
From Stress to Sick(le) and Back Again–Oxidative/Antioxidant Mechanisms, Genetic Modulation, and Cerebrovascular Disease in Children with Sickle Cell Anemia

Abstract
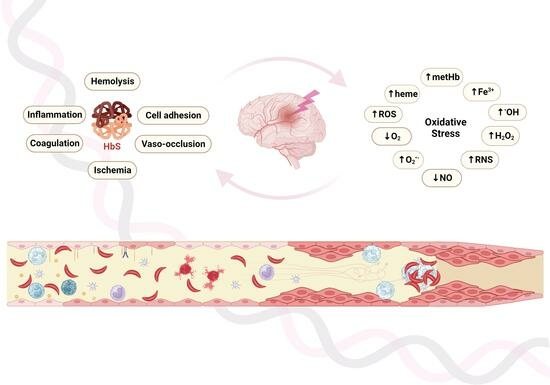
:
1. Introduction
2. Oxidation in RBCs, Hemoglobin, and the Vascular Milieu in SCA
3. The Vascular Endothelium, Endothelial Activation and Endothelial Dysfunction
3.1. Nitric Oxide Production and Regulation
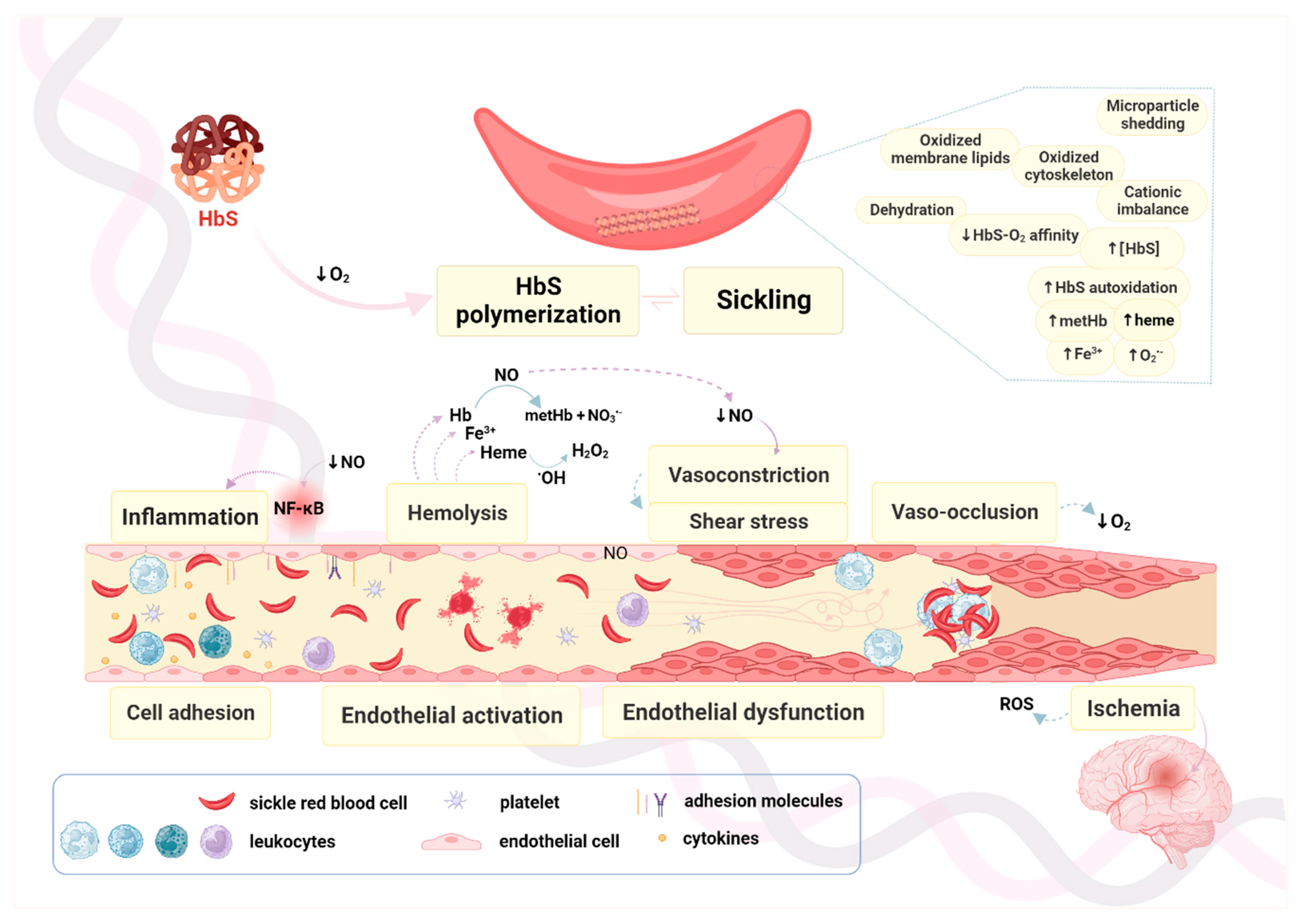
3.2. Endothelial Dysfunction vs. Endothelial Activation
3.3. Chronic Inflammation and ROS Production
3.4. Intravascular RBC Hemolysis and Production of RNS
3.5. Tissue Ischemia-Reperfusion Injury and ROS Production
4. Oxidative Pathways, Vasculopathy, and SCA
5. Cerebrovascular Disease and Hypoxia in SCA
6. Antioxidant Therapeutic Approaches in SCD Vasculopathy
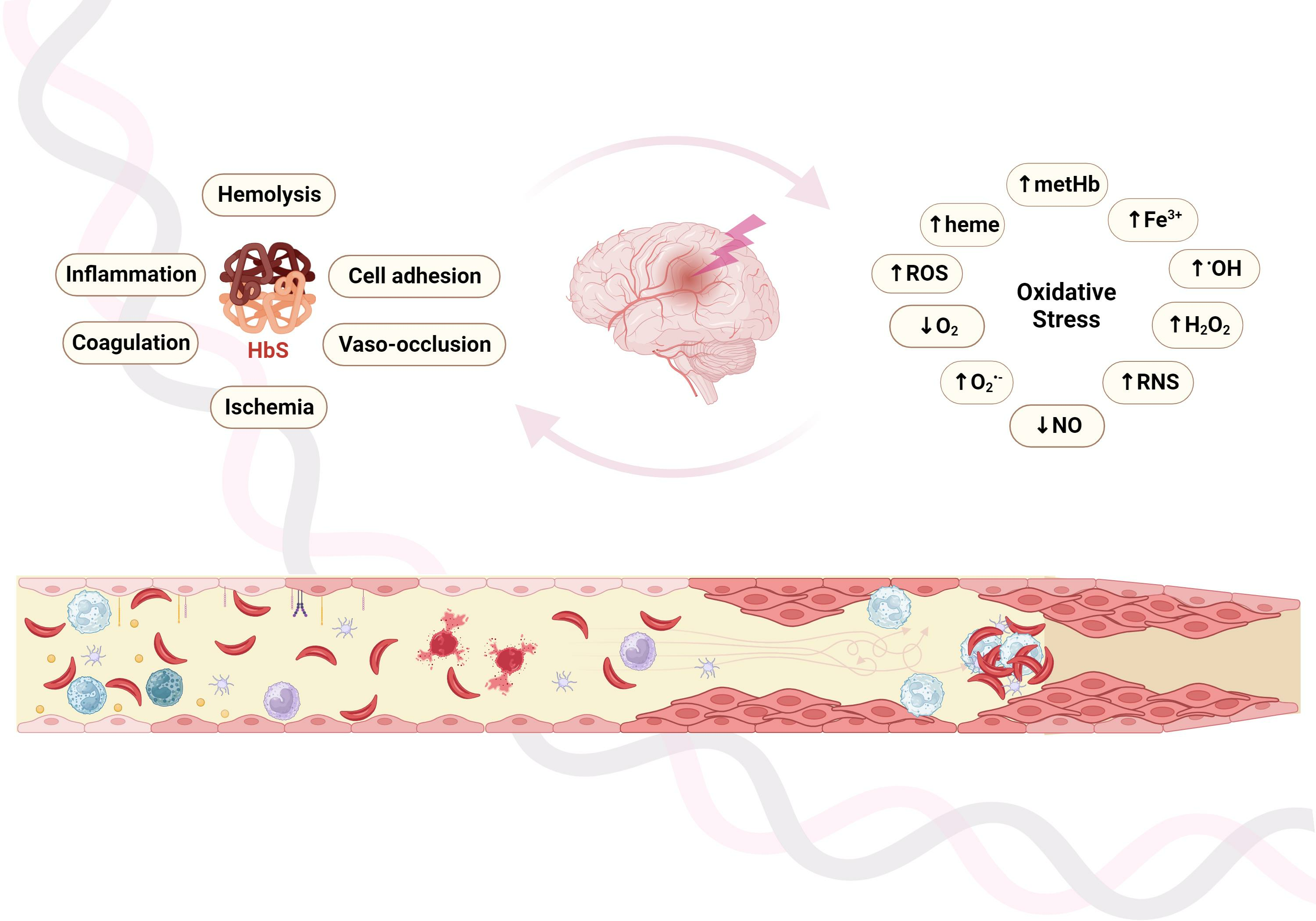
7. Genetic Modulation of Cerebrovascular Disease in SCA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Variant | Predicted Modulation | Reference |
|---|---|---|---|
| VCAM1 | G1238C | Stroke protection | [191] |
| VCAM1 | T1594C | Increased small-vessel stroke risk | [193] |
| VCAM1 | rs1409419_T Haplotype 7 | Increased stroke risk Increased stroke risk | [196] |
| NOS3 | intron 4_27 bp VNTR_4b Haplotype V Haplotype VII | Decreased SCI risk Decreased SCI risk Decreased CV risk | [196] |
| ITGA4 | rs113276800_CA rs3770138_T | Increased stroke risk Increased stroke risk Increased CV risk | [196] |
| IL4R | S503P | Increased large-vessel stroke risk | [193] |
| LDLR | Ncol +/− | Increased small-vessel stroke risk | [193] |
| ADRB2 | Q27E | Increased large-vessel stroke risk | [193] |
| AGT | GT repeats | Increased stroke risk | [190] |
| HLA | DRB1*0301 DRB1*0302 DQB1*0201 DRB1*1501 DQB1*0602 DPB1*0401 DPB1*1701 -A*0102 -A*2612 -A*3301 | Increased stroke risk Increased stroke risk Increased stroke risk Decreased stroke risk Decreased stroke risk Increased small-vessel stroke risk Increased small-vessel stroke risk Increased large-vessel stroke risk Increased large-vessel stroke risk Decreased large-vessel stroke k | [189,205] |
| TNF-α | -308G>A | Increased stroke risk | [193,195,206] |
| GOLGB1 | Y1212C | Decreased stroke risk | [198] |
| ENPP1 | K173Q | Decreased stroke risk Increased stroke risk Increased stroke risk | [198] [194] [199] |
| PON1 | Q192R | Increased stroke risk | [198] |
| HBA | -α3.7kb del | Decreased stroke risk | [195] |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Piel, F.B.; Tatem, A.J.; Huang, Z.; Gupta, S.; Williams, T.N.; Weatherall, D.J. Global Migration and the Changing Distribution of Sickle Haemoglobin: A Quantitative Study of Temporal Trends between 1960 and 2000. Lancet Glob. Health 2014, 2, e80–e89. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.J.; Lipowsky, H.H. Influence of Erythrocyte Aggregation on Leukocyte Margination in Postcapillary Expansions: A Lattice Boltzmann Analysis. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1460–H1471. [Google Scholar] [CrossRef] [PubMed]
- Debaun, M.R.; Kirkham, F.J. Central Nervous System Complications and Management in Sickle Cell Disease. Blood 2016, 127, 829–838. [Google Scholar]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle Cell Disease. Nat. Rev. Dis. Prim. 2018, 4, 18010. [Google Scholar] [CrossRef]
- Mandese, V.; Bigi, E.; Bruzzi, P.; Palazzi, G.; Predieri, B.; Lucaccioni, L.; Cellini, M.; Iughetti, L. Endocrine and Metabolic Complications in Children and Adolescents with Sickle Cell Disease: An Italian Cohort Study. BMC Pediatr. 2019, 19, 56. [Google Scholar] [CrossRef]
- Herrick, J.B. Peculiar Elongated and Sickle-Shaped Red Blood Corpuscles in a Case of Severe Anemia. Arch. Intern. Med. 1910, VI, 517–521. [Google Scholar] [CrossRef]
- Ingram, V.M. Gene Mutations in Human Haemoglobin: The Chemical Difference between Normal and Sickle Cell Haemoglobin. Nature 1957, 180, 326–328. [Google Scholar] [CrossRef]
- Dale, J.C.; Cochran, C.J.; Roy, L.; Jernigan, E.; Buchanan, G.R. Health-Related Quality of Life in Children and Adolescents with Sickle Cell Disease. J. Pediatr. Health Care 2011, 25, 208–215. [Google Scholar] [CrossRef]
- Kambasu, D.M.; Rujumba, J.; Lekuya, H.M.; Munube, D.; Mupere, E. Health-Related Quality of Life of Adolescents with Sickle Cell Disease in Sub-Saharan Africa: A Cross-Sectional Study. BMC Hematol. 2019, 19, 9. [Google Scholar] [CrossRef]
- Kato, G.J.; Hebbel, R.P.; Steinberg, M.H.; Gladwin, M.T. Vasculopathy in Sickle Cell Disease: Biology, Pathophysiology, Genetics, Translational Medicine and New Research Directions. Am. J. Hematol. 2009, 84, 618–625. [Google Scholar] [CrossRef]
- Voskou, S.; Aslan, M.; Fanis, P.; Phylactides, M.; Kleanthous, M. Oxidative Stress in β-Thalassaemia and Sickle Cell Disease. Redox Biol. 2015, 6, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Sposi, N.M.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants 2021, 10, 296. [Google Scholar] [CrossRef]
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle Cell Disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef] [PubMed]
- van Zwieten, R.; Verhoeven, A.J.; Roos, D. Inborn Defects in the Antioxidant Systems of Human Red Blood Cells. Free Radic. Biol. Med. 2014, 67, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Reiter, C.H.D.R.; Ang, X.U.W.; Antos, J.O.S.E.E.T.A.; Ogg, N.E.I.L.H.; Iii, R.I.O.C.A.; Chechter, A.L.A.N.N.S.; Ladwin, M.A.R.K.T.G. FreeHb Limits NO Availability in SCD. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Barabino, G.A.; Platt, M.O.; Kaul, D.K. Sickle Cell Biomechanics. Annu. Rev. Biomed. Eng. 2010, 12, 345–367. [Google Scholar] [CrossRef]
- Kaul, D.K.; Fabry, M.E.; Nagel, R.L. The Pathophysiology of Vascular Obstruction in the Sickle Syndromes. Blood Rev. 1996, 10, 29–44. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, C.; Manwani, D.; Frenette, P.S. Neutrophils, Platelets, and Inflammatory Pathways at the Nexus of Sickle Cell Disease Pathophysiology. Blood 2016, 127, 801–809. [Google Scholar] [CrossRef]
- Switzer, J.A.; Hess, D.C.; Nichols, F.T.; Adams, R.J. Pathophysiology and Treatment of Stroke in Sickle-Cell Disease: Present and Future. Lancet Neurol. 2006, 5, 501–512. [Google Scholar]
- Pries, A.R.; Kuebler, W.M. Normal Endothelium. In The Vascular Endothelium I. Handbook of Experimental Pharmacology; Moncada, S., Higgs, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 176/I. [Google Scholar] [CrossRef]
- Regan, E.R.; Aird, W.C. Dynamical Systems Approach to Endothelial Heterogeneity. Circ. Res. 2012, 111, 110–130. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The Vascular Endothelium and Human Diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef]
- Behrendt, D.; Ganz, P. Endothelial Function: From Vascular Biology to Clinical Aplications. Am. J. Cardiol. 2002, 90, 40L–48L. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Vanhoutte, P.M. Endothelium-Derived Relaxing and Contracting Factors. FASEB J. 1989, 3, 2007–2018. [Google Scholar] [CrossRef]
- Egashira, K.; Inou, T.; Hirooka, Y.; Kai, H.; Sugimachi, M.; Suzuki, S.; Kuga, T.; Urabe, Y.; Takeshita, A. Effects of Age on Endothelium-Dependent Vasodilation of Resistance Coronary Artery by Acetylcholine in Humans. Circulation 1993, 88, 77–81. [Google Scholar] [CrossRef]
- Egashira, K.; Inou, T.; Hirooka, Y.; Yamada, A.; Maruoka, Y.; Kai, H.; Sugimachi, M.; Suzuki, S.; Takeshita, A. Impaired Coronary Blood Flow Response to Acetylcholine in Patients with Coronary Risk Factors and Proximal Atherosclerotic Lesions. J. Clin. Investig. 1993, 91, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Zeiher, A.M.; Drexler, H.; Saurbier, B.; Just, H. Endothelium-Mediated Coronary Blood Flow Modulation in Humans: Effects of Age, Atherosclerosis, Hypercholesterolemia, and Hypertension. J. Clin. Investig. 1993, 92, 652–662. [Google Scholar] [CrossRef]
- Nabel, E.G.; Selwyn, A.P.; Ganz, P. Large Coronary Arteries in Humans Are Responsive to Changing Blood Flow: An Endothelium-Dependent Mechanism That Fails in Patients with Atherosclerosis. J. Am. Coll. Cardiol. 1990, 16, 349–356. [Google Scholar] [CrossRef]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Magagna, A.; Salvetti, A. Cyclooxygenase Inhibition Restores Nitric Oxide Activity in Essential Hypertension. Hypertension 1997, 29, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Félétou, M.; Boulanger, C.M.; Wu, H.F.; Levens, N.; Zhang, J.N.; Vanhoutte, P.M. Oxygen-Derived Free Radicals Mediate Endothelium-Dependent Contractions to Acetylcholine in Aortas from Spontaneously Hypertensive Rats. Br. J. Pharmacol. 2002, 136, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Belhassen, L.; Pelle, G.; Dubois-Rande, J.L.; Adnot, S. Improved Endothelial Function by the Thromboxane A2 Receptor Antagonist S 18886 in Patients with Coronary Artery Disease Treated with Aspirin. J. Am. Coll. Cardiol. 2003, 41, 1198–1204. [Google Scholar] [CrossRef]
- Flammer, A.J.; Lüscher, T.F. Human Endothelial Dysfunction: EDRFs. Pflüg. Arch. Eur. J. Physiol. 2010, 459, 1005–1013. [Google Scholar] [CrossRef]
- Porcu, P.; Emanueli, C.; Kapatsoris, M.; Chao, J.; Chao, L.; Madeddu, P. Reversal of Angiogenic Growth Factor Upregulation by Revascularization of Lower Limb Ischemia. Circulation 2002, 105, 67–72. [Google Scholar] [CrossRef]
- Tsui, J.C.S.; Baker, D.M.; Biecker, E.; Shaw, S.; Dashwood, M.R. Potential Role of Endothelin 1 in Ischaemia-Induced Angiogenesis in Critical Leg Ischaemia. Br. J. Surg. 2002, 89, 741–747. [Google Scholar] [CrossRef]
- Zetter, B.R. The Endothelial Cells of Large and Small Blood Vessels. Diabetes 1981, 30, 24–28. [Google Scholar] [CrossRef]
- Aird, W.C. Endothelial Cell Heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef] [PubMed]
- Risau, W. Differentiation of Endothelium. FASEB J. 1995, 9, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelium in Health and Disease. Pharmacol. Rep. 2008, 60, 139–143. [Google Scholar]
- Marui, N.; Offermann, M.K.; Swerlick, R.; Kunsch, C.; Rosen, C.A.; Ahmad, M.; Wayne Alexander, R.; Medford, R.M.; Kunsck, C.; Rosen, C.A.; et al. Vascular Cell Adhesion Molecule-1 (VCAM-1) Gene Transcription and Expression Are Regulated through an Antioxidant-Sensitive Mechanism in Human Vascular Endothelial Cells. J. Clin. Investig. 1993, 92, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Lanaro, C.; Franco-Penteado, C.F.; Albuqueque, D.M.; Saad, S.T.O.; Conran, N.; Costa, F.F. Altered Levels of Cytokines and Inflammatory Mediators in Plasma and Leukocytes of Sickle Cell Anemia Patients and Effects of Hydroxyurea Therapy. J. Leukoc. Biol. 2009, 85, 235–242. [Google Scholar] [CrossRef]
- Radke, R.M.; Diller, G.P.; Duck, M.; Orwat, S.; Hartmann, D.; Thum, T.; Baumgartner, H. Endothelial Function in Contemporary Patients with Repaired Coarctation of Aorta. Heart 2014, 100, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Solovey, A.A.; Solovey, A.N.; Harkness, J.; Hebbel, R.P. Modulation of Endothelial Cell Activation in Sickle Cell Disease: A Pilot Study. Blood 2001, 97, 1937–1941. [Google Scholar] [CrossRef]
- Sakamoto, T.M.; Lanaro, C.; Ozelo, M.C.; Garrido, V.T.; Olalla-Saad, S.T.; Conran, N.; Costa, F.F. Increased Adhesive and Inflammatory Properties in Blood Outgrowth Endothelial Cells from Sickle Cell Anemia Patients. Microvasc. Res. 2013, 90, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Mitchell, J.B. Chemical Biology of Nitric Oxide: Insights into Regulatory, Cytotoxic, and Cytoprotective Mechanisms of Nitric Oxide. Free Radic. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric Oxide Activates Guanylate Cyclase and Increases Guanosine 3 ’: 5 ’ -Cyclic Monophosphate Levels in Various Tissue Preparations Biochemistry. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Byrns, R.E.; Buga, G.M.; Wood, K.S. Endothelium-Derived Relaxing Factor From Pulmonary Artery and Vein Possesses Pharmacologic and Chemical Properties Identical to Those of Nitric Oxide Radical. Circ. Res. 1987, 61, 866–879. [Google Scholar] [CrossRef]
- Kato, G.J.; Martyr, S.; Blackwelder, W.C.; Nichols, J.S.; Coles, W.A.; Hunter, L.A.; Brennan, M.L.; Hazen, S.L.; Gladwin, M.T. Levels of Soluble Endothelium-Derived Adhesion Molecules in Patients with Sickle Cell Disease Are Associated with Pulmonary Hypertension, Organ Dysfunction, and Mortality. Br. J. Haematol. 2005, 130, 943–953. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric Oxide Synthase Isozymes Antibodies. Hypertension 1994, 23, 1121–1131. [Google Scholar] [CrossRef]
- Hemmens, B.; Mayer, B. Enzymology of Nitric Oxide Synthases. In Nitric Oxide Protocols; Titheradge, M.A., Ed.; Humana Press: Totowa, NJ, USA, 1998; pp. 1–32. ISBN 978-1-59259-749-9. [Google Scholar]
- García-Cardeña, G.; Fan, R.; Shah, V.; Sorrentino, R.; Cirino, G.; Papapetropoulos, A.; Sessa, W.C. Dynamic Activation of Endothelial Nitric Oxide Synthase by Hsp90. Nature 1998, 392, 821–824. [Google Scholar] [CrossRef]
- Pritchard, K.A.; Ackerman, A.W.; Gross, E.R.; Stepp, D.W.; Shi, Y.; Fontana, J.T.; Baker, J.E.; Sessa, W.C. Heat Shock Protein 90 Mediates the Balance of Nitric Oxide and Superoxide Anion from Endothelial Nitric-Oxide Synthase. J. Biol. Chem. 2001, 276, 17621–17624. [Google Scholar] [CrossRef]
- Song, Y.; Cardounel, A.J.; Zweier, J.L.; Xia, Y. Inhibition of Superoxide Generation from Neuronal Nitric Oxide Synthase by Heat Shock Protein 90: Implications in NOS Regulation. Biochemistry 2002, 41, 10616–10622. [Google Scholar] [CrossRef]
- Sowa, G.; Pypaert, M.; Sessa, W.C. Distinction between Signaling Mechanisms in Lipid Rafts vs. Caveolae. Proc. Natl. Acad. Sci. USA 2001, 98, 14072–14077. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, R.M.; Draznin, M.B.; Murad, F. Endothelium-Dependent Relaxation in Rat Aorta May Be Mediated through Cyclic GMP-Dependent Protein Phosphorylation. Nature 1983, 306, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Harbison, R.G.; Wood, K.S.; Kadowitz, P.J. Activation of Purified Soluble Guanylate Cyclase by Endothelium-Derived Relaxing Factor from Intrapulmonary Artery and Vein: Stimulation by Acetylcholine, Bradykinin and Arachidonic Acid. J. Pharmacol. Exp. Ther. 1986, 237, 893–900. [Google Scholar]
- Arndt, H.; Smith, C.W.; Granger, D.N. Leukocyte-Endothelial Cell Adhesion in Spontaneously Hypertensive and Normotensive Rats. Hypertension 1993, 21, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Zeiher, A.M. Nitric Oxide—An Endothelial Cell Survival Factor. Cell Death Differ. 1999, 6, 964–968. [Google Scholar] [CrossRef]
- Nunokawa, Y.; Tanaka, S. Interferon-γ Inhibits Proliferation of Rat Vascular Smooth Muscle Cells by Nitric Oxide Generation. Biochem. Biophys. Res. Commun. 1992, 188, 409–415. [Google Scholar] [CrossRef]
- Hogan, M.; Cerami, A.; Bucala, R. Advanced Glycosylation Endproducts Block the Antiproliferative Effect of Nitric Oxide: Role in the Vascular and Renal Complications of Diabetes Mellitus. J. Clin. Investig. 1992, 90, 1110–1115. [Google Scholar] [CrossRef]
- Murohara, T.; Asahara, T.; Silver, M.; Bauters, C.; Masuda, H.; Kalka, C.; Kearney, M.; Chen, D.; Chen, D.; Symes, J.F.; et al. Nitric Oxide Synthase Modulates Angiogenesis in Response to Tissue Ischemia. J. Clin. Investig. 1998, 101, 2567–2578. [Google Scholar] [CrossRef]
- Xia, N.; Förstermann, U.; Li, H. Resveratrol and Endothelial Nitric Oxide. Molecules 2014, 19, 16102–16121. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Vivar, J.; Kalyanaraman, B.; Martásek, P.; Hogg, N.; Masters, B.S.S.; Karoui, H.; Tordo, P.; Pritchard, K.A. Superoxide Generation by Endothelial Nitric Oxide Synthase: The Influence of Cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef]
- Xu, W.; Kaneko, F.T.; Zheng, S.; Comhair, S.A.A.; Janocha, A.J.; Goggans, T.; Thunnissen, F.B.J.M.; Farver, C.; Hazen, S.L.; Jennings, C.; et al. Increased Arginase II and Decreased NO Synthesis in Endothelial Cells of Patients with Pulmonary Arterial Hypertension. FASEB J. 2004, 18, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Shirodaria, C.; Leeson, P.; Antonopoulos, A.; Warrick, N.; Van-Assche, T.; Cunnington, C.; Tousoulis, D.; Pillai, R.; Ratnatunga, C.; et al. Association of Plasma Asymmetrical Dimethylarginine (ADMA) with Elevated Vascular Superoxide Production and Endothelial Nitric Oxide Synthase Uncoupling: Implications for Endothelial Function in Human Atherosclerosis. Eur. Heart J. 2009, 30, 1142–1150. [Google Scholar] [CrossRef]
- Chen, C.-A.; Wang, T.-Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.H.; Chen, Y.-R.; Druhan, L.J.; Zweier, J.L. S-Glutathionylation Uncouples ENOS and Regulates Its Cellular and Vascular Function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef]
- Zweier, J.L.; Chen, C.-A.; Druhan, L.J. S-Glutathionylation Reshapes Our Understanding of Endothelial Nitric Oxide Synthase Uncoupling and Nitric Oxide/Reactive Oxygen Species-Mediated Signaling. Antioxid. Redox Signal. 2011, 14, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Sachdev, V.; Jison, M.L.; Shizukuda, Y.; Plehn, J.F.; Minter, K.; Brown, B.; Coles, W.A.; Nichols, J.S.; Ernzt, I.; et al. Pulmonary Hypertension as a Risk Factor for Death in Patients with Sickle Cell Anemia. N. Engl. J. Med. 2004, 350, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Mattei, P.; Salvetti, A. Vasodilation to Acetylcholine in Primary and Secondary Forms of Human Hypertension. Hypertension 1993, 21, 929–933. [Google Scholar] [CrossRef]
- Hebbel, R.P.; Yamada, O.; Moldow, C.F.; Jacob, H.S.; White, J.G.; Eaton, J.W. Abnormal Adherence of Sickle Erythrocytes to Cultured Vascular Endothelium. Possible Mechanism for Microvascular Occlusion in Sickle Cell Disease. J. Clin. Investig. 1980, 65, 154–160. [Google Scholar] [CrossRef]
- Nath, K.A.; Shah, V.; Haggard, J.J.; Croatt, A.J.; Smith, L.A.; Hebbel, R.P.; Katusic, Z.S. Mechanisms of Vascular Instability in a Transgenic Mouse Model of Sickle Cell Disease. Am. J. Physiol.—Regul. Integr. Comp. Physiol. 2000, 279, 1949–1955. [Google Scholar] [CrossRef]
- Barone, F.C.; Feuerstein, G.Z. Inflammatory Mediators and Stroke: New Opportunities for Novel Therapeutics. J. Cereb. Blood Flow Metab. 1999, 19, 819–834. [Google Scholar] [CrossRef]
- del Zoppo, G.J.; Poeck, K.; Pessin, M.S.; Wolpert, S.M.; Furlan, A.J.; Ferbert, A.; Alberts, M.J.; Zivin, J.A.; Wechsler, L.; Busse, O. Recombinant Tissue Plasminogen Activator in Acute Thrombotic and Embolic Stroke. Ann. Neurol. 1992, 32, 78–86. [Google Scholar] [CrossRef]
- Kaul, D.K.; Finnegan, E.; Barabino, G.A. Sickle Red Cell-Endothelium Interactions. Microcirculation 2009, 16, 97–111. [Google Scholar] [CrossRef]
- Sultana, C.; Shen, Y.; Rattan, V.; Johnson, C.; Kalra, V.K. Interaction of Sickle Erythrocytes with Endothelial Cells in the Presence of Endothelial Cell Conditioned Medium Induces Oxidant Stress Leading to Transendothelial Migration of Monocytes. Blood 1998, 92, 3924–3935. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Conran, N.; Saad, S.T.O.; Costa, F.F.; Ikuta, T. Leukocyte Numbers Correlate with Plasma Levels of Granulocyte-Macrophage Colony-Stimulating Factor in Sickle Cell Disease. Ann. Hematol. 2007, 86, 255–261. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme Triggers TLR4 Signaling Leading to Endothelial Cell Activation and Vaso-Occlusion in Murine Sickle Cell Disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Schaer, C.A.; Deuel, J.W.; Bittermann, A.G.; Rubio, I.G.; Schoedon, G.; Spahn, D.R.; Wepf, R.A.; Vallelian, F.; Schaer, D.J. Mechanisms of Haptoglobin Protection against Hemoglobin Peroxidation Triggered Endothelial Damage. Cell Death Differ. 2013, 20, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Singla, S.; Sysol, J.R.; Dille, B.; Jones, N.; Chen, J.; Machado, R.F. Hemin Causes Lung Microvascular Endothelial Barrier Dysfunction by Necroptotic Cell Death. Am. J. Respir. Cell Mol. Biol. 2017, 57, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-Induced Neutrophil Extracellular Traps Contribute to the Pathogenesis of Sickle Cell Disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef]
- Ghosh, S.; Adisa, O.A.; Chappa, P.; Tan, F.; Jackson, K.A.; Archer, D.R.; Ofori-Acquah, S.F. Extracellular Hemin Crisis Triggers Acute Chest Syndrome in Sickle Mice. J. Clin. Investig. 2013, 123, 4809–4820. [Google Scholar] [CrossRef] [PubMed]
- Allali, S.; Rignault-Bricard, R.; de Montalembert, M.; Taylor, M.; Bouceba, T.; Hermine, O.; Maciel, T.T. HbS Promotes TLR4-Mediated Monocyte Activation and Proinflammatory Cytokine Production in Sickle Cell Disease. Blood 2022, 140, 1972–1982. [Google Scholar] [CrossRef]
- Kato, G.J.; McGowan, V.; Machado, R.F.; Little, J.A.; Taylor VI, J.; Morris, C.R.; Nichols, J.S.; Wang, X.; Poljakovic, M.; Morris, S.M.; et al. Lactate Dehydrogenase as a Biomarker of Hemolysis-Associated Nitric Oxide Resistance, Priapism, Leg Ulceration, Pulmonary Hypertension, and Death in Patients with Sickle Cell Disease. Blood 2006, 107, 2279–2285. [Google Scholar] [CrossRef]
- Kato, G.J. Anemia, Age, Desaturation and Impaired Neurocognition in Sickle Cell Anemia. Pediatr. Blood Cancer 2012, 59, 773–774. [Google Scholar] [CrossRef] [PubMed]
- Zorca, S.; Freeman, L.; Hildesheim, M.; Allen, D.; Remaley, A.T.; Taylor VI, J.G.; Kato, G.J. Lipid Levels in Sickle-Cell Disease Associated with Haemolytic Severity, Vascular Dysfunction and Pulmonary Hypertension. Br. J. Haematol. 2010, 149, 436–445. [Google Scholar] [CrossRef]
- O’Driscoll, S.; Height, S.E.; Dick, M.C.; Rees, D.C. Serum Lactate Dehydrogenase Activity as a Biomarker in Children with Sickle Cell Disease. Br. J. Haematol. 2008, 140, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Mecabo, G.; Yamamoto, M.; Biassi, T.P.; Figueiredo, M.S. Lactate Dehydrogenase Isoenzyme 3 and Hemolysis in Sickle Cell Anemia: A Possible Correlation. Blood 2015, 125, 3821–3822. [Google Scholar] [CrossRef]
- Kato, G.J.; Gladwin, M.T.; Steinberg, M.H. Deconstructing Sickle Cell Disease: Reappraisal of the Role of Hemolysis in the Development of Clinical Subphenotypes. Blood Rev. 2007, 21, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Pecker, L.H.; Ackerman, H.C. Cardiovascular Adaptations to Anemia and the Vascular Endothelium in Sickle Cell Disease Pathophysiology. In Sickle Cell Anemia: From Basic Science to Clinical Practice; Costa, F.F., Conran, N., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 129–175. ISBN 978-3-319-06713-1. [Google Scholar]
- Eltzschig, H.K.; Eckle, T. Ischemia and Reperfusion—From Mechanism to Translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Park, S.W.; Kim, M.; Brown, K.M.; D’Agati, V.D.; Lee, H.T. Paneth Cell-Derived IL-17A Causes Multi-Organ Dysfunction after Hepatic Ischemia and Reperfusion Injury. Hepatology 2011, 53, 1662–1675. [Google Scholar] [CrossRef]
- Osarogiagbon, U.R.; Choong, S.; Belcher, J.D.; Vercellotti, G.M.; Paller, M.S.; Hebbel, R.P. Reperfusion Injury Pathophysiology in Sickle Transgenic Mice. Blood 2000, 96, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P. I Ischemia-reperfusion Injury in Sickle Cell Anemia: Relationship to Acute Chest Syndrome, Endothelial Dysfunction, Arterial Vasculopathy, and Inflammatory Pain. Hematol. Oncol. Clin. N. Am. 2014, 28, 181–198. [Google Scholar] [CrossRef]
- Banda, M.A.; Lefer, D.J.; Granger, D.N. Postischemic Endothelium-Dependent Vascular Reactivity Is Preserved in Adhesion Molecule-Deficient Mice. Am. J. Physiol.—Heart Circ. Physiol. 1997, 273, 2721–2725. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G. Cellular and Molecular Mechanisms of Endothelial Cell Dysfunction. J. Clin. Investig. 1997, 100, 2153–2157. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.R. Reperfusion-Induced Changes in Capillary Perfusion and Filtration: Effects of Hypercholesterolemia. Am. J. Physiol. 1999, 277, H669–H675. [Google Scholar] [CrossRef]
- Kurose, I.; Anderson, D.C.; Miyasaka, M.; Tamatani, T.; Paulson, J.C.; Todd, R.F.; Rusche, J.R.; Granger, D.N. Molecular Determinants of Reperfusion-Induced Leukocyte Adhesion and Vascular Protein Leakage. Circ. Res. 1994, 74, 336–343. [Google Scholar] [CrossRef]
- Kurose, I.; Argenbright, L.W.; Wolf, R.; Lianxi, L.; Granger, D.N. Ischemia/Reperfusion-Induced Microvascular Dysfunction: Role of Oxidants and Lipid Mediators. Am. J. Physiol. 1997, 272, H2976–H2982. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell Death in Disease: Mechanisms and Emerging Therapeutic Concepts. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef]
- Ou, J.; Ou, Z.; Jones, D.W.; Holzhauer, S.; Hatoum, O.A.; Ackerman, A.W.; Weihrauch, D.W.; Gutterman, D.D.; Guice, K.; Oldham, K.T.; et al. L-4F, an Apolipoprotein A-1 Mimetic, Dramatically Improves Vasodilation in Hypercholesterolemia and Sickle Cell Disease. Circulation 2003, 107, 2337–2341. [Google Scholar] [CrossRef]
- Pritchard, K.A.; Ou, J.; Ou, Z.; Shi, Y.; Franciosi, J.P.; Signorino, P.; Kaul, S.; Ackland-Berglund, C.; Witte, K.; Holzhauer, S.; et al. Hypoxia-Induced Acute Lung Injury in Murine Models of Sickle Cell Disease. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2004, 286, 1–4. [Google Scholar] [CrossRef]
- Lowenstein, C.J.; Morrell, C.N.; Yamakuchi, M. Regulation of Weibel-Palade Body Exocytosis. Trends Cardiovasc. Med. 2005, 15, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Aslan, M.; Freeman, B.A. Redox-Dependent Impairment of Vascular Function in Sickle Cell Disease. Free Radic. Biol. Med. 2007, 43, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Pradhan-Sundd, T.; Pritchard, K.A.; Hillery, C.A. Redox Signaling in Sickle Cell Disease. Curr. Opin. Physiol. 2019, 9, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, R.; Van Solinge, W.W. The Energy-Less Red Blood Cell Is Lost: Erythrocyte Enzyme Abnormalities of Glycolysis. Blood 2005, 106, 4034–4042. [Google Scholar] [CrossRef]
- Mairbäurl, H.; Weber, R.E. Oxygen Transport by Hemoglobin. Compr. Physiol. 2012, 2, 1463–1489. [Google Scholar]
- Camus, S.M.; Gausserès, B.; Bonnin, P.; Loufrani, L.; Grimaud, L.; Charue, D.; De Moraes, J.A.; Renard, J.M.; Tedgui, A.; Boulanger, C.M.; et al. Erythrocyte Microparticles Can Induce Kidney Vaso-Occlusions in a Murine Model of Sickle Cell Disease. Blood 2012, 120, 5050–5058. [Google Scholar] [CrossRef]
- Wesseling, M.C.; Wagner-Britz, L.; Nguyen, D.B.; Asanidze, S.; Mutua, J.; Mohamed, N.; Hanf, B.; Ghashghaeinia, M.; Kaestner, L.; Bernhardt, I. Novel Insights in the Regulation of Phosphatidylserine Exposure in Human Red Blood Cells. Cell. Physiol. Biochem. 2016, 39, 1941–1954. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.; Rees, D.C.; Gibson, J.S. Role of Calcium in Phosphatidylserine Externalisation in Red Blood Cells from Sickle Cell Patients. Anemia 2011, 2011, 379894. [Google Scholar] [CrossRef]
- Kaestner, L.; Minetti, G. The Potential of Erythrocytes as Cellular Aging Models. Cell Death Differ. 2017, 24, 1475–1477. [Google Scholar] [CrossRef]
- Setty, Y.B.N.; Kulkarni, S.; Stuart, M.J. Role of Erythrocyte Phosphatidylserine in Sickle Red Cell-Endothelial Adhesion. Blood 2002, 99, 1564–1571. [Google Scholar] [CrossRef]
- Ghosh, S.; Flage, B.; Weidert, F.; Ofori-Acquah, S.F. P-Selectin Plays a Role in Haem-Induced Acute Lung Injury in Sickle Mice. Br. J. Haematol. 2019, 186, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular Hemolysis and the Pathophysiology of Sickle Cell Disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Vercellotti, G.M.; Zhang, P.; Nguyen, J.; Abdulla, F.; Chen, C.; Nguyen, P.; Nowotny, C.; Steer, C.J.; Smith, A.; Belcher, J.D. Hepatic Overexpression of Hemopexin Inhibits Inflammation and Vascular Stasis in Murine Models of Sickle Cell Disease. Mol. Med. 2016, 22, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hazra, R.; Ihunnah, C.A.; Weidert, F.; Flage, B.; Ofori-Acquah, S.F. Augmented NRF2 Activation Protects Adult Sickle Mice from Lethal Acute Chest Syndrome. Br. J. Haematol. 2018, 182, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhu, X.; Ward, C.M.; Starlard-Davenport, A.; Takezaki, M.; Berry, A.; Ward, A.; Wilder, C.; Neunert, C.; Kutlar, A.; et al. MIR-144-Mediated NRF2 Gene Silencing Inhibits Fetal Hemoglobin Expression in Sickle Cell Disease. Exp. Hematol. 2019, 70, 85–96. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Abdulla, F.; Zhang, P.; Nguyen, H.; Nguyen, P.; Killeen, T.; Miescher, S.M.; Brinkman, N.; et al. Haptoglobin and Hemopexin Inhibit Vaso-Occlusion and Inflammation in Murine Sickle Cell Disease: Role of Heme Oxygenase-1 Induction. PLoS ONE 2018, 13, e0196455. [Google Scholar] [CrossRef]
- Belcher, J.D.; Gomperts, E.; Nguyen, J.; Chen, C.; Abdulla, F.; Kiser, Z.M.; Gallo, D.; Levy, H.; Otterbein, L.E.; Vercellotti, G.M. Oral Carbon Monoxide Therapy in Murine Sickle Cell Disease: Beneficial Effects on Vasoocclusion, Inflammation and Anemia. PLoS ONE 2018, 13, e0205194. [Google Scholar] [CrossRef]
- Xu, H.; Wandersee, N.J.; Guo, Y.H.; Jones, D.W.; Holzhauer, S.L.; Hanson, M.S.; Machogu, E.; Brousseau, D.C.; Hogg, N.; Densmore, J.C.; et al. Sickle Cell Disease Increases High Mobility Group Box 1: A Novel Mechanism of Inflammation. Blood 2014, 124, 3978–3981. [Google Scholar] [CrossRef]
- Bennewitz, M.F.; Tutuncuoglu, E.; Gudapati, S.; Brzoska, T.; Watkins, S.C.; Monga, S.P.; Pradhan-Sundd, T.; Sundd, P. P-Selectin-Deficient Mice to Study Pathophysiology of Sickle Cell Disease. Blood Adv. 2020, 4, 266–273. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, H.; Weihrauch, D.; Jones, D.W.; Jing, X.; Shi, Y.; Gourlay, D.; Oldham, K.T.; Hillery, C.A.; Pritchard, K.A. Inhibition of Myeloperoxidase Decreases Vascular Oxidative Stress and Increases Vasodilatation in Sickle Cell Disease Mice. J. Lipid Res. 2013, 54, 3009–3015. [Google Scholar] [CrossRef]
- Biswal, S.; Rizwan, H.; Pal, S.; Sabnam, S.; Parida, P.; Pal, A. Oxidative Stress, Antioxidant Capacity, Biomolecule Damage, and Inflammation Symptoms of Sickle Cell Disease in Children. Hematology 2019, 24, 1–9. [Google Scholar] [CrossRef]
- Castilhos, L.G.; de Oliveira, J.S.; Adefegha, S.A.; Magni, L.P.; Doleski, P.H.; Abdalla, F.H.; de Andrade, C.M.; Leal, D.B.R. Increased Oxidative Stress Alters Nucleosides Metabolite Levels in Sickle Cell Anemia. Redox Rep. 2017, 22, 451–459. [Google Scholar] [CrossRef]
- Turhan, A.; Weiss, L.A.; Mohandas, N.; Coller, B.S.; Frenette, P.S. Primary Role for Adherent Leukocytes in Sickle Cell Vascular Occlusion: A New Paradigm. Proc. Natl. Acad. Sci. USA 2002, 99, 3047–3051. [Google Scholar] [CrossRef]
- Kassim, A.A.; DeBaun, M.R. Sickle Cell Disease, Vasculopathy, and Therapeutics. Annu. Rev. Med. 2013, 64, 451–466. [Google Scholar] [CrossRef]
- Ohene-Frempong, K.; Weiner, S.J.; Sleeper, L.A.; Miller, S.T.; Embury, S.; Moohr, J.W.; Wethers, D.L.; Pegelow, C.H.; Gill, F.M. Cerebrovascular Accidents in Sickle Cell Disease: Rates and Risk Factors. Blood 1998, 91, 288–294. [Google Scholar] [PubMed]
- Nickel, R.S.; Hsu, L.L. Clinical Manifestations of Sickle Cell Anemia: Infants and Children. In Sickle Cell Anemia: From Basic Science to Clinical Practice; Costa, F.F., Conran, N., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 213–229. ISBN 978-3-319-06713-1. [Google Scholar]
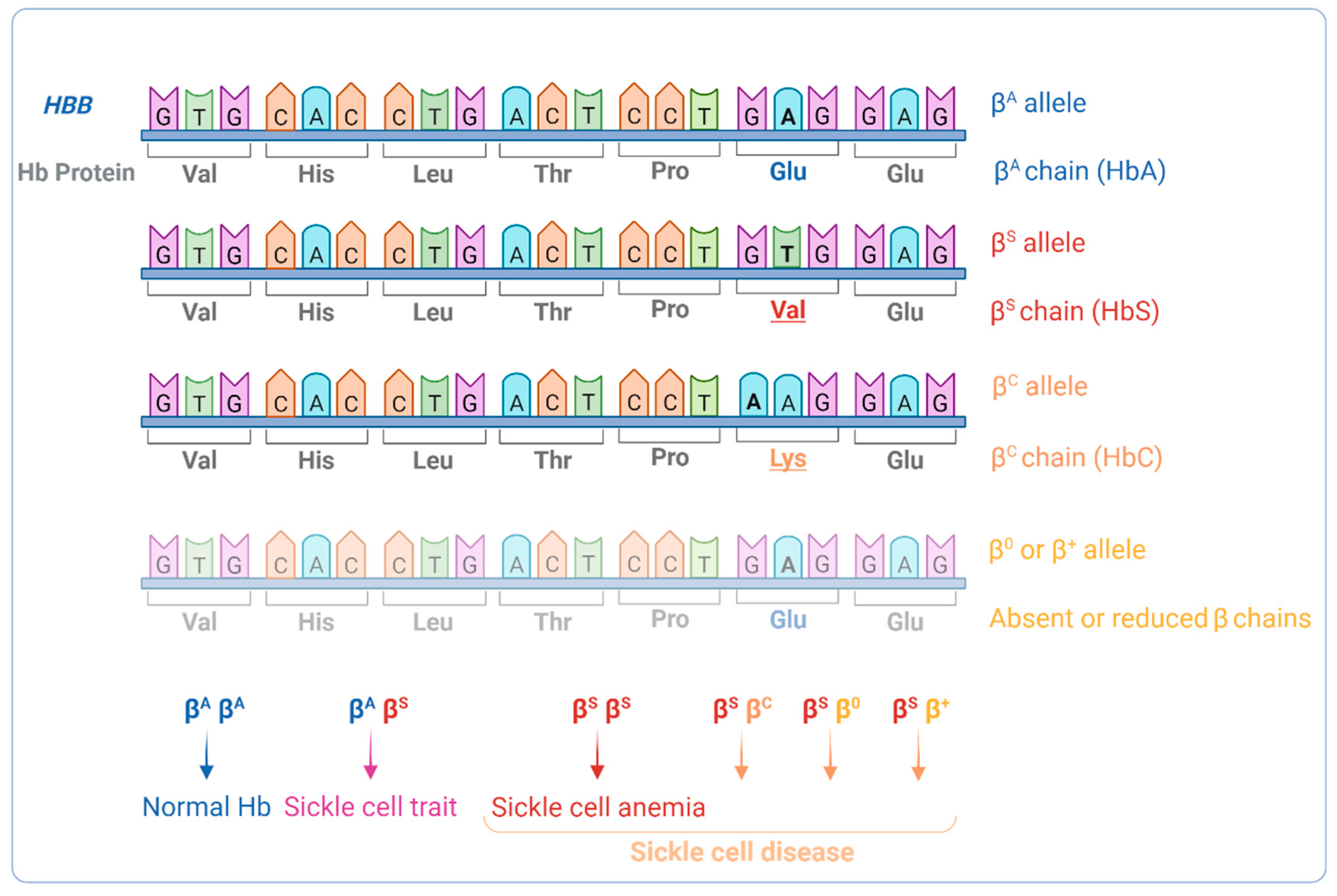
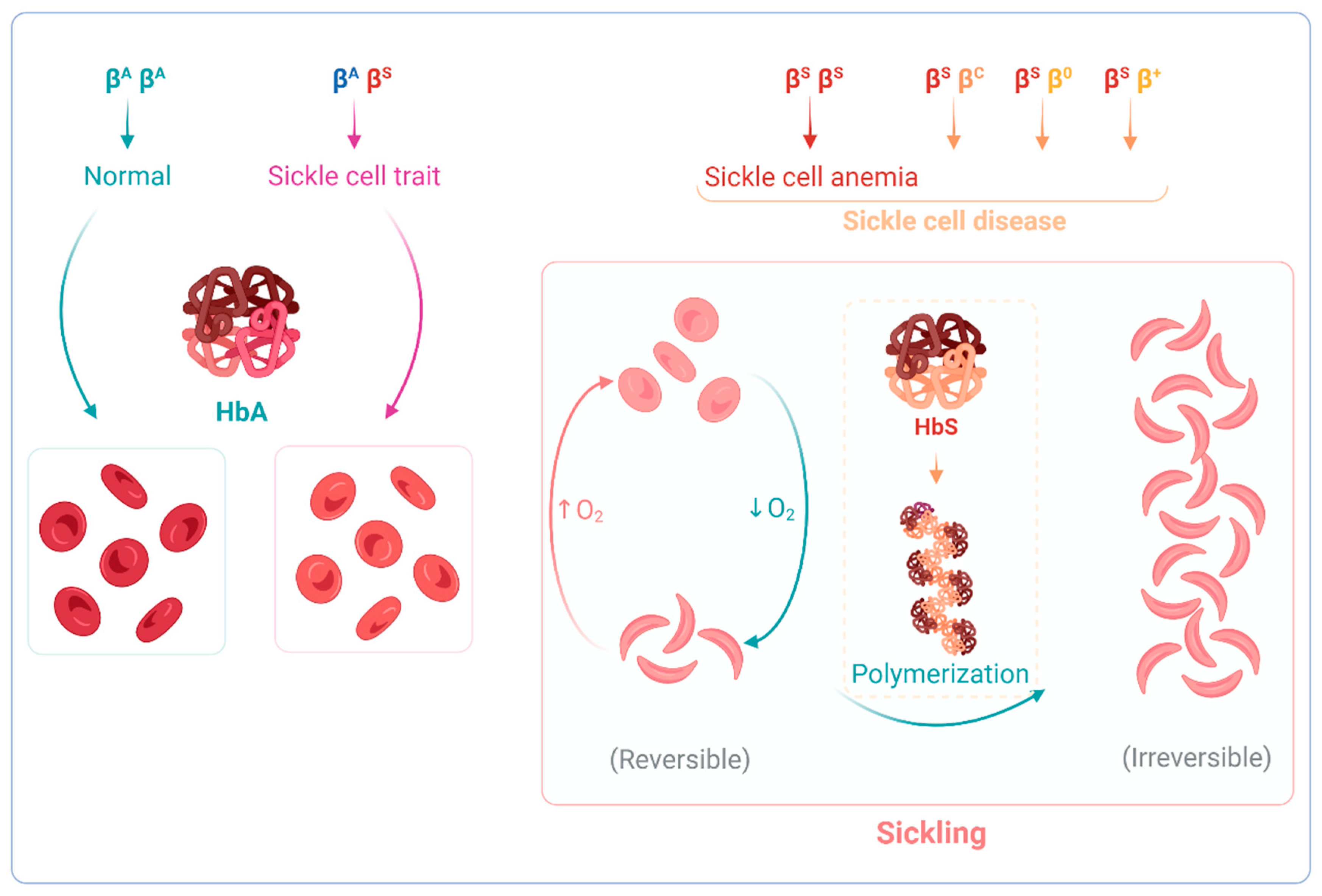
- Steinberg, M.H. Genetic Etiologies for Phenotypic Diversity in Sickle Cell Anemia. Sci. World J. 2009, 9, 46–67. [Google Scholar] [CrossRef]
- Schatz, J.; White, D.A.; Moinuddin, A.; Armstrong, M.; DeBaun, M.R. Lesion Burden and Cognitive Morbidity in Children with Sickle Cell Disease. J. Child Neurol. 2002, 17, 891–895. [Google Scholar] [CrossRef]
- Armstrong, F.D.; Thompson, R.J.; Wang, W.; Zimmerman, R.; Pegelow, C.H.; Miller, S.; Moser, F.; Bello, J.; Hurtig, A. Cognitive Functioning and Brain Magnetic Resonance Imaging in Children With Sickle Cell Disease. Pediatrics 1996, 97, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, C. Defining Stroke Risk in Children with Sickle Cell Anaemia. Br. J. Haematol. 2004, 128, 751–766. [Google Scholar] [CrossRef]
- Powars, D.; Wilson, B.; Imbus, C.; Pegelow, C.; Allen, J. The Natural History of Stroke in Sickle Cell Disease. Am. J. Med. 1978, 65, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Powars, D.R.; Chan, L.S.; Hiti, A.; Ramicone, E.; Johnson, C. Outcome of Sickle Cell Anemia: A 4-Decade Observational Study of 1056 Patients. Medicine 2005, 84, 363–376. [Google Scholar] [CrossRef] [PubMed]
- DeBaun, M.R.; Sarnaik, S.A.; Rodeghier, M.J.; Minniti, C.P.; Howard, T.H.; Iyer, R.V.; Inusa, B.; Telfer, P.T.; Kirby-Allen, M.; Quinn, C.T.; et al. Associated Risk Factors for Silent Cerebral Infarcts in Sickle Cell Anemia: Low Baseline Hemoglobin, Sex, and Relative High Systolic Blood Pressure. Blood 2012, 119, 3684–3690. [Google Scholar] [CrossRef]
- Moser, F.G.; Miller, S.T.; Bello, J.A.; Pegelow, C.H.; Zimmerman, R.A.; Wang, W.C.; Ohene-Frempong, K.; Schwartz, A.; Vichinsky, E.P.; Gallagher, D.; et al. The Spectrum of Brain MR Abnormalities in Sickle-Cell Disease: A Report from the Cooperative Study of Sickle Cell Disease. Am. J. Neuroradiol. 1996, 17, 965–972. [Google Scholar]
- Pegelow, C.H.; Macklin, E.A.; Moser, F.G.; Wang, W.C.; Bello, J.A.; Miller, S.T.; Vichinsky, E.P.; DeBaun, M.R.; Guarini, L.; Zimmerman, R.A.; et al. Longitudinal Changes in Brain Magnetic Resonance Imaging Findings in Children with Sickle Cell Disease. Blood 2002, 99, 3014–3018. [Google Scholar] [CrossRef]
- Steen, R.G.; Hankins, G.M.; Xiong, X.; Wang, W.C.; Beil, K.; Langston, J.W.; Helton, K.J. Prospective Brain Imaging Evaluation of Children with Sickle Cell Trait: Initial Observations. Radiology 2003, 228, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.J.; Nichols, F.T.; Figueroa, R.; McKie, V.; Lott, T. Transcranial Doppler Correlation with Cerebral Angiography in Sickle Cell Disease. Stroke 1992, 23, 1073–1077. [Google Scholar] [CrossRef]
- Deane, C.R.; Goss, D.; Bartram, J.; Pohl, K.R.E.; Height, S.E.; Sibtain, N.; Jarosz, J.; Thein, S.L.; Rees, D.C. Extracranial Internal Carotid Arterial Disease in Children with Sickle Cell Anemia. Haematologica 2010, 95, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Stockman, J.A.; Nigro, M.A.; Mishkin, M.M.; Oski, F.A. Occlusion of Large Cerebral Vessels in Sickle-Cell Anemia. N. Engl. J. Med. 1972, 287, 846–849. [Google Scholar] [CrossRef]
- Russell, M.O.; Goldberg, H.I.; Reis, L.; Friedman, S.; Slater, R.; Reivich, M.; Schwartz, E. Transfusion Therapy for Cerebrovascular Abnormalities in Sickle Cell Disease. J. Pediatr. 1976, 88, 382–387. [Google Scholar] [CrossRef]
- Momjian-Mayor, I.; Baron, J.C. The Pathophysiology of Watershed Infarction in Internal Carotid Artery Disease: Review of Cerebral Perfusion Studies. Stroke 2005, 36, 567–577. [Google Scholar] [CrossRef]
- Guilliams, K.P.; Fields, M.E.; Ragan, D.K.; Chen, Y.; Eldeniz, C.; Hulbert, M.L.; Binkley, M.M.; Rhodes, J.N.; Shimony, J.S.; McKinstry, R.C.; et al. Large-Vessel Vasculopathy in Children With Sickle Cell Disease: A Magnetic Resonance Imaging Study of Infarct Topography and Focal Atrophy. Pediatr. Neurol. 2017, 69, 49–57. [Google Scholar] [CrossRef]
- Fields, M.E.; Guilliams, K.P.; Ragan, D.K.; Binkley, M.M.; Eldeniz, C.; Chen, Y.; Hulbert, M.L.; McKinstry, R.C.; Shimony, J.S.; Vo, K.D.; et al. Regional Oxygen Extraction Predicts Border Zone Vulnerability to Stroke in Sickle Cell Disease. Neurology 2018, 90, e1134–e1144. [Google Scholar] [CrossRef]
- Powers, W.J.; Grubb, R.L.; Darriet, D.; Raichle, M.E. Cerebral Blood Flow and Cerebral Metabolic Rate of Oxygen Requirements for Cerebral Function and Viability in Humans. J. Cereb. Blood Flow Metab. 1985, 5, 600–608. [Google Scholar] [CrossRef]
- Derdeyn, C.P.; Videen, T.O.; Yundt, K.D.; Fritsch, S.M.; Carpenter, D.A.; Grubb, R.L.; Powers, W.J. Variability of Cerebral Blood Volume and Oxygen Extraction: Stages of Cerebral Haemodynamic Impairment Revisited. Brain 2002, 125, 595–607. [Google Scholar] [CrossRef]
- Heiss, W.D.; Huber, M.; Fink, G.R.; Herholz, K.; Pietrzyk, U.; Wagner, R.; Wienhard, K. Progressive Derangement of Periinfarct Viable Tissue in Ischemic Stroke. J. Cereb. Blood Flow Metab. 1992, 12, 193–203. [Google Scholar] [CrossRef]
- Grubb Robert, L., Jr.; Derdeyn, C.P.; Fritsch, S.M.; Carpenter, D.A.; Yundt, K.D.; Videen, T.O.; Spitznagel, E.L.; Powers, W.J. Importance of Hemodynamic Factors in the Prognosis of Symptomatic Carotid Occlusion. JAMA 1998, 280, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Gevers, S.; Nederveen, A.J.; Fijnvandraat, K.; Van Den Berg, S.M.; Van Ooij, P.; Heijtel, D.F.; Heijboer, H.; Nederkoorn, P.J.; Engelen, M.; Van Osch, M.J.; et al. Arterial Spin Labeling Measurement of Cerebral Perfusion in Children with Sickle Cell Disease. J. Magn. Reson. Imaging 2012, 35, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Kassim, A.A.; Pruthi, S.; Day, M.; Rodeghier, M.; Gindville, M.C.; Brodsky, M.A.; Debaun, M.R.; Jordan, L.C. Silent Cerebral Infarcts and Cerebral Aneurysms Are Prevalent in Adults with Sickle Cell Anemia. Blood 2016, 127, 2038–2040. [Google Scholar] [CrossRef]
- DeBaun, M.R.; Schatz, J.; Siegel, M.J.; Koby, M.; Craft, S.; Resar, L.; Chu, J.-Y.; Launius, G.; Dadash-Zadeh, M.; Lee, R.B.; et al. Cognitive Screening Examinations for Silent Cerebral Infarcts in Sickle Cell Disease. Neurology 1998, 50, 1678–1682. [Google Scholar] [CrossRef]
- Bernaudin, F.; Verlhac, S.; Fréard, F.; Roudot-Thoraval, F.; Benkerrou, M.; Thuret, I.; Mardini, R.; Vannier, J.P.; Ploix, E.; Romero, M.; et al. Multicenter Prospective Study of Children With Sickle Cell Disease: Radiographic and Psychometric Correlation. J. Child Neurol. 2000, 15, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Baldeweg, T.; Hogan, A.M.; Saunders, D.E.; Telfer, P.; Gadian, D.G.; Vargha-Khadem, F.; Kirkham, F.J. Detecting White Matter Injury in Sickle Cell Disease Using Voxel-Based Morphometry. Ann. Neurol. 2006, 59, 662–672. [Google Scholar] [CrossRef]
- Balci, A.; Karazincir, S.; Beyoglu, Y.; Cingiz, C.; Davran, R.; Gali, E.; Okuyucu, E.; Egilmez, E. Quantitative Brain Diffusion-Tensor MRI Findings in Patients with Sickle Cell Disease. Am. J. Roentgenol. 2012, 198, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Fulling, K.H.; Nelson, J.S. Sickle Cell Anemia and Central Nervous System Infarction: A Neuropathological Study. Ann. Neurol. 1986, 20, 684–690. [Google Scholar] [CrossRef]
- Ford, A.L.; Ragan, D.K.; Fellah, S.; Binkley, M.M.; Fields, M.E.; Guilliams, K.P.; An, H.; Jordan, L.C.; McKinstry, R.C.; Lee, J.M.; et al. Silent Infarcts in Sickle Cell Disease Occur in the Border Zone Region and Are Associated with Low Cerebral Blood Flow. Blood 2018, 132, 1714–1723. [Google Scholar] [CrossRef]
- Light, J.; Boucher, M.; Baskin-Miller, J.; Winstead, M. Managing the Cerebrovascular Complications of Sickle Cell Disease: Current Perspectives. J. Blood Med. 2023, 14, 279–293. [Google Scholar] [CrossRef]
- Telen, M.J. Curative vs Targeted Therapy for SCD: Does It Make More Sense to Address the Root Cause than Target Downstream Events? Blood 2020, 4, 3457–3465. [Google Scholar] [CrossRef] [PubMed]
- Cokic, V.P.; Smith, R.D.; Beleslin-Cokic, B.B.; Njoroge, J.M.; Miller, J.L.; Gladwin, M.T.; Schechter, A.N. Hydroxyurea Induces Fetal Hemoglobin by the Nitric Oxide-Dependent Activation of Soluble Guanylyl Cyclase. J. Clin. Investig. 2003, 111, 231–239. [Google Scholar] [CrossRef]
- Cokic, V.P.; Beleslin-Cokic, B.B.; Noguchi, C.T.; Schechter, A.N. Hydroxyurea Increases ENOS Protein Levels through Inhibition of Proteasome Activity. Nitric Oxide—Biol. Chem. 2007, 16, 371–378. [Google Scholar] [CrossRef]
- Charache, S.; Terrin, M.L.; Moore, R.D.; Dover, G.J.; Barton, F.B.; Eckert, S.; McMahon, R.P.; Bonds, D.R. Effect of Hydroxyurea on the Frequency of Painful Crises in Sickle Cell Anemia. N. Engl. J. Med. 1995, 332, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, C.D.; Files, B.A.; Luo, Z.; Miller, S.T.; Kalpatthi, R.; Iyer, R.; Seaman, P.; Lebensburger, J.; Alvarez, O.; Thompson, B.; et al. Impact of Hydroxyurea on Clinical Events in the BABY HUG Trial. Blood J. Am. Soc. Hematol. 2012, 120, 4304–4310. [Google Scholar] [CrossRef]
- Steinberg, M.; Barton, F.B.; Castro, O.; Ballas, S.K. Effect of Hydroxyurea on Mortality and Morbidity in Adult Sickle Cell Anemia: Risks and Benefits up to 9 Years of Treatment. JAMA 2003, 289, 1645–1651. [Google Scholar] [CrossRef]
- Ware, R.E.; Davis, B.R.; Schultz, W.H.; Brown, R.C.; Aygun, B.; Sarnaik, S.; Odame, I.; Fuh, B.; George, A.; Owen, W.; et al. Hydroxycarbamide versus Chronic Transfusion for Maintenance of Transcranial Doppler Flow Velocities in Children with Sickle Cell Anaemia—TCD with Transfusions Changing to Hydroxyurea (TWiTCH): A Multicentre, Open-Label, Phase 3, Non-Inferiority Trial. Lancet 2016, 387, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, C.D.; Wigfall, D.R.; Ware, R.E. Enalapril and Hydroxyurea Therapy for Children with Sickle Nephropathy. Pediatr. Blood Cancer 2005, 45, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Voskaridou, E.; Christoulas, D.; Bilalis, A.; Plata, E.; Varvagiannis, K.; Stamatopoulos, G.; Sinopoulou, K.; Balassopoulou, A.; Loukopoulos, D.; Terpos, E.; et al. The Effect of Prolonged Administration of Hydroxyurea on Morbidity and Mortality in Adult Patients with Sickle Cell Syndromes: Results of a 17-Year, Single-Center Trial (LaSHS). Blood 2010, 115, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Santana, S.S.; Pitanga, T.N.; de Santana, J.M.; Zanette, D.L.; Vieira, J.d.J.; Yahouédéhou, S.C.M.A.; Adanho, C.S.A.; Viana, S. de M.; Luz, N.F.; Borges, V.M.; et al. Hydroxyurea Scavenges Free Radicals and Induces the Expression of Antioxidant Genes in Human Cell Cultures Treated with Hemin. Front. Immunol. 2020, 11, 1488. [Google Scholar] [CrossRef]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l -Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Hebbel, R.P.; Hedlund, B.E. Sickle Hemoglobin Oxygen Affinity-Shifting Strategies Have Unequal Cerebrovascular Risks. Am. J. Hematol. 2018, 93, 321–325. [Google Scholar] [CrossRef]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef]
- Gabbianelli, M.; Testa, U.; Massa, A.; Pelosi, E.; Sposi, N.M.; Riccioni, R.; Luchetti, L.; Peschle, C. Hemoglobin Switching in Unicellular Erythroid Culture of Sibling Erythroid Burst-Forming Units: Kit Ligand Induces a Dose-Dependent Fetal Hemoglobin Reactivation Potentiated by Sodium Butyrate. Blood 2000, 95, 3555–3561. [Google Scholar] [CrossRef]
- Matsui, N.M.; Varki, A.; Embury, S.H. Heparin Inhibits the Flow Adhesion of Sickle Red Blood Cells to P-Selectin. Blood 2002, 100, 3790–3796. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Patton, J.T.; Sarkar, A.; Ernst, B.; Magnani, J.L.; Frenette, P.S. GMI-1070, a Novel Pan-Selectin Antagonist, Reverses Acute Vascular Occlusions in Sickle Cell Mice. Blood 2010, 116, 1779–1786. [Google Scholar] [CrossRef]
- Wun, T.; Soulieres, D.; Frelinger, A.L.; Krishnamurti, L.; Novelli, E.M.; Kutlar, A.; Ataga, K.I.; Knupp, C.L.; Mcmahon, L.E.; Strouse, J.J.; et al. A Double-Blind, Randomized, Multicenter Phase 2 Study of Prasugrel versus Placebo in Adult Patients with Sickle Cell Disease. J. Hematol. Oncol. 2013, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Styles, L.; Heiselman, D.; Heath, L.E.; Moser, B.A.; Small, D.S.; Jakubowski, J.A.; Zhou, C.; Redding-Lallinger, R.; Heeney, M.M.; Quinn, C.T.; et al. Prasugrel in Children with Sickle Cell Disease: Pharmacokinetic and Pharmacodynamic Data from an Open-Label, Adaptive-Design, Dose-Ranging Study. J. Pediatr. Hematol. Oncol. 2015, 37, 1–9. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Krishnamoorthy, S.; Gupta, D.; Lancelot, M.; Moore, N.; Sarnaik, S.; Hobbs, W.E.; Light, D.R.; Hines, P. VLA-4 Blockade by Natalizumab Inhibits Sickle Reticulocyte and Leucocyte Adhesion during Simulated Blood Flow. Br. J. Haematol. 2016, 174, 970–982. [Google Scholar] [CrossRef]
- Hoppe, C.; Kuypers, F.; Hagar, W.; Vichinsky, E. A Pilot Study of the Short-Term Use of Simvastatin in Sickle Cell Disease: Effects on Markers of Vascular Dysfunction. Br. J. Haematol. 2011, 153, 655–663. [Google Scholar] [CrossRef]
- Kapoor, S.; Little, J.A.; Pecker, L.H. Advances in the Treatment of Sickle Cell Disease. Mayo Clin. Proc. 2018, 93, 1810–1824. [Google Scholar] [CrossRef]
- Telen, M.J.; Malik, P.; Vercellotti, G.M. Therapeutic Strategies for Sickle Cell Disease: Towards a Multi-Agent Approach. Nat. Rev. Drug Discov. 2019, 18, 139–158. [Google Scholar] [CrossRef]
- Telen, M.J. Beyond Hydroxyurea: New and Old Drugs in the Pipeline for Sickle Cell Disease. Blood 2016, 127, 810–819. [Google Scholar] [CrossRef]
- Valadi, N.; Silva, G.S.; Bowman, L.S.; Ramsingh, D.; Vicari, P.; Filho, A.C.; Massaro, A.R.; Kutlar, A.; Nichols, F.T.; Adams, R.J. Transcranial Doppler Ultrasonography in Adults with Sickle Cell Disease. Neurology 2006, 67, 572–574. [Google Scholar] [CrossRef]
- Ngo, D.A.; Steinberg, M.H. Genomic Approaches to Identifying Targets for Treating β Hemoglobinopathies. BMC Med. Genom. 2015, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Sommet, J.; Alberti, C.; Couque, N.; Verlhac, S.; Haouari, Z.; Mohamed, D.; Missud, F.; Holvoet, L.; Elmaleh, M.; Ithier, G.; et al. Clinical and Haematological Risk Factors for Cerebral Macrovasculopathy in a Sickle Cell Disease Newborn Cohort: A Prospective Study. Br. J. Haematol. 2016, 172, 966–977. [Google Scholar] [CrossRef]
- Jordan, L.C.; Casella, J.F.; Debaun, M.R. Prospects for Primary Stroke Prevention in Children with Sickle Cell Anaemia. Br. J. Haematol. 2012, 157, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Gardner, K.; Thein, S.L. Genetic Factors Modifying Sickle Cell Disease Severity. In Sickle Cell Anemia: From Basic Science to Clinical Practice; Costa, F.F., Conran, N., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 371–397. ISBN 978-3-319-06713-1. [Google Scholar]
- Styles, L.A.; Hoppe, C.; Klitz, W.; Vichinsky, E.; Lubin, B.; Trachtenberg, E. Evidence for HLA-Related Susceptibility for Stroke in Children with Sickle Cell Disease. Blood J. Am. Soc. Hematol. 2000, 95, 3562–3567. [Google Scholar]
- Tang, D.C.; Prauner, R.; Liu, W.; Kim, K.; Hirsch, R.P.; Driscoll, M.C.; Rodgers, G.P. Polymorphisms within the Angiotensinogen Gene (GT-Repeat) and the Risk of Stroke in Pediatric Patients with Sickle Cell Disease: A Case-Control Study. Am. J. Hematol. 2001, 68, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.G.; Tang, D.C.; Savage, S.A.; Leitman, S.F.; Heller, S.I.; Serjeant, G.R.; Rodgers, G.P.; Chanock, S.J. Variants in the VCAM1 Gene and Risk for Symptomatic Stroke in Sickle Cell Disease. Blood 2002, 100, 4303–4309. [Google Scholar] [CrossRef]
- Driscoll, C.C.; Hurlet, A.; Styles, L.; McKie, V.; Files, B.; Olivieri, N.; Pegelow, C.; Berman, B.; Drachtman, R.; Patel, K.; et al. Stroke Risk in Siblings with Sickle Cell Anemia. Blood 2003, 101, 2401–2404. [Google Scholar] [CrossRef]
- Hoppe, C.; Klitz, W.; Cheng, S.; Apple, R.; Steiner, L.; Robles, L.; Girard, T.; Vichinsky, E.; Styles, L. Gene Interactions and Stroke Risk in Children with Sickle Cell Anemia. Blood 2004, 103, 2391–2396. [Google Scholar] [CrossRef]
- Belisário, A.R.; Sales, R.R.; Toledo, N.E.; Velloso-Rodrigues, C.; Silva, C.M.; Viana, M.B. Association between ENPP1 K173Q and Stroke in a Newborn Cohort of 395 Brazilian Children with Sickle Cell Anemia. Blood 2015, 126, 1259–1260. [Google Scholar] [CrossRef]
- Belisário, A.R.; Nogueira, F.L.; Rodrigues, R.S.; Toledo, N.E.; Cattabriga, A.L.M.; Velloso-Rodrigues, C.; Duarte, F.O.C.; Silva, C.M.; Viana, M.B. Association of Alpha-Thalassemia, TNF-Alpha (-308G>A) and VCAM-1 (c.1238G>C) Gene Polymorphisms with Cerebrovascular Disease in a Newborn Cohort of 411 Children with Sickle Cell Anemia. Blood Cells Mol. Dis. 2015, 54, 44–50. [Google Scholar] [CrossRef]
- Silva, M.; Vargas, S.; Coelho, A.; Ferreira, E.; Mendonça, J.; Vieira, L.; Maia, R.; Dias, A.; Ferreira, T.; Morais, A.; et al. Biomarkers and Genetic Modulators of Cerebral Vasculopathy in Sub-Saharan Ancestry Children with Sickle Cell Anemia. Blood Cells Mol. Dis. 2020, 83, 102436. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.M.; Frohlich, D.M.; Howard, T.A.; Schultz, W.H.; Driscoll, C.; Nagasubramanian, R.; Mortier, N.A.; Kimble, A.C.; Aygun, B.; Adams, R.J.; et al. Genetic Predictors for Stroke in Children with Sickle Cell Anemia. Blood 2011, 117, 6681–6684. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.M.; Sheehan, V.; Linder, H.; Howard, T.A.; Wang, Y.D.; Hoppe, C.C.; Aygun, B.; Adams, R.J.; Neale, G.A.; Ware, R.E. Genetic Mapping and Exome Sequencing Identify 2 Mutations Associated with Stroke Protection in Pediatric Patients with Sickle Cell Anemia. Blood 2013, 121, 3237–3245. [Google Scholar] [CrossRef] [PubMed]
- Martella, M.; Quaglia, N.; Frigo, A.C.; Basso, G.; Colombatti, R.; Sainati, L. Association between a Combination of Single Nucleotide Polymorphisms and Large Vessel Cerebral Vasculopathy in African Children with Sickle Cell Disease. Blood Cells Mol. Dis. 2016, 61, 1–3. [Google Scholar] [CrossRef]
- Bitoungui, V.J.N.; Pule, G.D.; Hanchard, N.; Ngogang, J.; Wonkam, A. Beta-Globin Gene Haplotypes Among Cameroonians and Review of the Global Distribution: Is There a Case for a Single Sickle Mutation Origin in Africa? Omi. J. Integr. Biol. 2015, 19, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, T.; Nicholls, S.J.; Topol, E.J.; Zhang, R.; Yang, X.; Schmitt, D.; Fu, X.; Shao, M.; Brennan, D.M.; Ellis, S.G.; et al. Relationship of Paraoxonase 1 (PON1) Gene Polymorphisms and Functional Activity With Systemic Oxidative Stress and Cardiovascular Risk. JAMA 2008, 299, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Luu, H.N.; Kingah, P.L.; North, K.; Boerwinkle, E.; Volcik, K.A. Interaction of Folate Intake and the Paraoxonase Q192R Polymorphism with Risk of Incident Coronary Heart Disease and Ischemic Stroke: The Atherosclerosis Risk in Communities Study. Ann. Epidemiol. 2011, 21, 815–823. [Google Scholar] [CrossRef]
- Coelho, A.; Dias, A.; Morais, A.; Nunes, B.; Ferreira, E.; Picanço, I.; Faustino, P.; Lavinha, J. Genetic Variation in CD36, HBA, NOS3 and VCAM1 Is Associated with Chronic Haemolysis Level in Sickle Cell Anaemia: A Longitudinal Study. Eur. J. Haematol. 2014, 92, 237–243. [Google Scholar] [CrossRef]
- Li, N.; Zhou, H.; Tang, Q. Red Blood Cell Distribution Width: A Novel Predictive Indicator for Cardiovascular and Cerebrovascular Diseases. Dis. Markers 2017, 2017. [Google Scholar] [CrossRef]
- Hoppe, C.; Klitz, W.; Noble, J.; Vigil, L.; Vichinsky, E.; Styles, L. Distinct HLA Associations by Stroke Subtype in Children with Sickle Cell Anemia. Blood J. Am. Soc. Hematol. 2003, 101, 2865–2869. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, C.; Klitz, W.; Harlingue, K.D.; Cheng, S.; Grow, M.; Steiner, L.; Noble, J.; Adams, R.; Styles, L. Confirmation of an Association Between the TNF (-308 ) Promoter Polymorphism and Stroke Risk in Children With Sickle Cell Anemia. Stroke 2007, 38, 2241–2246. [Google Scholar] [CrossRef]
- Lang, D.; Reuter, S.; Buzescu, T.; August, C.; Heidenreich, S. Heme-Induced Heme Oxygenase-1 (HO-1) in Human Monocytes Inhibits Apoptosis despite Caspase-3 up-Regulation. Int. Immunol. 2005, 17, 155–165. [Google Scholar] [CrossRef]
- Belcher, J.D.; Mahaseth, H.; Welch, T.E.; Otterbein, L.E.; Hebbel, R.P.; Vercellotti, G.M. Heme Oxygenase-1 Is a Modulator of Inflammation and Vaso-Occlusion in Transgenic Sickle Mice. J. Clin. Investig. 2006, 116, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Bryant, C.J.; Nguyen, J.; Bowlin, P.R.; Kielbik, M.C.; Bischof, J.C.; Hebbel, R.P.; Vercellotti, G.M. Transgenic Sickle Mice Have Vascular Inflammation. Blood 2003, 101, 3953–3959. [Google Scholar] [CrossRef]
- Silva, M.; Coelho, A.; Vargas, S.; Faustino, P. VCAM1, HMOX1 and NOS3 Differential Endothelial Expression May Impact Sickle Cell Anemia Vasculopathy. Blood Cells Mol. Dis. 2022, 93, 102639. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Xu, X.; Belmadani, S.; Park, Y.; Tang, Z.; Feldman, A.M.; Chilian, W.M.; Zhang, C. TNF-α Contributes to Endothelial Dysfunction by Upregulating Arginase in Ischemia/Reperfusion Injury. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Channon, K.M. Synthesis and Recycling of Tetrahydrobiopterin in Endothelial Function and Vascular Disease. Nitric Oxide—Biol. Chem. 2011, 25, 81–88. [Google Scholar] [CrossRef]
- da Guarda, C.C.; Santiago, R.P.; Pitanga, T.N.; Santana, S.S.; Zanette, D.L.; Borges, V.M.; Goncalves, M.S. Heme Changes HIF-α, ENOS and Nitrite Production in HUVECs after Simvastatin, HU, and Ascorbic Acid Therapies. Microvasc. Res. 2016, 106, 128–136. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, M.; Faustino, P. From Stress to Sick(le) and Back Again–Oxidative/Antioxidant Mechanisms, Genetic Modulation, and Cerebrovascular Disease in Children with Sickle Cell Anemia. Antioxidants 2023, 12, 1977. https://doi.org/10.3390/antiox12111977
Silva M, Faustino P. From Stress to Sick(le) and Back Again–Oxidative/Antioxidant Mechanisms, Genetic Modulation, and Cerebrovascular Disease in Children with Sickle Cell Anemia. Antioxidants. 2023; 12(11):1977. https://doi.org/10.3390/antiox12111977
Chicago/Turabian StyleSilva, Marisa, and Paula Faustino. 2023. "From Stress to Sick(le) and Back Again–Oxidative/Antioxidant Mechanisms, Genetic Modulation, and Cerebrovascular Disease in Children with Sickle Cell Anemia" Antioxidants 12, no. 11: 1977. https://doi.org/10.3390/antiox12111977