.; Tanner, J.M.; Kearns, D.; Cahoon, J.M.; Pettey, D.; Losee, J.; Duncan, B.; Gale, D.; et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes 2012, 61, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Cantalupo, A.; Sasset, L.; Gargiulo, A.; Rubinelli, L.; Del Gaudio, I.; Benvenuto, D.; Wadsack, C.; Jiang, X.C.; Bucci, M.R.; Di Lorenzo, A. Endothelial Sphingolipid De Novo Synthesis Controls Blood Pressure by Regulating Signal Transduction and NO via Ceramide. Hypertension 2020, 75, 1279–1288. [Google Scholar] [CrossRef]
- Barsacchi, R.; Perrotta, C.; Bulotta, S.; Moncada, S.; Borgese, N.; Clementi, E. Activation of endothelial nitric-oxide synthase by tumor necrosis factor-alpha: A novel pathway involving sequential activation of neutral sphingomyelinase, phosphatidylinositol-3’ kinase, and Akt. Mol. Pharmacol. 2003, 63, 886–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulotta, S.; Barsacchi, R.; Rotiroti, D.; Borgese, N.; Clementi, E. Activation of the endothelial nitric-oxide synthase by tumor necrosis factor-alpha. A novel feedback mechanism regulating cell death. J. Biol. Chem. 2001, 276, 6529–6536. [Google Scholar] [CrossRef] [Green Version]
- Freed, J.K.; Beyer, A.M.; LoGiudice, J.A.; Hockenberry, J.C.; Gutterman, D.D. Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ. Res. 2014, 115, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.R.; Visioli, F.; Frei, B.; Hagen, T.M. Age-related changes in endothelial nitric oxide synthase phosphorylation and nitric oxide dependent vasodilation: Evidence for a novel mechanism involving sphingomyelinase and ceramide-activated phosphatase 2A. Aging Cell 2006, 5, 391–400. [Google Scholar] [CrossRef]
- Fu, M.; Li, Z.; Tan, T.; Guo, W.; Xie, N.; Liu, Q.; Zhu, H.; Xie, X.; Lei, H. Akt/eNOS signaling pathway mediates inhibition of endothelial progenitor cells by palmitate-induced ceramide. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H11–H17. [Google Scholar] [CrossRef] [Green Version]
- Mehra, V.C.; Jackson, E.; Zhang, X.M.; Jiang, X.C.; Dobrucki, L.W.; Yu, J.; Bernatchez, P.; Sinusas, A.J.; Shulman, G.I.; Sessa, W.C.; et al. Ceramide-activated phosphatase mediates fatty acid-induced endothelial VEGF resistance and impaired angiogenesis. Am. J. Pathol. 2014, 184, 1562–1576. [Google Scholar] [CrossRef] [Green Version]
- Hino, H.; Takaki, K.; Mochida, S. Inhibitor-1 and -2 of PP2A have preference between PP2A complexes. Biochem. Biophys. Res. Commun. 2015, 467, 297–302. [Google Scholar] [CrossRef]
- Bharath, L.P.; Ruan, T.; Li, Y.; Ravindran, A.; Wan, X.; Nhan, J.K.; Walker, M.L.; Deeter, L.; Goodrich, R.; Johnson, E.; et al. Ceramide-Initiated Protein Phosphatase 2A Activation Contributes to Arterial Dysfunction In Vivo. Diabetes 2015, 64, 3914–3926. [Google Scholar] [CrossRef] [Green Version]
- Chun, L.; Junlin, Z.; Aimin, W.; Niansheng, L.; Benmei, C.; Minxiang, L. Inhibition of ceramide synthesis reverses endothelial dysfunction and atherosclerosis in streptozotocin-induced diabetic rats. Diabetes Res. Clin. Pract. 2011, 93, 77–85. [Google Scholar] [CrossRef]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxid. Med. Cell. Longev. 2020, 2020, 5245308. [Google Scholar] [CrossRef]
- Schissel, S.L.; Tweedie-Hardman, J.; Rapp, J.H.; Graham, G.; Williams, K.J.; Tabas, I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low-density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J. Clin. Investig. 1996, 98, 1455–1464. [Google Scholar] [CrossRef]
- Abbas, A.; Aukrust, P.; Russell, D.; Krohg-Sorensen, K.; Almas, T.; Bundgaard, D.; Bjerkeli, V.; Sagen, E.L.; Michelsen, A.E.; Dahl, T.B.; et al. Matrix metalloproteinase 7 is associated with symptomatic lesions and adverse events in patients with carotid atherosclerosis. PLoS ONE 2014, 9, e84935. [Google Scholar] [CrossRef] [Green Version]
- Ruuth, M.; Nguyen, S.D.; Vihervaara, T.; Hilvo, M.; Laajala, T.D.; Kondadi, P.K.; Gistera, A.; Lahteenmaki, H.; Kittila, T.; Huusko, J.; et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur. Heart J. 2018, 39, 2562–2573. [Google Scholar] [CrossRef] [Green Version]
- Zelnik, I.D.; Kim, J.L.; Futerman, A.H. The Complex Tail of Circulating Sphingolipids in Atherosclerosis and Cardiovascular Disease. J. Lipid Atheroscler. 2021, 10, 268–281. [Google Scholar] [CrossRef]
- Walters, M.J.; Wrenn, S.P. Effect of sphingomyelinase-mediated generation of ceramide on aggregation of low-density lipoprotein. Langmuir 2008, 24, 9642–9647. [Google Scholar] [CrossRef]
- Sneck, M.; Nguyen, S.D.; Pihlajamaa, T.; Yohannes, G.; Riekkola, M.L.; Milne, R.; Kovanen, P.T.; Oorni, K. Conformational changes of apoB-100 in SMase-modified LDL mediate formation of large aggregates at acidic pH. J. Lipid Res. 2012, 53, 1832–1839. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yang, X.; Xing, S.; Bian, F.; Yao, W.; Bai, X.; Zheng, T.; Wu, G.; Jin, S. Endogenous ceramide contributes to the transcytosis of oxLDL across endothelial cells and promotes its subendothelial retention in vascular wall. Oxid. Med. Cell. Longev. 2014, 2014, 823071. [Google Scholar] [CrossRef]
- Sun, S.W.; Zu, X.Y.; Tuo, Q.H.; Chen, L.X.; Lei, X.Y.; Li, K.; Tang, C.K.; Liao, D.F. Caveolae and caveolin-1 mediate endocytosis and transcytosis of oxidized low density lipoprotein in endothelial cells. Acta Pharmacol. Sin. 2010, 31, 1336–1342. [Google Scholar] [CrossRef] [Green Version]
- Auge, N.; Andrieu, N.; Negre-Salvayre, A.; Thiers, J.C.; Levade, T.; Salvayre, R. The sphingomyelin-ceramide signaling pathway is involved in oxidized low density lipoprotein-induced cell proliferation. J. Biol. Chem. 1996, 271, 19251–19255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Pararasa, C.; Dunston, C.R.; Bailey, C.J.; Griffiths, H.R. Palmitate promotes monocyte atherogenicity via de novo ceramide synthesis. Free Radic. Biol. Med. 2012, 53, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.X.; Tabas, I. Sphingomyelinase enhances low density lipoprotein uptake and ability to induce cholesteryl ester accumulation in macrophages. J. Biol. Chem. 1991, 266, 24849–24858. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.; Tabas, I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J. Lipid Res. 2009, 50, S382–S387. [Google Scholar] [CrossRef] [Green Version]
- Deigner, H.P.; Claus, R.; Bonaterra, G.A.; Gehrke, C.; Bibak, N.; Blaess, M.; Cantz, M.; Metz, J.; Kinscherf, R. Ceramide induces aSMase expression: Implications for oxLDL-induced apoptosis. FASEB J. 2001, 15, 807–814. [Google Scholar] [CrossRef]
- Zhao, M.; Pan, W.; Shi, R.Z.; Bai, Y.P.; You, B.Y.; Zhang, K.; Fu, Q.M.; Schuchman, E.H.; He, X.X.; Zhang, G.G. Acid Sphingomyelinase Mediates Oxidized-LDL Induced Apoptosis in Macrophage via Endoplasmic Reticulum Stress. J. Atheroscler. Thromb. 2016, 23, 1111–1125. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.; Bennett, M. The emerging role of vascular smooth muscle cell apoptosis in atherosclerosis and plaque stability. Am. J. Nephrol. 2006, 26, 531–535. [Google Scholar] [CrossRef]
- Boyle, J.J. Vascular smooth muscle cell apoptosis in atherosclerosis. Int. J. Exp. Pathol. 1999, 80, 197–203. [Google Scholar] [CrossRef]
- Loidl, A.; Claus, R.; Ingolic, E.; Deigner, H.P.; Hermetter, A. Role of ceramide in activation of stress-associated MAP kinases by minimally modified LDL in vascular smooth muscle cells. Biochim. Biophys. Acta 2004, 1690, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Sangiorgi, G.; Rumberger, J.A.; Severson, A.; Edwards, W.D.; Gregoire, J.; Fitzpatrick, L.A.; Schwartz, R.S. Arterial calcification and not lumen stenosis is highly correlated with atherosclerotic plaque burden in humans: A histologic study of 723 coronary artery segments using nondecalcifying methodology. J. Am. Coll. Cardiol. 1998, 31, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.C.; Littlewood, T.D.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Bennett, M.R. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ. Res. 2008, 102, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Liao, L.; Zhou, Q.; Song, Y.; Wu, W.; Yu, H.; Wang, S.; Chen, Y.; Ye, M.; Lu, L. Ceramide mediates Ox-LDL-induced human vascular smooth muscle cell calcification via p38 mitogen-activated protein kinase signaling. PLoS ONE 2013, 8, e82379. [Google Scholar] [CrossRef]
- Song, Y.; Hou, M.; Li, Z.; Luo, C.; Ou, J.S.; Yu, H.; Yan, J.; Lu, L. TLR4/NF-kappaB/Ceramide signaling contributes to Ox-LDL-induced calcification of human vascular smooth muscle cells. Eur. J. Pharmacol. 2017, 794, 45–51. [Google Scholar] [CrossRef]
- Luong, T.T.D.; Tuffaha, R.; Schuchardt, M.; Moser, B.; Schelski, N.; Boehme, B.; Gollmann-Tepekoylu, C.; Schramm, C.; Holfeld, J.; Pieske, B.; et al. Acid sphingomyelinase promotes SGK1-dependent vascular calcification. Clin. Sci. 2021, 135, 515–534. [Google Scholar] [CrossRef]
- Devlin, C.M.; Leventhal, A.R.; Kuriakose, G.; Schuchman, E.H.; Williams, K.J.; Tabas, I. Acid sphingomyelinase promotes lipoprotein retention within early atheromata and accelerates lesion progression. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Park, T.S.; Panek, R.L.; Mueller, S.B.; Hanselman, J.C.; Rosebury, W.S.; Robertson, A.W.; Kindt, E.K.; Homan, R.; Karathanasis, S.K.; Rekhter, M.D. Inhibition of sphingomyelin synthesis reduces atherogenesis in apolipoprotein E-knockout mice. Circulation 2004, 110, 3465–3471. [Google Scholar] [CrossRef] [Green Version]
- Hojjati, M.R.; Li, Z.; Zhou, H.; Tang, S.; Huan, C.; Ooi, E.; Lu, S.; Jiang, X.C. Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J. Biol. Chem. 2005, 280, 10284–10289. [Google Scholar] [CrossRef] [Green Version]
- Park, T.S.; Hu, Y.; Noh, H.L.; Drosatos, K.; Okajima, K.; Buchanan, J.; Tuinei, J.; Homma, S.; Jiang, X.C.; Abel, E.D.; et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J. Lipid Res. 2008, 49, 2101–2112. [Google Scholar] [CrossRef]
- Yu, Z.; Peng, Q.; Li, S.; Hao, H.; Deng, J.; Meng, L.; Shen, Z.; Yu, W.; Nan, D.; Bai, Y.; et al. Myriocin and d-PDMP ameliorate atherosclerosis in ApoE−/− mice via reducing lipid uptake and vascular inflammation. Clin. Sci. 2020, 134, 439–458. [Google Scholar] [CrossRef] [Green Version]
- Tarasov, K.; Ekroos, K.; Suoniemi, M.; Kauhanen, D.; Sylvanne, T.; Hurme, R.; Gouni-Berthold, I.; Berthold, H.K.; Kleber, M.E.; Laaksonen, R.; et al. Molecular lipids identify cardiovascular risk and are efficiently lowered by simvastatin and PCSK9 deficiency. J. Clin. Endocrinol. Metab. 2014, 99, E45–E52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laaksonen, R.; Ekroos, K.; Sysi-Aho, M.; Hilvo, M.; Vihervaara, T.; Kauhanen, D.; Suoniemi, M.; Hurme, R.; Marz, W.; Scharnagl, H.; et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur. Heart J. 2016, 37, 1967–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Wang, X.; Pang, J.; Zhang, Y.; Zhang, H.; Xu, Z.; Chen, Q.; Ling, W. Associations between plasma ceramides and mortality in patients with coronary artery disease. Atherosclerosis 2020, 314, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, M. Plasma ceramides and cardiac risk. Eur. Heart J. 2017, 38, 1359–1360. [Google Scholar] [CrossRef]
- Cheng, J.M.; Suoniemi, M.; Kardys, I.; Vihervaara, T.; de Boer, S.P.; Akkerhuis, K.M.; Sysi-Aho, M.; Ekroos, K.; Garcia-Garcia, H.M.; Oemrawsingh, R.M.; et al. Plasma concentrations of molecular lipid species in relation to coronary plaque characteristics and cardiovascular outcome: Results of the ATHEROREMO-IVUS study. Atherosclerosis 2015, 243, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Bonapace, S.; Lunardi, G.; Canali, G.; Dugo, C.; Vinco, G.; Calabria, S.; Barbieri, E.; Laaksonen, R.; Bonnet, F.; et al. Associations between specific plasma ceramides and severity of coronary-artery stenosis assessed by coronary angiography. Diabetes Metab. 2020, 46, 150–157. [Google Scholar] [CrossRef]
- Spiegel, S.; Merrill, A.H., Jr. Sphingolipid metabolism and cell growth regulation. FASEB J. 1996, 10, 1388–1397. [Google Scholar] [CrossRef]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef]
- Hla, T.; Venkataraman, K.; Michaud, J. The vascular S1P gradient-cellular sources and biological significance. Biochim. Biophys. Acta 2008, 1781, 477–482. [Google Scholar] [CrossRef]
- Ishimaru, K.; Yoshioka, K.; Kano, K.; Kurano, M.; Saigusa, D.; Aoki, J.; Yatomi, Y.; Takuwa, N.; Okamoto, Y.; Proia, R.L.; et al. Sphingosine kinase-2 prevents macrophage cholesterol accumulation and atherosclerosis by stimulating autophagic lipid degradation. Sci. Rep. 2019, 9, 18329. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, K.; Thangada, S.; Michaud, J.; Oo, M.L.; Ai, Y.; Lee, Y.M.; Wu, M.; Parikh, N.S.; Khan, F.; Proia, R.L.; et al. Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem. J. 2006, 397, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Olivera, A.; Allende, M.L.; Proia, R.L. Shaping the landscape: Metabolic regulation of S1P gradients. Biochim. Biophys. Acta 2013, 1831, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Narayanaswamy, P.; Shinde, S.; Sulc, R.; Kraut, R.; Staples, G.; Thiam, C.H.; Grimm, R.; Sellergren, B.; Torta, F.; Wenk, M.R. Lipidomic “deep profiling”: An enhanced workflow to reveal new molecular species of signaling lipids. Anal. Chem. 2014, 86, 3043–3047. [Google Scholar] [CrossRef]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [Green Version]
- Morozov, V.I.; Sakuta, G.A.; Kalinski, M.I. Sphingosine-1-phosphate: Distribution, metabolism and role in the regulation of cellular functions. Ukr. Biokhim. Zh. 2013, 85, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: Signaling inside and out. FEBS Lett. 2000, 476, 55–57. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.; Hla, T.; Lynch, K.R.; Spiegel, S.; Moolenaar, W.H. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 2010, 62, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Ishii, I.; Fukushima, N.; Ye, X.; Chun, J. Lysophospholipid receptors: Signaling and biology. Annu. Rev. Biochem. 2004, 73, 321–354. [Google Scholar] [CrossRef] [Green Version]
- Gaire, B.P.; Lee, C.H.; Sapkota, A.; Lee, S.Y.; Chun, J.; Cho, H.J.; Nam, T.G.; Choi, J.W. Identification of Sphingosine 1-Phosphate Receptor Subtype 1 (S1P(1)) as a Pathogenic Factor in Transient Focal Cerebral Ischemia. Mol. NeuroBiol. 2018, 55, 2320–2332. [Google Scholar] [CrossRef]
- Kuang, Y.; Li, X.; Liu, X.; Wei, L.; Chen, X.; Liu, J.; Zhuang, T.; Pi, J.; Wang, Y.; Zhu, C.; et al. Vascular endothelial S1pr1 ameliorates adverse cardiac remodelling via stimulating reparative macrophage proliferation after myocardial infarction. Cardiovasc. Res. 2021, 117, 585–599. [Google Scholar] [CrossRef]
- Siehler, S.; Manning, D.R. Pathways of transduction engaged by sphingosine 1-phosphate through G protein-coupled receptors. Biochim. Biophys. Acta 2002, 1582, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Belyantseva, I.A.; Skoura, A.; Frolenkov, G.I.; Starost, M.F.; Dreier, J.L.; Lidington, D.; Bolz, S.S.; Friedman, T.B.; Hla, T.; et al. Deafness and stria vascularis defects in S1P2 receptor-null mice. J. Biol. Chem. 2007, 282, 10690–10696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burczyk, M.; Burkhalter, M.D.; Blatte, T.; Matysik, S.; Caron, M.G.; Barak, L.S.; Philipp, M. Phenotypic regulation of the sphingosine 1-phosphate receptor miles apart by G protein-coupled receptor kinase 2. Biochemistry 2015, 54, 765–775. [Google Scholar] [CrossRef] [Green Version]
- Gonda, K.; Okamoto, H.; Takuwa, N.; Yatomi, Y.; Okazaki, H.; Sakurai, T.; Kimura, S.; Sillard, R.; Harii, K.; Takuwa, Y. The novel sphingosine 1-phosphate receptor AGR16 is coupled via pertussis toxin-sensitive and -insensitive G-proteins to multiple signalling pathways. Biochem. J. 1999, 337 Pt 1, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Takasugi, H.; Ohnuma, S.; Koide, Y.; Sakurai, A.; Takeda, S.; Hasegawa, T.; Sasamori, J.; Konno, T.; Hayashi, K.; et al. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: Investigation based on a new S1P3 receptor antagonist. Mol. Pharmacol. 2010, 77, 704–713. [Google Scholar] [CrossRef]
- Windh, R.T.; Lee, M.J.; Hla, T.; An, S.; Barr, A.J.; Manning, D.R. Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the G(i), G(q), and G(12) families of heterotrimeric G proteins. J. Biol. Chem. 1999, 274, 27351–27358. [Google Scholar] [CrossRef] [Green Version]
- Golfier, S.; Kondo, S.; Schulze, T.; Takeuchi, T.; Vassileva, G.; Achtman, A.H.; Graler, M.H.; Abbondanzo, S.J.; Wiekowski, M.; Kremmer, E.; et al. Shaping of terminal megakaryocyte differentiation and proplatelet development by sphingosine-1-phosphate receptor S1P4. FASEB J. 2010, 24, 4701–4710. [Google Scholar] [CrossRef]
- Niedernberg, A.; Scherer, C.R.; Busch, A.E.; Kostenis, E. Comparative analysis of human and rat S1P(5) (edg8): Differential expression profiles and sensitivities to antagonists. Biochem. Pharmacol. 2002, 64, 1243–1250. [Google Scholar] [CrossRef]
- Graler, M.H.; Grosse, R.; Kusch, A.; Kremmer, E.; Gudermann, T.; Lipp, M. The sphingosine 1-phosphate receptor S1P4 regulates cell shape and motility via coupling to Gi and G12/13. J. Cell. Biochem. 2003, 89, 507–519. [Google Scholar] [CrossRef]
- Bogatcheva, N.V.; Verin, A.D. Reprint of “The role of cytoskeleton in the regulation of vascular endothelial barrier function” [Microvascular Research 76 (2008) 202-207]. Microvasc. Res. 2009, 77, 64–69. [Google Scholar] [CrossRef]
- English, D.; Garcia, J.G.; Brindley, D.N. Platelet-released phospholipids link haemostasis and angiogenesis. Cardiovasc. Res. 2001, 49, 588–599. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Thangada, S.; Claffey, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha’afi, R.I.; Hla, T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 1999, 99, 301–312. [Google Scholar] [CrossRef] [Green Version]
- An, S.; Zheng, Y.; Bleu, T. Sphingosine 1-phosphate-induced cell proliferation, survival, and related signaling events mediated by G protein-coupled receptors Edg3 and Edg5. J. Biol. Chem. 2000, 275, 288–296. [Google Scholar] [CrossRef] [Green Version]
- Schaphorst, K.L.; Chiang, E.; Jacobs, K.N.; Zaiman, A.; Natarajan, V.; Wigley, F.; Garcia, J.G. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L258–L267. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701. [Google Scholar] [CrossRef]
- Singleton, P.A.; Dudek, S.M.; Chiang, E.T.; Garcia, J.G. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J. 2005, 19, 1646–1656. [Google Scholar] [CrossRef]
- Gonzalez, E.; Kou, R.; Michel, T. Rac1 modulates sphingosine 1-phosphate-mediated activation of phosphoinositide 3-kinase/Akt signaling pathways in vascular endothelial cells. J. Biol. Chem. 2006, 281, 3210–3216. [Google Scholar] [CrossRef] [Green Version]
- Schnoor, M.; Stradal, T.E.; Rottner, K. Cortactin: Cell Functions of A Multifaceted Actin-Binding Protein. Trends Cell Biol. 2018, 28, 79–98. [Google Scholar] [CrossRef]
- Li, Y.; Uruno, T.; Haudenschild, C.; Dudek, S.M.; Garcia, J.G.; Zhan, X. Interaction of cortactin and Arp2/3 complex is required for sphingosine-1-phosphate-induced endothelial cell remodeling. Exp. Cell Res. 2004, 298, 107–121. [Google Scholar] [CrossRef]
- Sander, E.E.; ten Klooster, J.P.; van Delft, S.; van der Kammen, R.A.; Collard, J.G. Rac downregulates Rho activity: Reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 1999, 147, 1009–1022. [Google Scholar] [CrossRef]
- Igarashi, J.; Michel, T. S1P and eNOS regulation. Biochim. Biophys. Acta 2008, 1781, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Dantas, A.P.; Igarashi, J.; Michel, T. Sphingosine 1-phosphate and control of vascular tone. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H2045–H2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, J.; Bernier, S.G.; Michel, T. Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase. differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. J. Biol. Chem. 2001, 276, 12420–12426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolotina, V.M.; Najibi, S.; Palacino, J.J.; Pagano, P.J.; Cohen, R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 1994, 368, 850–853. [Google Scholar] [CrossRef]
- Wilkerson, B.A.; Grass, G.D.; Wing, S.B.; Argraves, W.S.; Argraves, K.M. Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: High density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J. Biol. Chem. 2012, 287, 44645–44653. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Van Brocklyn, J.R.; Thangada, S.; Liu, C.H.; Hand, A.R.; Menzeleev, R.; Spiegel, S.; Hla, T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 1998, 279, 1552–1555. [Google Scholar] [CrossRef]
- Krump-Konvalinkova, V.; Yasuda, S.; Rubic, T.; Makarova, N.; Mages, J.; Erl, W.; Vosseler, C.; Kirkpatrick, C.J.; Tigyi, G.; Siess, W. Stable knock-down of the sphingosine 1-phosphate receptor S1P1 influences multiple functions of human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 546–552. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, T.; Estrada-Hernandez, T.; Paik, J.H.; Wu, M.T.; Venkataraman, K.; Brinkmann, V.; Claffey, K.; Hla, T. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J. Biol. Chem. 2003, 278, 47281–47290. [Google Scholar] [CrossRef]
- Dudek, S.M.; Camp, S.M.; Chiang, E.T.; Singleton, P.A.; Usatyuk, P.V.; Zhao, Y.; Natarajan, V.; Garcia, J.G. Pulmonary endothelial cell barrier enhancement by FTY720 does not require the S1P1 receptor. Cell. Signal. 2007, 19, 1754–1764. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Fan, L.; Wei, L.; Gao, H.; Zhang, R.; Tao, L.; Cao, F.; Wang, H. FTY720 protects cardiac microvessels of diabetes: A critical role of S1P1/3 in diabetic heart disease. PLoS ONE 2012, 7, e42900. [Google Scholar] [CrossRef] [Green Version]
- Burg, N.; Swendeman, S.; Worgall, S.; Hla, T.; Salmon, J.E. Sphingosine 1-Phosphate Receptor 1 Signaling Maintains Endothelial Cell Barrier Function and Protects Against Immune Complex-Induced Vascular Injury. Arthritis Rheumatol. 2018, 70, 1879–1889. [Google Scholar] [CrossRef]
- Liu, H.; Peng, H.; Chen, S.; Liu, Y.; Xiang, H.; Chen, R.; Chen, W.; Zhao, S.; Chen, P.; Lu, H. S1PR2 antagonist protects endothelial cells against high glucose-induced mitochondrial apoptosis through the Akt/GSK-3beta signaling pathway. Biochem. Biophys. Res. Commun. 2017, 490, 1119–1124. [Google Scholar] [CrossRef]
- Chen, S.; Yang, J.; Xiang, H.; Chen, W.; Zhong, H.; Yang, G.; Fang, T.; Deng, H.; Yuan, H.; Chen, A.F.; et al. Role of sphingosine-1-phosphate receptor 1 and sphingosine-1-phosphate receptor 2 in hyperglycemia-induced endothelial cell dysfunction. Int. J. Mol. Med. 2015, 35, 1103–1108. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, K.; Lee, Y.M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Hammad, S.M.; Pierce, J.S.; Soodavar, F.; Smith, K.J.; Al Gadban, M.M.; Rembiesa, B.; Klein, R.L.; Hannun, Y.A.; Bielawski, J.; Bielawska, A. Blood sphingolipidomics in healthy humans: Impact of sample collection methodology. J. Lipid Res. 2010, 51, 3074–3087. [Google Scholar] [CrossRef] [Green Version]
- Tran-Dinh, A.; Diallo, D.; Delbosc, S.; Varela-Perez, L.M.; Dang, Q.B.; Lapergue, B.; Burillo, E.; Michel, J.B.; Levoye, A.; Martin-Ventura, J.L.; et al. HDL and endothelial protection. Br. J. Pharmacol. 2013, 169, 493–511. [Google Scholar] [CrossRef] [Green Version]
- Murata, N.; Sato, K.; Kon, J.; Tomura, H.; Yanagita, M.; Kuwabara, A.; Ui, M.; Okajima, F. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem. J. 2000, 352 Pt 3, 809–815. [Google Scholar] [CrossRef]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnstrom, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef]
- Kurano, M.; Tsukamoto, K.; Ohkawa, R.; Hara, M.; Iino, J.; Kageyama, Y.; Ikeda, H.; Yatomi, Y. Liver involvement in sphingosine 1-phosphate dynamism revealed by adenoviral hepatic overexpression of apolipoprotein M. Atherosclerosis 2013, 229, 102–109. [Google Scholar] [CrossRef]
- Arkensteijn, B.W.; Berbee, J.F.; Rensen, P.C.; Nielsen, L.B.; Christoffersen, C. The apolipoprotein m-sphingosine-1-phosphate axis: Biological relevance in lipoprotein metabolism, lipid disorders and atherosclerosis. Int. J. Mol. Sci. 2013, 14, 4419–4431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nofer, J.R.; Assmann, G. Atheroprotective effects of high-density lipoprotein-associated lysosphingolipids. Trends Cardiovasc. Med. 2005, 15, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zeng, Y.; Zhu, X.; Tan, Y.; Li, Y.; Li, Q.; Yi, G. ApoM-S1P Modulates Ox-LDL-Induced Inflammation Through the PI3K/Akt Signaling Pathway in HUVECs. Inflammation 2019, 42, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and Their Role in Atherosclerosis: Pathophysiology and Transcriptome Analysis. Biomed. Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Kurano, M.; Yatomi, Y. Sphingosine 1-Phosphate and Atherosclerosis. J. Atheroscler. Thromb. 2018, 25, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Feuerborn, R.; Besser, M.; Poti, F.; Burkhardt, R.; Weissen-Plenz, G.; Ceglarek, U.; Simoni, M.; Proia, R.L.; Freise, H.; Nofer, J.R. Elevating Endogenous Sphingosine-1-Phosphate (S1P) Levels Improves Endothelial Function and Ameliorates Atherosclerosis in Low Density Lipoprotein Receptor-Deficient (LDL-R−/−) Mice. Thromb. Haemost. 2018, 118, 1470–1480. [Google Scholar] [CrossRef] [Green Version]
- Feuerborn, R.; Becker, S.; Poti, F.; Nagel, P.; Brodde, M.; Schmidt, H.; Christoffersen, C.; Ceglarek, U.; Burkhardt, R.; Nofer, J.R. High density lipoprotein (HDL)-associated sphingosine 1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 activity and survivin expression. Atherosclerosis 2017, 257, 29–37. [Google Scholar] [CrossRef]
- Duenas, A.I.; Aceves, M.; Fernandez-Pisonero, I.; Gomez, C.; Orduna, A.; Crespo, M.S.; Garcia-Rodriguez, C. Selective attenuation of Toll-like receptor 2 signalling may explain the atheroprotective effect of sphingosine 1-phosphate. Cardiovasc. Res. 2008, 79, 537–544. [Google Scholar] [CrossRef]
- Skoura, A.; Michaud, J.; Im, D.S.; Thangada, S.; Xiong, Y.; Smith, J.D.; Hla, T. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Graler, M.H. Sphingosine 1-phosphate and its type 1 G protein-coupled receptor: Trophic support and functional regulation of T lymphocytes. J. Leukoc. Biol. 2004, 76, 30–35. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Miano, J.M.; Fisher, E.A.; Majesky, M.W. Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation 2021, 143, 2110–2116. [Google Scholar] [CrossRef]
- Kluk, M.J.; Hla, T. Role of the sphingosine 1-phosphate receptor EDG-1 in vascular smooth muscle cell proliferation and migration. Circ. Res. 2001, 89, 496–502. [Google Scholar] [CrossRef] [Green Version]
- Tamama, K.; Tomura, H.; Sato, K.; Malchinkhuu, E.; Damirin, A.; Kimura, T.; Kuwabara, A.; Murakami, M.; Okajima, F. High-density lipoprotein inhibits migration of vascular smooth muscle cells through its sphingosine 1-phosphate component. Atherosclerosis 2005, 178, 19–23. [Google Scholar] [CrossRef]
- Shimizu, T.; Nakazawa, T.; Cho, A.; Dastvan, F.; Shilling, D.; Daum, G.; Reidy, M.A. Sphingosine 1-phosphate receptor 2 negatively regulates neointimal formation in mouse arteries. Circ. Res. 2007, 101, 995–1000. [Google Scholar] [CrossRef] [Green Version]
- Lockman, K.; Hinson, J.S.; Medlin, M.D.; Morris, D.; Taylor, J.M.; Mack, C.P. Sphingosine 1-phosphate stimulates smooth muscle cell differentiation and proliferation by activating separate serum response factor co-factors. J. Biol. Chem. 2004, 279, 42422–42430. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Diez, M.; Rodriguez, C.; Badimon, L.; Martinez-Gonzalez, J. Prostacyclin induction by high-density lipoprotein (HDL) in vascular smooth muscle cells depends on sphingosine 1-phosphate receptors: Effect of simvastatin. Thromb. Haemost. 2008, 100, 119–126. [Google Scholar] [CrossRef]
- Tolle, M.; Pawlak, A.; Schuchardt, M.; Kawamura, A.; Tietge, U.J.; Lorkowski, S.; Keul, P.; Assmann, G.; Chun, J.; Levkau, B.; et al. HDL-associated lysosphingolipids inhibit NAD(P)H oxidase-dependent monocyte chemoattractant protein-1 production. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1542–1548. [Google Scholar] [CrossRef]
- Pan, D.; Wu, W.; Zuo, G.; Xie, X.; Li, H.; Ren, X.; Kong, C.; Zhou, W.; Zhang, Z.; Waterfall, M.; et al. Sphingosine 1-phosphate receptor 2 promotes erythrocyte clearance by vascular smooth muscle cells in intraplaque hemorrhage through MFG-E8 production. Cell. Signal. 2022, 98, 110419. [Google Scholar] [CrossRef]
- Nofer, J.R.; Bot, M.; Brodde, M.; Taylor, P.J.; Salm, P.; Brinkmann, V.; van Berkel, T.; Assmann, G.; Biessen, E.A. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007, 115, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keul, P.; Tolle, M.; Lucke, S.; von Wnuck Lipinski, K.; Heusch, G.; Schuchardt, M.; van der Giet, M.; Levkau, B. The sphingosine-1-phosphate analogue FTY720 reduces atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingenberg, R.; Nofer, J.R.; Rudling, M.; Bea, F.; Blessing, E.; Preusch, M.; Grone, H.J.; Katus, H.A.; Hansson, G.K.; Dengler, T.J. Sphingosine-1-phosphate analogue FTY720 causes lymphocyte redistribution and hypercholesterolemia in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2392–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poti, F.; Gualtieri, F.; Sacchi, S.; Weissen-Plenz, G.; Varga, G.; Brodde, M.; Weber, C.; Simoni, M.; Nofer, J.R. KRP-203, sphingosine 1-phosphate receptor type 1 agonist, ameliorates atherosclerosis in LDL-R−/− mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1505–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keul, P.; Peters, S.; von Wnuck Lipinski, K.; Schroder, N.H.; Nowak, M.K.; Duse, D.A.; Polzin, A.; Weske, S.; Graler, M.H.; Levkau, B. Sphingosine-1-Phosphate (S1P) Lyase Inhibition Aggravates Atherosclerosis and Induces Plaque Rupture in ApoE−/− Mice. Int. J. Mol. Sci. 2022, 23, 9606. [Google Scholar] [CrossRef]
- von Eckardstein, A.; Hersberger, M.; Rohrer, L. Current understanding of the metabolism and biological actions of HDL. Curr Opin. Clin. Nutr. Metab. Care 2005, 8, 147–152. [Google Scholar] [CrossRef]
- Nofer, J.R.; van der Giet, M.; Tolle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Godecke, A.; Ishii, I.; Kleuser, B.; et al. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Investig. 2004, 113, 569–581. [Google Scholar] [CrossRef]
- Theilmeier, G.; Schmidt, C.; Herrmann, J.; Keul, P.; Schafers, M.; Herrgott, I.; Mersmann, J.; Larmann, J.; Hermann, S.; Stypmann, J.; et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 2006, 114, 1403–1409. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Gamble, J.R.; Rye, K.A.; Wang, L.; Hii, C.S.; Cockerill, P.; Khew-Goodall, Y.; Bert, A.G.; Barter, P.J.; Vadas, M.A. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 14196–14201. [Google Scholar] [CrossRef]
- Sattler, K.; Levkau, B. Sphingosine-1-phosphate as a mediator of high-density lipoprotein effects in cardiovascular protection. Cardiovasc. Res. 2009, 82, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Sattler, K.J.; Elbasan, S.; Keul, P.; Elter-Schulz, M.; Bode, C.; Graler, M.H.; Brocker-Preuss, M.; Budde, T.; Erbel, R.; Heusch, G.; et al. Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res. Cardiol. 2010, 105, 821–832. [Google Scholar] [CrossRef]
- Austen, W.G.; Edwards, J.E.; Frye, R.L.; Gensini, G.G.; Gott, V.L.; Griffith, L.S.; McGoon, D.C.; Murphy, M.L.; Roe, B.B. A reporting system on patients evaluated for coronary artery disease. Report of the Ad Hoc Committee for Grading of Coronary Artery Disease, Council on Cardiovascular Surgery, American Heart Association. Circulation 1975, 51, 5–40. [Google Scholar] [CrossRef] [Green Version]
- Argraves, K.M.; Sethi, A.A.; Gazzolo, P.J.; Wilkerson, B.A.; Remaley, A.T.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Yeatts, S.D.; Nicholas, K.S.; Barth, J.L.; et al. S1P, dihydro-S1P and C24:1-ceramide levels in the HDL-containing fraction of serum inversely correlate with occurrence of ischemic heart disease. Lipids Health Dis. 2011, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Sattler, K.; Lehmann, I.; Graler, M.; Brocker-Preuss, M.; Erbel, R.; Heusch, G.; Levkau, B. HDL-bound sphingosine 1-phosphate (S1P) predicts the severity of coronary artery atherosclerosis. Cell. Physiol. Biochem. 2014, 34, 172–184. [Google Scholar] [CrossRef]
- WHO. Cardiovascular Diseases (CVDs). Available online: https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 10 November 2022).
- Peters, L.; Kuebler, W.M.; Simmons, S. Sphingolipids in Atherosclerosis: Chimeras in Structure and Function. Int. J. Mol. Sci. 2022, 23, 1948. [Google Scholar] [CrossRef]
- Hartmann, D.; Lucks, J.; Fuchs, S.; Schiffmann, S.; Schreiber, Y.; Ferreiros, N.; Merkens, J.; Marschalek, R.; Geisslinger, G.; Grosch, S. Long chain ceramides and very long chain ceramides have opposite effects on human breast and colon cancer cell growth. Int. J. Biochem. Cell Biol. 2012, 44, 620–628. [Google Scholar] [CrossRef]
- Argraves, K.M.; Gazzolo, P.J.; Groh, E.M.; Wilkerson, B.A.; Matsuura, B.S.; Twal, W.O.; Hammad, S.M.; Argraves, W.S. High density lipoprotein-associated sphingosine 1-phosphate promotes endothelial barrier function. J. Biol. Chem. 2008, 283, 25074–25081. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Species | Structure | Function | Ref. |
|---|---|---|---|
| Cer(d18:1/16:0) | 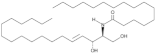 | Increased circulating levels associated with higher CV mortality risk | [142,143,144] |
| Cer(d18:1/18:0) | 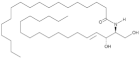 | Increased circulating levels associated with higher CV mortality risk | [142,143,144] |
| Cer(d18:1/24:1) |  | Increased circulating levels associated with higher CV mortality risk | [142,143,144] |
| Cer(d18:1/24:0) | 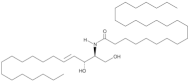 | Increased circulating levels associated with lower CV mortality risk | [142,143,144] |
| Cer(d18:1/16:0) | 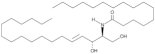 | Increased plasma levels associated with higher necrotic core fraction of AS plaques and higher Major Cardiovascular Adverse Events (MACE) rate | [146] |
| Cer(d18:1/20:0) | 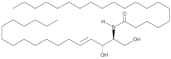 | Increased plasma levels associated with higher severity of stenosis of the left anterior descending coronary artery in CAD patients | [147] |
| Cer(d18:1/22:0) | 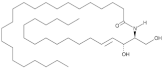 | Increased plasma levels associated with higher severity of stenosis of the left anterior descending coronary artery in CAD patients | [147] |
| Cer(d18:1/24:0) | 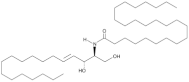 | Increased plasma levels associated with higher severity of stenosis of the left anterior descending coronary artery in CAD patients | [147] |
| Species | Target | Effects | Ref |
|---|---|---|---|
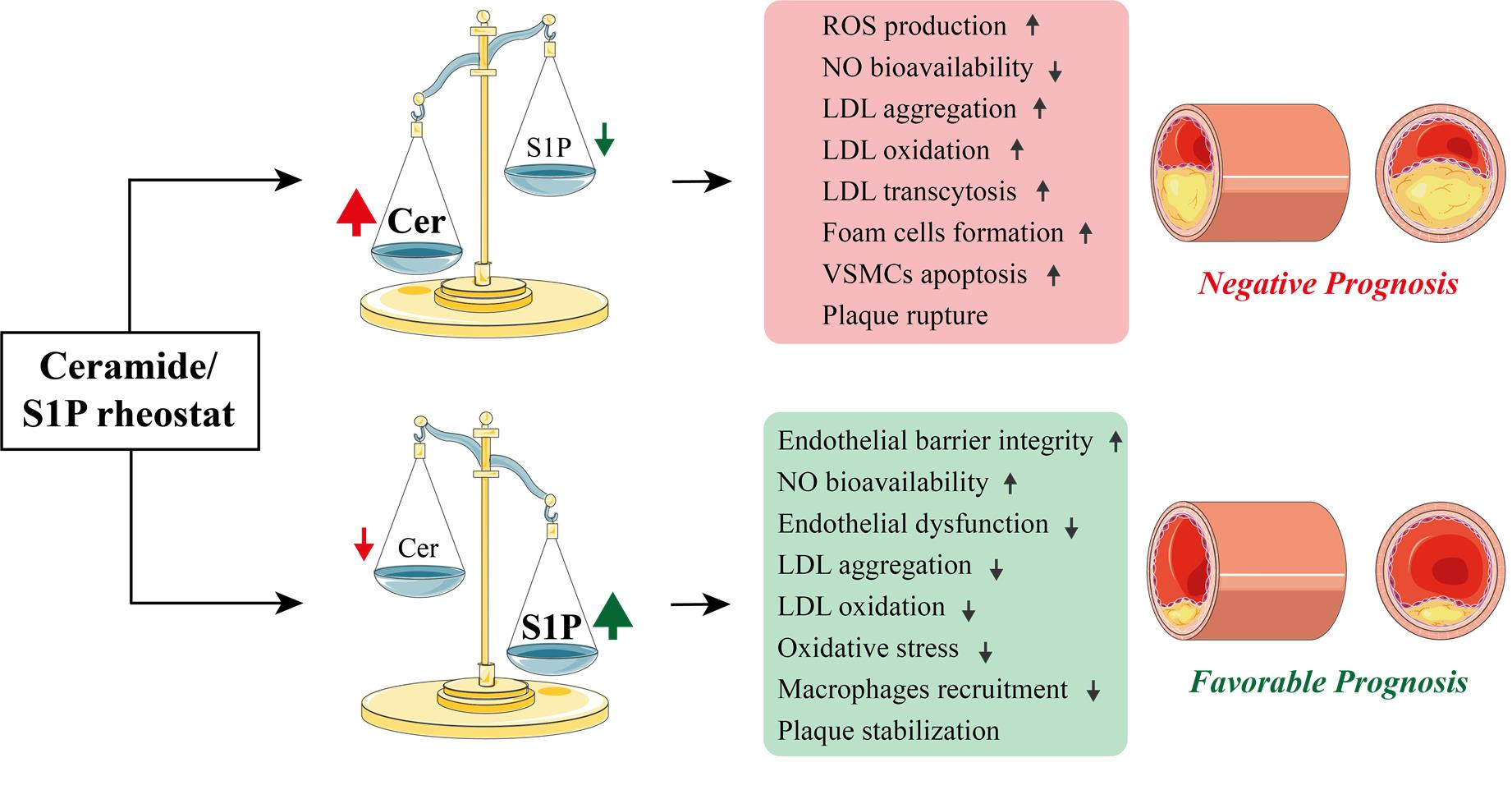
| Ceramide | Endothelial Cells | Increase of ROS production | [69,71,72,84,85,86,96,97] |
| Increase in TNF-α | [87,88,89,90] | ||
| Increase in eNOS and NO oxidation | [70,86,101,102,104,107,108,109,111] | ||
| Lipoproteins | Increase in LDL aggregation | [114,115,116,117] | |
| Increase in conformational changes in ApoB100 | [118,119] | ||
| Increase in transcytosis of oxLDL | [120,121,122] | ||
| Monocytes | Increase of CD11b and CD36 | [123] | |
| VSMCs | Increase in apoptosis | [10,129,130,132] | |
| Release of proinflammatory cytokines (IL-6) | [10] | ||
| Increase osteogenic differentiation of VSMC, leading to their vascular calcification | [132,133,134,135,136] | ||
| S1P | Endothelial Cells | Increase EC spreading by promoting F-actin polymerization and myosin light chain phosphorylation | [130,179,180,181] |
| Increase vascular relaxation by modulating NO and inhibiting eNOS | [182,183,184,185,186,187] | ||
| Increase cell contractility and VE-cadherin degradation leading to stabilization of endothelial cell-cell junctions | [173,188,189,190] | ||
| Lipoproteins | Bound to HDL by apoM. Absence of apoM leads to a dramatic decrease in S1P | [200,201,202] | |
| Decrease in inflammatory state induced by oxLDL | [203,204] | ||
| Monocytes | Inhibition of monocytes recruitment and apoptosis by activation of STAT3 via binding to S1PR1 or S1PR3 | [208,209] | |
| Enhancement of lymphocyte recruitment to sites of inflammation via binding to S1PR2 | [210,211,212] | ||
| VSMCs | Reduction of migration by binding to S1PR2 | [216,217] | |
| Suppression of NADPH oxidase and ROS production by binding to S1PR2 or S1PR3 | [219] | ||
| Inhibition of MCP-1 production | [220,221] | ||
| Increase erythrophagocytosis by binding to S1PR2 | [221] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccoli, M.; Cirillo, F.; Ghiroldi, A.; Rota, P.; Coviello, S.; Tarantino, A.; La Rocca, P.; Lavota, I.; Creo, P.; Signorelli, P.; et al. Sphingolipids and Atherosclerosis: The Dual Role of Ceramide and Sphingosine-1-Phosphate. Antioxidants 2023, 12, 143. https://doi.org/10.3390/antiox12010143
Piccoli M, Cirillo F, Ghiroldi A, Rota P, Coviello S, Tarantino A, La Rocca P, Lavota I, Creo P, Signorelli P, et al. Sphingolipids and Atherosclerosis: The Dual Role of Ceramide and Sphingosine-1-Phosphate. Antioxidants. 2023; 12(1):143. https://doi.org/10.3390/antiox12010143
Chicago/Turabian StylePiccoli, Marco, Federica Cirillo, Andrea Ghiroldi, Paola Rota, Simona Coviello, Adriana Tarantino, Paolo La Rocca, Ivana Lavota, Pasquale Creo, Paola Signorelli, and et al. 2023. "Sphingolipids and Atherosclerosis: The Dual Role of Ceramide and Sphingosine-1-Phosphate" Antioxidants 12, no. 1: 143. https://doi.org/10.3390/antiox12010143