




Tricarboxylic Acid Cycle Regulation of Metabolic Program, Redox System, and Epigenetic Remodeling for Bone Health and Disease

 ,
,  and
and
Abstract
1. Introduction
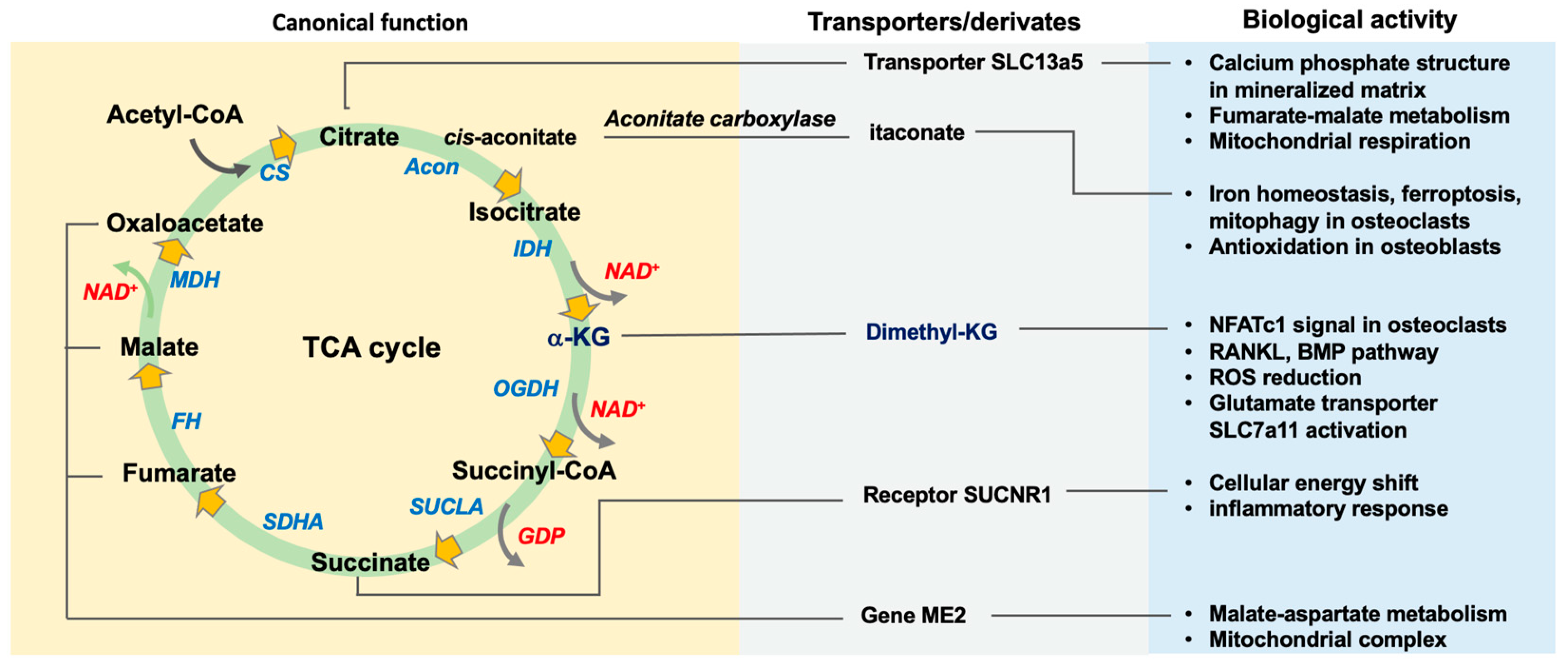
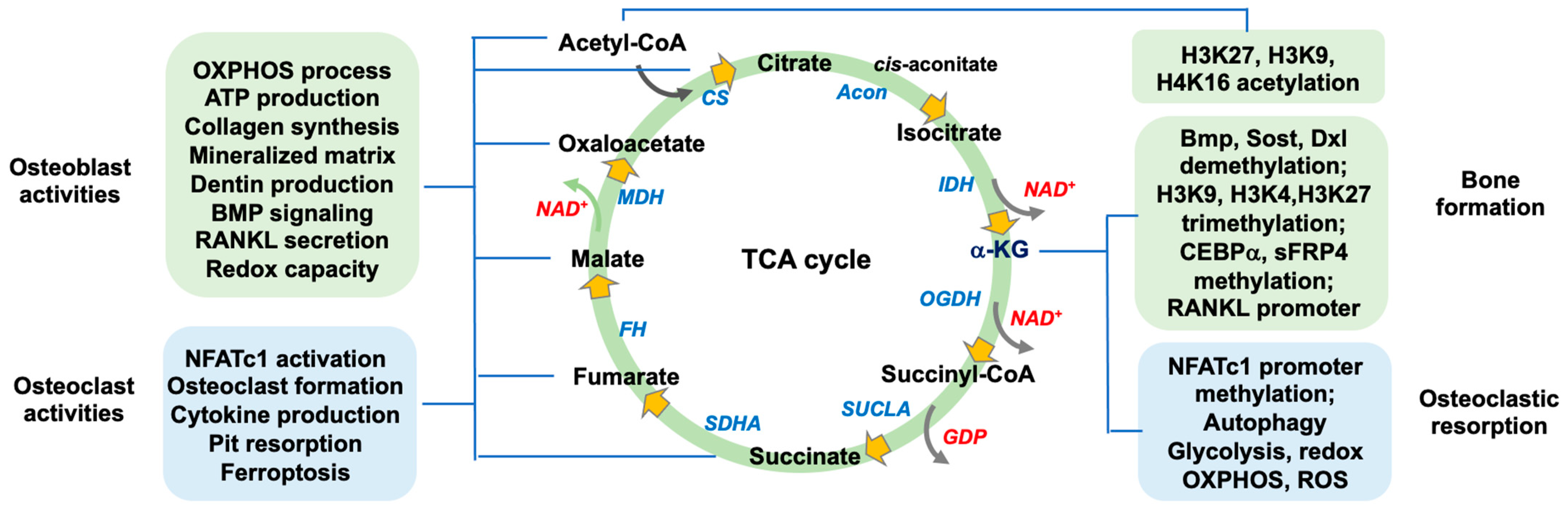
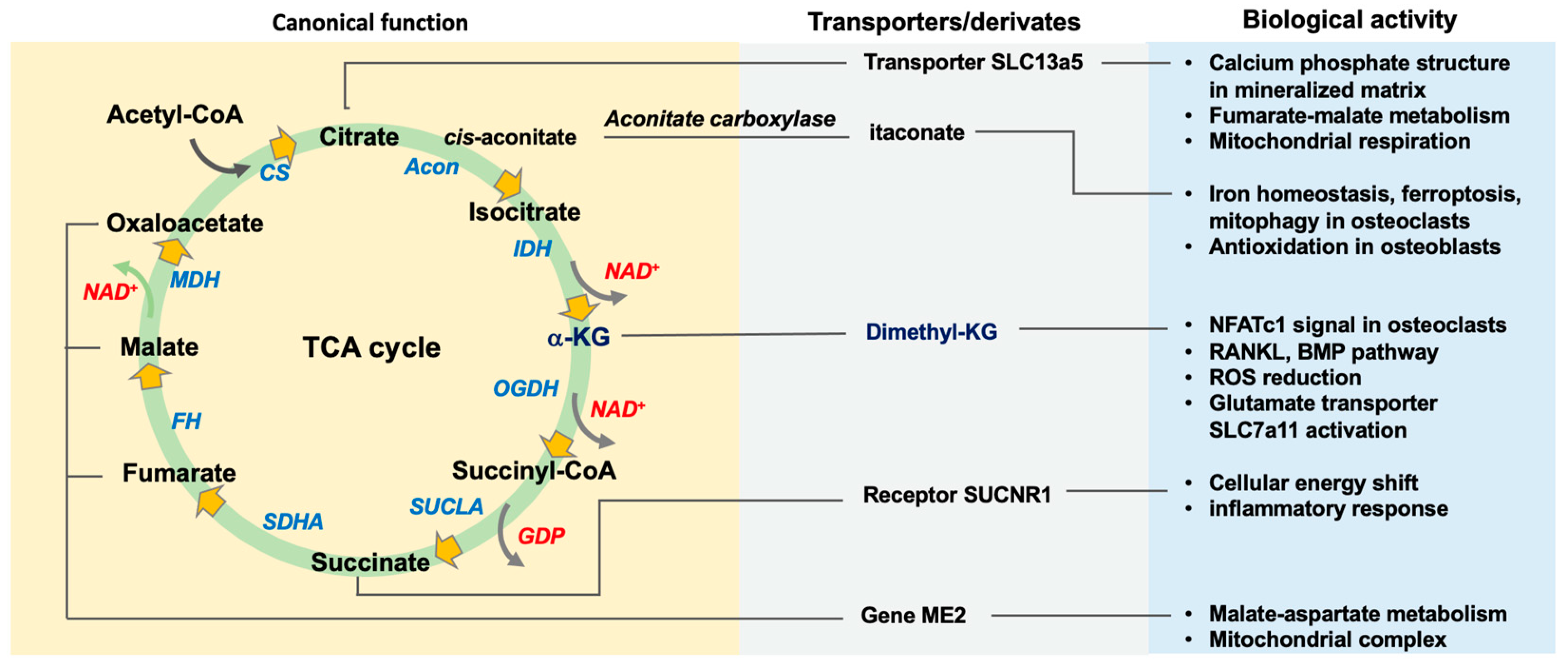
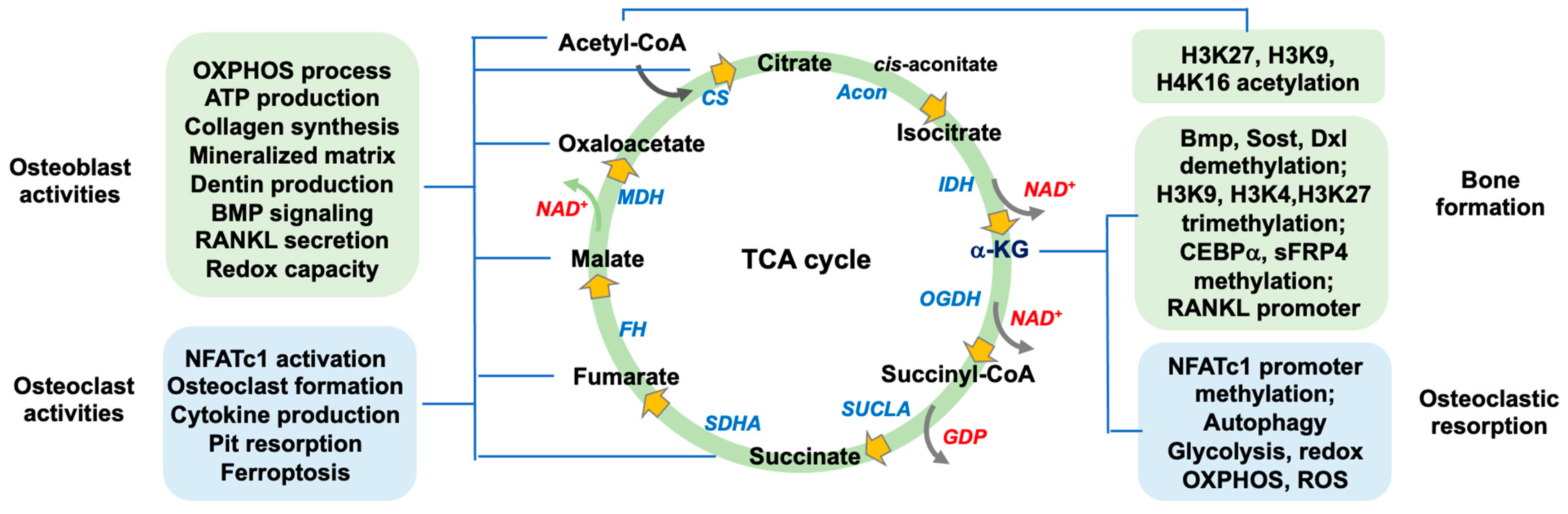
2. The Function of TCA Cycle Metabolism in Bone Mass
2.1. Canonical Actions of TCA Cycle in Osteogenic or Osteoclastogenic Capacity
2.1.1. Citrate, Citrate Synthase, and Acetyl-CoA
2.1.2. Aconitase, Cis-Aconitate, and Itaconate
2.1.3. Isocitrate Dehydrogenase and α-Ketoglutarate
2.1.4. Succinate and Succinate Dehydrogenase
2.1.5. Fumarate, Malate, and Oxaloacetate
2.1.6. Acetyl-CoA
2.2. Glutamate Metabolic Control of the TCA Cycle
3. The TCA Cycle Regulates Redox Homeostasis
4. Noncanonical Actions of TCA Cycle Metabolism in Bone Formation and Resorption
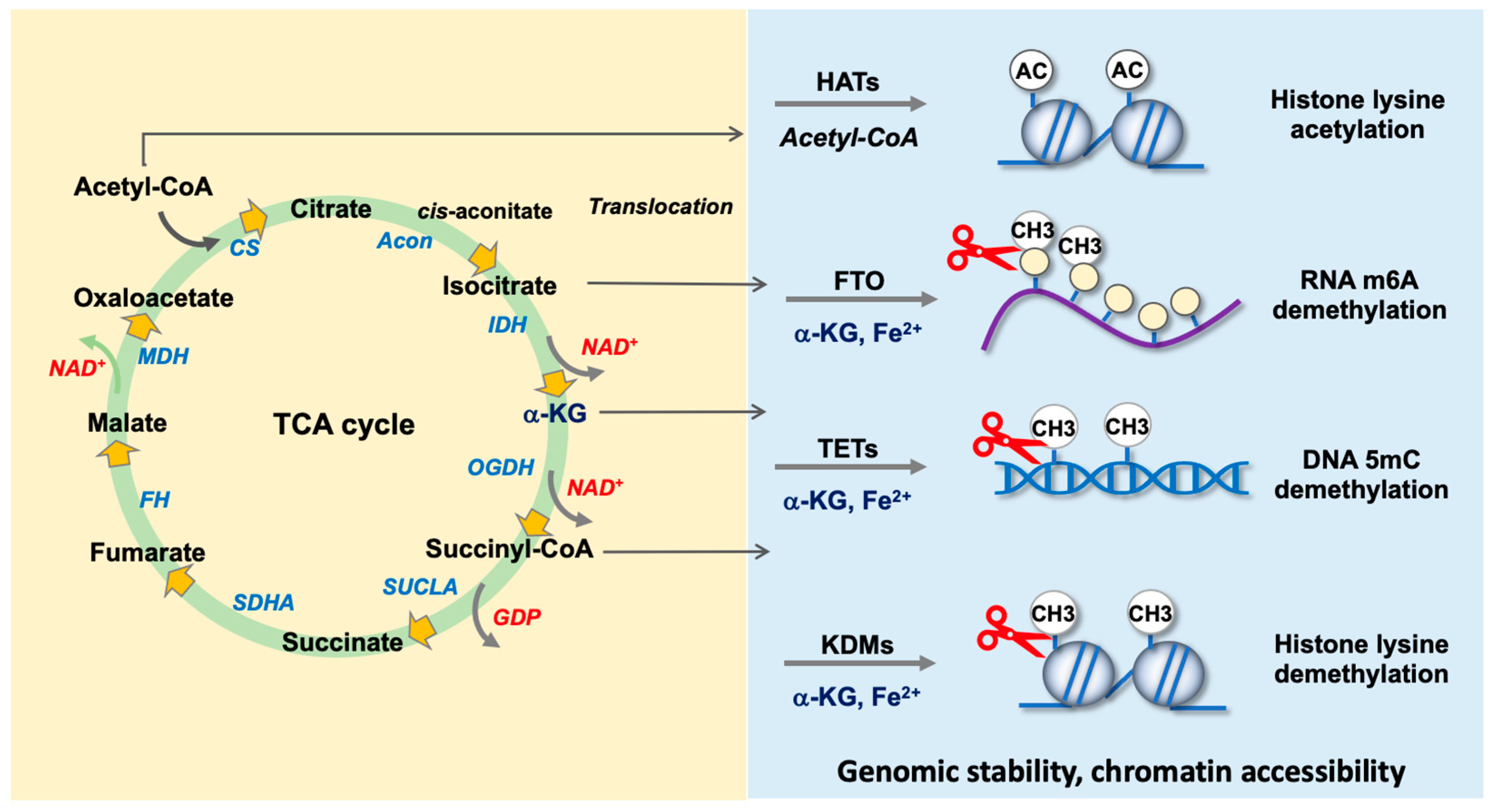
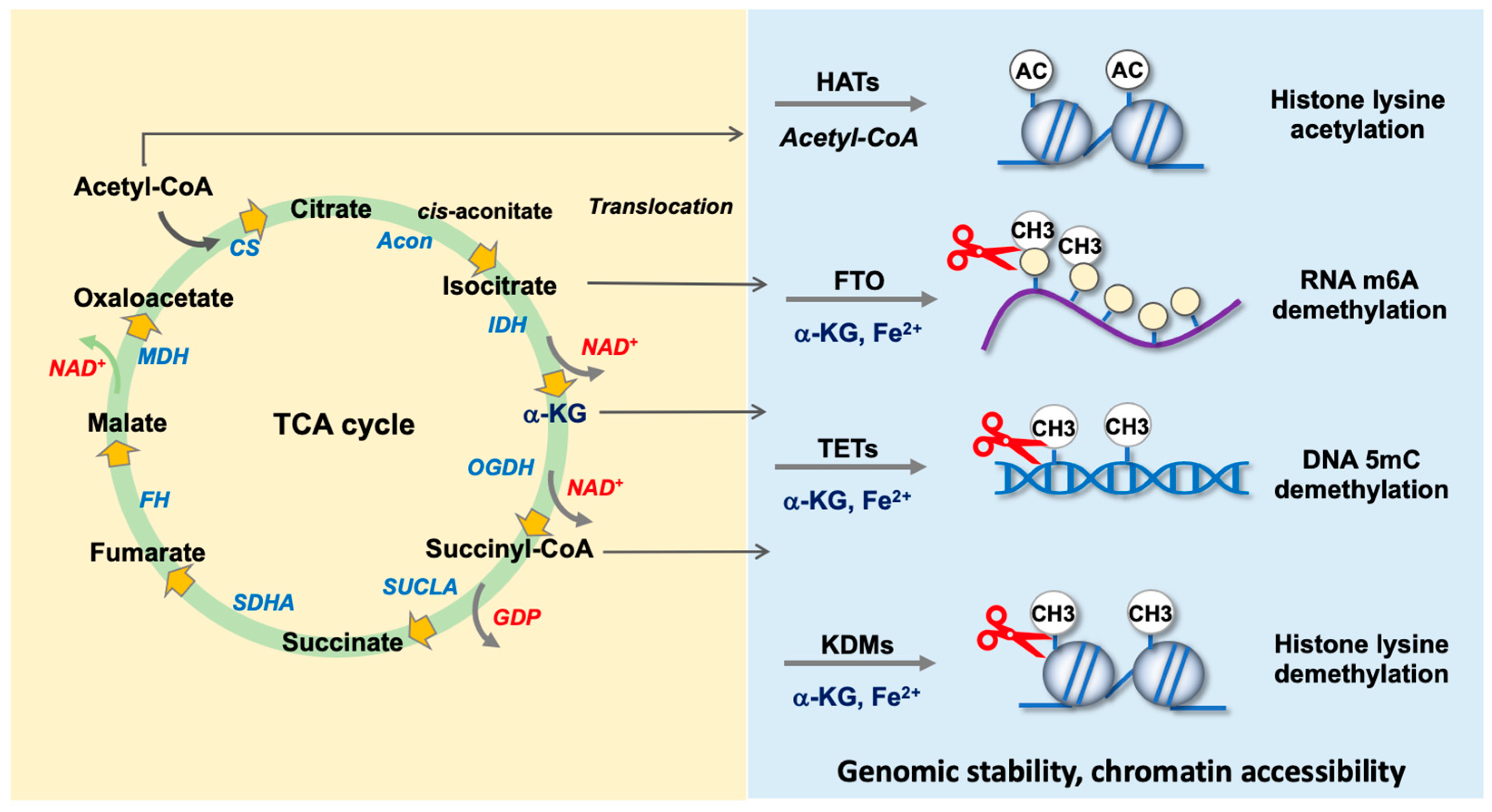
4.1. Nuclear Translocation of TCA Cycle Enzymes
4.2. TCA Cycle Intermediates as Co-Factors for Epigenetic Regulators
4.2.1. Acetyl-CoA as a Co-Factor for Histone Acetyltransferases
4.2.2. α-KG Is Required by DNA Demethylase TETs
4.2.3. α-KG as a Co-Factor for RNA m6A Demethylases FTO and ALKBH5
4.2.4. α-KG Is Indispensable in Histone Demethylase-Mediated Histone Demethylation
4.2.5. KDM4A and KDM4B
4.2.6. KDM5B and KDM5C
4.2.7. KDM6A and KDM6B
4.2.8. KDM7A
5. TCA Cycle Metabolites for Counteracting Osteoporotic Disorders
5.1. Perspectives of Remedial Options
5.2. Limitations of TCA Cycle Metabolites for Slowing Osteoporosis
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leser, J.M.; Torre, O.M.; Gould, N.R.; Guo, Q.; Buck, H.V.; Kodama, J.; Otsuru, S.; Stains, J.P. Osteoblast-lineage calcium/calmodulin-dependent kinase 2 delta and gamma regulates bone mass and quality. Proc. Natl. Acad. Sci. USA 2023, 120, e2304492120. [Google Scholar] [CrossRef]
- Engelmann, J.; Zarrer, J.; Gensch, V.; Riecken, K.; Berenbrok, N.; Luu, T.V.; Beitzen-Heineke, A.; Vargas-Delgado, M.E.; Pantel, K.; Bokemeyer, C.; et al. Regulation of bone homeostasis by MERTK and TYRO3. Nat. Commun. 2022, 13, 7689. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, B.Y.; Lee, J.S.; Jeong, Y.M.; Cho, H.J.; Park, E.; Kim, D.; Kim, S.S.; Kim, B.T.; Choi, Y.J.; et al. UBAP2 plays a role in bone homeostasis through the regulation of osteoblastogenesis and osteoclastogenesis. Nat. Commun. 2023, 14, 3668. [Google Scholar] [CrossRef] [PubMed]
- Vilaca, T.; Eastell, R.; Schini, M. Osteoporosis in men. Lancet Diabetes Endocrinol. 2022, 10, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.D.; Shane, E. Postmenopausal Osteoporosis. N. Engl. J. Med. 2023, 389, 1979–1991. [Google Scholar] [CrossRef] [PubMed]
- Reid, I.R.; Billington, E.O. Drug therapy for osteoporosis in older adults. Lancet 2022, 399, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Soós, B.; Szentpétery, Á.; Raterman, H.G.; Lems, W.F.; Bhattoa, H.P.; Szekanecz, Z. Effects of targeted therapies on bone in rheumatic and musculoskeletal diseases. Nat. Rev. Rheumatol. 2022, 18, 249–257. [Google Scholar] [CrossRef]
- Marques-Carvalho, A.; Kim, H.N.; Almeida, M. The role of reactive oxygen species in bone cell physiology and pathophysiology. Bone Rep. 2023, 19, 101664. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698. [Google Scholar] [CrossRef]
- Piroli, G.G.; Manuel, A.M.; McCain, R.S.; Smith, H.H.; Ozohanics, O.; Mellid, S.; Cox, J.H.; Cotham, W.E.; Walla, M.D.; Cascón, A.; et al. Defective function of α-ketoglutarate dehydrogenase exacerbates mitochondrial ATP deficits during complex I deficiency. Redox Biol. 2023, 67, 102932. [Google Scholar] [CrossRef]
- Nanadikar, M.S.; Vergel Leon, A.M.; Guo, J.; van Belle, G.J.; Jatho, A.; Philip, E.S.; Brandner, A.F.; Böckmann, R.A.; Shi, R.; Zieseniss, A.; et al. IDH3γ functions as a redox switch regulating mitochondrial energy metabolism and contractility in the heart. Nat. Commun. 2023, 14, 2123. [Google Scholar] [CrossRef]
- Löffler, J.; Noom, A.; Ellinghaus, A.; Dienelt, A.; Kempa, S.; Duda, G.N. A comprehensive molecular profiling approach reveals metabolic alterations that steer bone tissue regeneration. Commun. Biol. 2023, 6, 327. [Google Scholar] [CrossRef]
- Madhu, V.; Hernandaz-Meadows, M.; Coleman, A.; Sao, K.; Inguito, K.; Haslam, O.; Boneski, P.K.; Sesaki, H.; Collins, J.A.; Risbud, M.V. OPA1 protects intervertebral disc and knee joint health in aged mice by maintaining the structure and metabolic functions of mitochondria. bioRxiv, 2024; Preprint. [Google Scholar] [CrossRef]
- Wu, Y.L.; Lin, Z.J.; Li, C.C.; Lin, X.; Shan, S.K.; Guo, B.; Zheng, M.H.; Li, F.; Yuan, L.Q.; Li, Z.H. Epigenetic regulation in metabolic diseases: Mechanisms and advances in clinical study. Signal Transduct. Target Ther. 2023, 8, 98. [Google Scholar] [CrossRef]
- Roig-Soriano, J.; Griñán-Ferré, C.; Espinosa-Parrilla, J.F.; Abraham, C.R.; Bosch, A.; Pallàs, M.; Chillón, M. AAV-mediated expression of secreted and transmembrane αKlotho isoforms rescues relevant aging hallmarks in senescent SAMP8 mice. Aging Cell 2022, 21, e13581. [Google Scholar] [CrossRef]
- Yang, C.; Dong, Z.; Ling, Z.; Chen, Y. The crucial mechanism and therapeutic implication of RNA methylation in bone pathophysiology. Aging Res. Rev. 2022, 79, 101641. [Google Scholar] [CrossRef]
- Yin, B.; Yu, F.; Wang, C.; Li, B.; Liu, M.; Ye, L. Epigenetic control of mesenchymal stem cell fate decision via histone methyltransferase Ash1l. Stem Cells 2019, 37, 115–127. [Google Scholar] [CrossRef]
- Liu, X.; Si, W.; He, L.; Yang, J.; Peng, Y.; Ren, J.; Liu, X.; Jin, T.; Yu, H.; Zhang, Z.; et al. The existence of a nonclassical TCA cycle in the nucleus that wires the metabolic-epigenetic circuitry. Signal Transduct. Target Ther. 2021, 6, 375. [Google Scholar] [CrossRef]
- Jaccard, A.; Wyss, T.; Maldonado-Pérez, N.; Rath, J.A.; Bevilacqua, A.; Peng, J.J.; Lepez, A.; Von Gunten, C.; Franco, F.; Kao, K.C.; et al. Reductive carboxylation epigenetically instructs T cell differentiation. Nature 2023, 621, 849–856. [Google Scholar] [CrossRef]
- Ly, C.H.; Lynch, G.S.; Ryall, J.G. A metabolic roadmap for somatic stem cell fate. Cell Metab. 2020, 31, 1052–1067. [Google Scholar] [CrossRef]
- Bonnay, F.; Veloso, A.; Steinmann, V.; Köcher, T.; Abdusselamoglu, M.D.; Bajaj, S.; Rivelles, E.; Landskron, L.; Esterbauer, H.; Zinzen, R.P.; et al. Oxidative metabolism drives immortalization of neural stem cells during tumorigenesis. Cell 2020, 182, 1490–1507.e19. [Google Scholar] [CrossRef]
- Rohatgi, N.; Zou, W.; Li, Y.; Cho, K.; Collins, P.L.; Tycksen, E.; Pandey, G.; DeSelm, C.J.; Patti, G.J.; Dey, A.; et al. BAP1 promotes osteoclast function by metabolic reprogramming. Nat. Commun. 2023, 14, 5923. [Google Scholar] [CrossRef]
- Wu, X.; Dai, H.; Yu, S.; Zhao, Y.; Long, Y.; Li, W.; Tu, J. Citrate regulates extracellular matrix mineralization during osteoblast differentiation in vitro. J. Inorg. Biochem. 2021, 214, 111269. [Google Scholar] [CrossRef]
- Dirckx, N.; Zhang, Q.; Chu, E.Y.; Tower, R.J.; Li, Z.; Guo, S.; Yuan, S.; Khare, P.A.; Zhang, C.; Verardo, A.; et al. A specialized metabolic pathway partitions citrate in hydroxyapatite to impact mineralization of bones and teeth. Proc. Natl. Acad. Sci. USA 2022, 119, e2212178119. [Google Scholar] [CrossRef]
- Villaseñor, A.; Aedo-Martín, D.; Obeso, D.; Erjavec, I.; Rodríguez-Coira, J.; Buendía, I.; Ardura, J.A.; Barbas, C.; Gortazar, A.R. Metabolomics reveals citric acid secretion in mechanically-stimulated osteocytes is inhibited by high glucose. Sci. Rep. 2019, 19, 2295. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Y.; Dai, H.; Tian, X.; Cui, Z.K.; Chen, Z.; Hu, L.; Song, Q.; Liu, A.; Zhang, Z.; et al. Bone and plasma citrate is reduced in osteoporosis. Bone 2018, 114, 189–197. [Google Scholar] [CrossRef]
- Wei, J.; Yang, Y.; Guo, D.; Xu, S.; Huang, H.; Zhang, D.; Xie, J.; Zhou, X. Osteoblasts induce glucose-derived ATP perturbations in chondrocytes through noncontact communication. Acta Biochim. Biophys. Sin. 2022, 54, 625–636. [Google Scholar] [CrossRef]
- Monteiro, C.; Ferreira de Oliveira, J.M.P.; Pinho, F.; Bastos, V.; Oliveira, H.; Peixoto, F.; Santos, C. Biochemical and transcriptional analyses of cadmium-induced mitochondrial dysfunction and oxidative stress in human osteoblasts. J. Toxicol. Environ. Health A 2018, 81, 705–717. [Google Scholar] [CrossRef]
- Ni, S.; Yuan, Y.; Qian, Z.; Zhong, Z.; Lv, T.; Kuang, Y.; Yu, B. Hypoxia inhibits RANKL-induced ferritinophagy and protects osteoclasts from ferroptosis. Free Radic. Biol. Med. 2021, 169, 271–282. [Google Scholar] [CrossRef]
- Bonadonna, M.; Altamura, S.; Tybl, E.; Palais, G.; Qatato, M.; Polycarpou-Schwarz, M.; Schneider, M.; Kalk, C.; Rüdiger, W.; Ertl, A.; et al. Iron regulatory protein (IRP)-mediated iron homeostasis is critical for neutrophil development and differentiation in the bone marrow. Sci. Adv. 2022, 8, eabq4469. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, Z.; Liu, C.; Sun, P.; Liu, P.; Li, X. ABCG2 is an itaconate exporter that limits antibacterial innate immunity by alleviating TFEB-dependent lysosomal biogenesis. Cell Metab. 2024, 36, 498–510.e11. [Google Scholar] [CrossRef]
- Song, J.; Zhang, Y.; Frieler, R.A.; Andren, A.; Wood, S.; Tyrrell, D.J.; Sajjakulnukit, P.; Deng, J.C.; Lyssiotis, C.A.; Mortensen, R.M.; et al. Itaconate suppresses atherosclerosis by activating a Nrf2-dependent antiinflammatory response in macrophages in mice. J. Clin. Investig. 2023, 134, e173034. [Google Scholar] [CrossRef]
- Ramalho, T.; Assis, P.A.; Ojelabi, O.; Tan, L.; Carvalho, B.; Gardinassi, L.; Campos, O.; Lorenzi, P.L.; Fitzgerald, K.A.; Haynes, C.; et al. Itaconate impairs immune control of Plasmodium by enhancing mtDNA-mediated PD-L1 expression in monocyte-derived dendritic cells. Cell Metab. 2024, 36, 484–497. [Google Scholar] [CrossRef]
- Maassen, S.; Coenen, B.; Ioannidis, M.; Harber, K.; Grijpstra, P.; Van den Bossche, J.; van den Bogaart, G. Itaconate promotes a wound resolving phenotype in pro-inflammatory macrophages. Redox Biol. 2023, 59, 102591. [Google Scholar] [CrossRef]
- Lippross, S.; Beckmann, R.; Streubesand, N.; Ayub, F.; Tohidnezhad, M.; Campbell, G.; Kan, Y.W.; Horst, F.; Sönmez, T.T.; Varoga, D.; et al. Nrf2 deficiency impairs fracture healing in mice. Calcif. Tissue Int. 2014, 95, 349–361. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, B.; Pan, X.; Huang, H.; Xie, Z.; Ma, Y.; Hu, B.; Wang, J.; Chen, Z.; Shi, P. Octyl itaconate inhibits osteoclastogenesis by suppressing Hrd1 and activating Nrf2 signaling. FASEB J. 2019, 33, 12929–12940. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, Z.; She, C.; Lin, Y.; Hong, Y.; Shi, L.; Zhang, Y.; Cao, P.; Xu, X. Four-octyl itaconate activates Nrf2 cascade to protect osteoblasts from hydrogen peroxide-induced oxidative injury. Cell Death Dis. 2020, 11, 772. [Google Scholar] [CrossRef]
- Kubo, Y.; Wruck, C.J.; Fragoulis, A.; Drescher, W.; Pape, H.C.; Lichte, P.; Fischer, H.; Tohidnezhad, M.; Hildebrand, F.; Pufe, T.; et al. Role of Nrf2 in fracture healing: Clinical aspects of oxidative stress. Calcif. Tissue Int. 2019, 105, 341–352. [Google Scholar] [CrossRef]
- Chae, U.; Park, N.R.; Kim, E.S.; Choi, J.Y.; Yim, M.; Lee, H.S.; Lee, S.R.; Lee, S.; Park, J.W.; Lee, D.S. IDH2-deficient mice develop spinal deformities with aging. Physiol. Res. 2018, 67, 487–494. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, S.H.; Lee, J.H.; Park, J.W.; Kim, J.E. IDH2 deficiency increases bone mass with reduced osteoclastogenesis by limiting RANKL expression in osteoblasts. Bone 2019, 129, 115056. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, P.; Liu, Y.; Wu, Y.; Chen, Y.; Guo, Y.; Zhang, S.; Zheng, X.; Zhou, L.; Liu, W.; et al. Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat. Commun. 2020, 11, 5596. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, W.; Liu, T.; Tan, Y.; Chen, C.; Zhao, J.; Geng, H.; Ma, C. The physiological metabolite α-ketoglutarate ameliorates osteoarthritis by regulating mitophagy and oxidative stress. Redox Biol. 2023, 62, 102663. [Google Scholar] [CrossRef]
- Stegen, S.; Moermans, K.; Stockmans, I.; Thienpont, B.; Carmeliet, G. The serine synthesis pathway drives osteoclast differentiation through epigenetic regulation of NFATc1 expression. Nat. Metab. 2024, 6, 141–152. [Google Scholar] [CrossRef]
- Lee, S.; Kim, H.S.; Kim, M.J.; Min, K.Y.; Choi, W.S.; You, J.S. Glutamine metabolite α-ketoglutarate acts as an epigenetic co-factor to interfere with osteoclast differentiation. Bone 2021, 145, 115836. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, J.; Yu, Y.; Ni, Y.; Wei, Y.; Cheng, Y.; Han, L.; Xiao, L.; Ma, X.; Wei, H.; et al. Mitochondrial transfer regulates cell fate through metabolic remodeling in osteoporosis. Adv. Sci. 2023, 10, e2204871. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, F.; Thomas, S.C.; Zhang, Y.; Paul, B.; Sakilam, S.; Chae, S.; Li, P.; Almeter, C.; Kamer, A.R.; et al. Targeting the succinate receptor effectively inhibits periodontitis. Cell Rep. 2022, 40, 111389. [Google Scholar] [CrossRef]
- Lee, W.C.; Ji, X.; Nissim, I.; Long, F. Malic enzyme couples mitochondria with aerobic glycolysis in osteoblasts. Cell Rep. 2020, 32, 108108. [Google Scholar] [CrossRef]
- Michaletti, A.; Gioia, M.; Tarantino, U.; Zolla, L. Effects of microgravity on osteoblast mitochondria: A proteomic and metabolomics profile. Sci. Rep. 2017, 7, 15376. [Google Scholar] [CrossRef]
- Sánchez-de-Diego, C.; Pedrazza, L.; Pimenta-Lopes, C.; Martinez-Martinez, A.; Dahdah, N.; Valer, J.A.; Garcia-Roves, P.; Rosa, J.L.; Ventura, F. NRF2 function in osteocytes is required for bone homeostasis and drives osteocytic gene expression. Redox Biol. 2021, 40, 101845. [Google Scholar] [CrossRef]
- Jaramillo, J.; Taylor, C.; McCarley, R.; Berger, M.; Busse, E.; Sammarco, M.C. Oxaloacetate enhances and accelerates regeneration in young mice by promoting proliferation and mineralization. Front. Cell Dev. Biol. 2023, 11, 1117836. [Google Scholar] [CrossRef]
- Tower, R.J.; Busse, E.; Jaramillo, J.; Lacey, M.; Hoffseth, K.; Guntur, A.R.; Simkin, J.; Sammarco, M.C. Spatial transcriptomics reveals metabolic changes underly age-dependent declines in digit regeneration. Elife 2022, 11, e71542. [Google Scholar] [CrossRef]
- Guo, Q.; Kang, H.; Wang, J.; Dong, Y.; Peng, R.; Zhao, H.; Wu, W.; Guan, H.; Li, F. Inhibition of ACLY leads to suppression of osteoclast differentiation and function via regulation of histone acetylation. J. Bone Miner. Res. 2021, 36, 2065–2080. [Google Scholar] [CrossRef]
- Pouikli, A.; Maleszewska, M.; Parekh, S.; Yang, M.; Nikopoulou, C.; Bonfiglio, J.J.; Mylonas, C.; Sandoval, T.; Schumacher, A.L.; Hinze, Y.; et al. Hypoxia promotes osteogenesis by facilitating acetyl-CoA-mediated mitochondrial-nuclear communication. EMBO J. 2022, 41, e111239. [Google Scholar] [CrossRef]
- Stegen, S.; Rinaldi, G.; Loopmans, S.; Stockmans, I.; Moermans, K.; Thienpont, B.; Fendt, S.M.; Carmeliet, P.; Carmeliet, G. Glutamine metabolism controls chondrocyte identity and function. Dev. Cell 2020, 53, 530–544.e8. [Google Scholar] [CrossRef]
- Watanabe, K.; Iida, M.; Harada, S.; Kato, S.; Kuwabara, K.; Kurihara, A.; Takeuchi, A.; Sugiyama, D.; Okamura, T.; Suzuki, A.; et al. Metabolic profiling of charged metabolites in association with menopausal status in Japanese community-dwelling midlife women: Tsuruoka Metabolomic Cohort Study. Maturitas 2022, 155, 54–62. [Google Scholar] [CrossRef]
- Yu, Y.; Newman, H.; Shen, L.; Sharma, D.; Hu, G.; Mirando, A.J.; Zhang, H.; Knudsen, E.; Zhang, G.F.; Hilton, M.J.; et al. Glutamine metabolism regulates proliferation and lineage allocation in skeletal stem cells. Cell Metab. 2019, 29, 966–978.e4. [Google Scholar] [CrossRef]
- Liu, X.; Yan, Z.; Cai, J.; Wang, D.; Yang, Y.; Ding, Y.; Shao, X.; Hao, X.; Luo, E.; Guo, X.E.; et al. Glucose- and glutamine-dependent bioenergetics sensitize bone mechanoresponse after unloading by modulating osteocyte calcium dynamics. J. Clin. Investig. 2023, 133, e164508. [Google Scholar] [CrossRef]
- Guo, Q.; Zhao, H.; Dong, Z.; Cheng, H.; Zhu, M.; Fang, Z. Inhibiting glutaminase exerts opposite effects on ovariectomy-induced and age-related reductions in murine bone mass. Aging Dis. 2024. [Google Scholar] [CrossRef]
- Peng, R.; Dong, Y.; Zheng, M.; Kang, H.; Wang, P.; Zhu, M.; Song, K.; Wu, W.; Li, F. IL-17 promotes osteoclast-induced bone loss by regulating glutamine-dependent energy metabolism. Cell Death Dis. 2024, 15, 111. [Google Scholar] [CrossRef]
- Huang, X.; Lan, Y.; Shen, J.; Zhao, X.; Zhou, Y.; Wu, W.; Mao, J.; Wu, Y.; Xie, Z.; Chen, Z. M2 macrophages secrete glutamate-containing extracellular vesicles to alleviate osteoporosis by reshaping osteoclast precursor fate. Mol. Ther. 2024, 32, 1158–1177. [Google Scholar] [CrossRef]
- Go, M.; Shin, E.; Jang, S.Y.; Nam, M.; Hwang, G.S.; Lee, S.Y. BCAT1 promotes osteoclast maturation by regulating branched-chain amino acid metabolism. Exp. Mol. Med. 2022, 54, 825–833. [Google Scholar] [CrossRef]
- Vujic, A.; Koo, A.N.M.; Prag, H.A.; Krieg, T. Mitochondrial redox and TCA cycle metabolite signaling in the heart. Free Radic. Biol. Med. 2021, 166, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, S.; Tórtora, V.; Pignataro, F.; Sastre, S.; Castro, I.; Chiribao, M.L.; Robello, C.; Zeida, A.; Santos, J.; Castro, L. Redox sensitive human mitochondrial aconitase and its interaction with frataxin: In vitro and in silico studies confirm that it takes two to tango. Free Radic. Biol. Med. 2023, 197, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Chalifoux, O.; Faerman, B.; Mailloux, R.J. Mitochondrial hydrogen peroxide production by pyruvate dehydrogenase and α-ketoglutarate dehydrogenase in oxidative eustress and oxidative distress. J. Biol. Chem. 2023, 299, 105399. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.R.; Kong, M.J.; Han, S.J.; Kim, J.I.; Park, K.M. Isocitrate dehydrogenase 2 deficiency aggravates prolonged high-fat diet intake-induced hypertension. Redox Biol. 2020, 34, 101548. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Go, Y.; Kim, D.Y.; Lee, S.H.; Kim, O.H.; Jeon, Y.H.; Kwon, T.K.; Bae, J.H.; Song, D.K.; Rhyu, I.J.; et al. Isocitrate dehydrogenase 2 protects mice from high-fat diet-induced metabolic stress by limiting oxidative damage to the mitochondria from brown adipose tissue. Exp. Mol. Med. 2020, 52, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.G.; Yang, M.; Prag, H.A.; Blanco, G.R.; Nikitopoulou, E.; Segarra-Mondejar, M.; Powell, C.A.; Young, T.; Burger, N.; Miljkovic, J.L.; et al. Disruption of the TCA cycle reveals an ATF4-dependent integration of redox and amino acid metabolism. Elife 2021, 10, e72593. [Google Scholar] [CrossRef] [PubMed]
- Altea-Manzano, P.; Vandekeere, A.; Edwards-Hicks, J.; Roldan, M.; Abraham, E.; Lleshi, X.; Guerrieri, A.N.; Berardi, D.; Wills, J.; Junior, J.M.; et al. Reversal of mitochondrial malate dehydrogenase 2 enables anaplerosis via redox rescue in respiration-deficient cells. Mol. Cell. 2022, 82, 4537–4547.e7. [Google Scholar] [CrossRef] [PubMed]
- Stegen, S.; Carmeliet, G. Metabolic regulation of skeletal cell fate and function. Nat. Rev. Endocrinol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Li, X.; Wang, Q.; Yu, L.; Yang, P.; Chen, W.; Yang, X.; Zhou, J.; Geng, D. Redox signaling and antioxidant defense in osteoclasts. Free Radic. Biol. Med. 2024, 212, 403–414. [Google Scholar] [CrossRef]
- Kubo, Y.; Beckmann, R.; Fragoulis, A.; Conrads, C.; Pavanram, P.; Nebelung, S.; Wolf, M.; Wruck, C.J.; Jahr, H.; Pufe, T. Nrf2/ARE signaling directly regulates SOX9 to potentially alter age-dependent cartilage degeneration. Antioxidants. 2022, 11, 263. [Google Scholar] [CrossRef]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as signaling organelles control mammalian stem cell fate. Cell Stem Cell 2021, 28, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Long, Q.; Wu, H.; Zhou, Y.; Duan, L.; Yuan, H.; Ding, Y.; Huang, Y.; Wu, Y.; Huang, J.; et al. Nuclear localization of mitochondrial TCA cycle enzymes modulates pluripotency via histone acetylation. Nat. Commun. 2022, 13, 7414. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhu, F.; Liu, X.; Lu, Y.; Yao, K.; Tian, N.; Tong, L.; Figge, D.A.; Wang, X.; Han, Y.; et al. Non-oxidative pentose phosphate pathway controls regulatory T cell function by integrating metabolism and epigenetics. Nat. Metab. 2022, 4, 559–574. [Google Scholar] [CrossRef]
- Kafkia, E.; Andres-Pons, A.; Ganter, K.; Seiler, M.; Smith, T.S.; Andrejeva, A.; Jouhten, P.; Pereira, F.; Franco, C.; Kuroshchenkova, A.; et al. Operation of a TCA cycle subnetwork in the mammalian nucleus. Sci. Adv. 2022, 8, eabq5206. [Google Scholar] [CrossRef] [PubMed]
- Tournaire, G.; Loopmans, S.; Stegen, S.; Rinaldi, G.; Eelen, G.; Torrekens, S.; Moermans, K.; Carmeliet, P.; Ghesquière, B.; Thienpont, B.; et al. Skeletal progenitors preserve proliferation and self-renewal upon inhibition of mitochondrial respiration by rerouting the TCA cycle. Cell Rep. 2022, 40, 111105. [Google Scholar] [CrossRef] [PubMed]
- Michalak, E.M.; Burr, M.L.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Koutelou, E.; Dent, S.Y.R. Now open: Evolving insights to the roles of lysine acetylation in chromatin organization and function. Mol. Cell. 2022, 82, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Traube, F.R.; Özdemir, D.; Sahin, H.; Scheel, C.; Glück, A.F.; Geserich, A.S.; Oganesian, S.; Kostidis, S.; Iwan, K.; Rahimoff, R.; et al. Redirected nuclear glutamate dehydrogenase supplies Tet3 with α-ketoglutarate in neurons. Nat. Commun. 2021, 12, 4100. [Google Scholar] [CrossRef] [PubMed]
- Ming-Chin Lee, K.; Achuthan, A.A.; De Souza, D.P.; Lupancu, T.J.; Binger, K.J.; Lee, M.K.S.; Xu, Y.; McConville, M.J.; de Weerd, N.A.; Dragoljevic, D.; et al. Type I interferon antagonism of the JMJD3-IRF4 pathway modulates macrophage activation and polarization. Cell Rep. 2022, 39, 110719. [Google Scholar] [CrossRef]
- Wei, J.; Yu, X.; Yang, L.; Liu, X.; Gao, B.; Huang, B.; Dou, X.; Liu, J.; Zou, Z.; Cui, X.L.; et al. FTO mediates LINE1 m6A demethylation and chromatin regulation in mESCs and mouse development. Science 2022, 376, 968–973. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Anmangandla, A.; Ren, Y.; Fu, Q.; Zhang, S.; Lin, H. The Acyl-CoA specificity of human lysine acetyltransferase KAT2A. Biochemistry 2022, 61, 1874–1882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, K.; Xu, J.; Chen, X.; Sheng, C.; Zhang, D.; Yang, Y.; Sun, L.; Zhao, H.; Wang, X.; et al. Acetyltransferases CBP/p300 control transcriptional switch of β-Catenin and Stat1 promoting osteoblast differentiation. J. Bone Miner. Res. 2023, 38, 1885–1899. [Google Scholar] [CrossRef]
- Abe, Y.; Kofman, E.R.; Ouyang, Z.; Cruz-Becerra, G.; Spann, N.J.; Seidman, J.S.; Troutman, T.D.; Stender, J.D.; Taylor, H.; Fan, W.; et al. A TLR4/TRAF6-dependent signaling pathway mediates NCoR coactivator complex formation for inflammatory gene activation. Proc. Natl. Acad. Sci. USA 2024, 121, e2316104121. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, M.; Huynh, N.C.; Okamoto, K.; Muro, R.; Terashima, A.; Kurikawa, Y.; Komatsu, N.; Pluemsakunthai, W.; Nitta, T.; Abe, T.; et al. Stepwise cell fate decision pathways during osteoclastogenesis at single-cell resolution. Nat. Metab. 2020, 2, 1382–1390. [Google Scholar] [CrossRef]
- Pezoa, S.A.; Artinger, K.B.; Niswander, L.A. GCN5 acetylation is required for craniofacial chondrocyte maturation. Dev. Biol. 2020, 464, 24–34. [Google Scholar] [CrossRef]
- Chen, J.; Liu, D.; Chen, B.; Yang, Y.; Zhu, H.; Li, D.; Liu, K.; Zhu, L.; Liu, H.; Li, M.; et al. The histone acetyltransferase Mof regulates Runx2 and Osterix for osteoblast differentiation. Cell Tissue Res. 2023, 393, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, H.; Tang, M.; Guo, C.; Deng, A.; Li, J.; Wang, Y.; Xiao, L.; Yang, G. High methylation of lysine acetyltransferase 6B is associated with the Cobb angle in patients with congenital scoliosis. J. Transl. Med. 2020, 18, 210. [Google Scholar] [CrossRef]
- Wang, L.; You, X.; Ruan, D.; Shao, R.; Dai, H.Q.; Shen, W.; Xu, G.L.; Liu, W.; Zou, W. TET enzymes regulate skeletal development through increasing chromatin accessibility of RUNX2 target genes. Nat. Commun. 2022, 13, 4709. [Google Scholar] [CrossRef]
- Kubo, Y.; Gonzalez, J.A.H.; Beckmann, R.; Weiler, M.; Pahlavani, H.; Saldivar, M.C.; Szymanski, K.; Rosenhain, S.; Fragoulis, A.; Leeflang, S.; et al. Nuclear factor erythroid 2-related factor 2 (Nrf2) deficiency causes age-dependent progression of female osteoporosis. BMC Musculoskelet. Disord. 2022, 23, 1015. [Google Scholar] [CrossRef]
- Thaler, R.; Khani, F.; Sturmlechner, I.; Dehghani, S.S.; Denbeigh, J.M.; Zhou, X.; Pichurin, O.; Dudakovicm, A.; Jerez, S.S.; Zhong, J.; et al. Vitamin C epigenetically controls osteogenesis and bone mineralization. Nat. Commun. 2022, 13, 5883. [Google Scholar] [CrossRef] [PubMed]
- Dusadeemeelap, C.; Rojasawasthien, T.; Matsubara, T.; Kokabu, S.; Addison, W.N. Inhibition of TET-mediated DNA demethylation suppresses osteoblast differentiation. FASEB J. 2022, 36, e22153. [Google Scholar] [CrossRef] [PubMed]
- Cakouros, D.; Hemming, S.; Gronthos, K.; Liu, R.; Zannettino, A.; Shi, S.; Gronthos, S. Specific functions of TET1 and TET2 in regulating mesenchymal cell lineage determination. Epigenetics Chromatin. 2019, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tao, H.; Zhang, H.; Xia, Y.; Bai, J.; Ge, G.; Li, W.; Zhang, W.; Xiao, L.; Xu, Y.; et al. TET2 regulates osteoclastogenesis by modulating autophagy in OVX-induced bone loss. Autophagy 2022, 18, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Sun, J.; Zhang, W.; Wang, X.; Xu, Y.; Peng, Y.; Zhang, L.; Xiong, W.; Liu, Y.; Liu, H. Novel insights into the roles of N6-methyladenosine (m6A) modification and autophagy in human diseases. Int. J. Biol. Sci. 2023, 19, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.S.; Zhang, M.; Chen, P.; Xiong, X.F.; Liu, P.Q.; Wang, H.B.; Wang, J.J.; Shen, J. The m6A demethylase FTO promotes the osteogenesis of mesenchymal stem cells by downregulating PPARG. Acta Pharmacol. Sin. 2022, 43, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Riddle, R.C.; Yang, Q.; Rosen, C.R.; Guttridge, D.C.; Dirckx, N.; Faugere, M.C.; Farber, C.R.; Clemens, T.L. The RNA demethylase FTO is required for maintenance of bone mass and functions to protect osteoblasts from genotoxic damage. Proc. Natl. Acad. Sci. USA 2019, 116, 17980–17989. [Google Scholar] [CrossRef]
- Zhuang, J.; Ning, H.; Wang, M.; Zhao, W.; Jing, Y.; Liu, X.; Zu, J.; Kong, P.; Wang, X.; Sun, C.; et al. Downregulated fat mass and obesity-associated protein inhibits bone resorption and osteoclastogenesis by nuclear factor-kappa B inactivation. Cell Signal 2021, 87, 110137. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, P.; Li, J.; Xie, Z.; Cen, S.; Li, M.; Liu, W.; Ye, G.; Zheng, G.; Ma, M.; et al. The N6-methyladenosine demethylase ALKBH5 negatively regulates the osteogenic differentiation of mesenchymal stem cells through PRMT6. Cell Death Dis. 2021, 12, 578. [Google Scholar] [CrossRef]
- Feng, L.; Fan, Y.; Zhou, J.; Li, S.; Zhang, X. The RNA demethylase ALKBH5 promotes osteoblast differentiation by modulating Runx2 mRNA stability. FEBS Lett. 2021, 595, 2007–2014. [Google Scholar] [CrossRef]
- Qi, Q.; Wang, Y.; Wang, X.; Yang, J.; Xie, Y.; Zhou, J.; Li, X.; Wang, B. Histone demethylase KDM4A regulates adipogenic and osteogenic differentiation via epigenetic regulation of C/EBPα and canonical Wnt signaling. Cell. Mol. Life Sci. 2020, 77, 2407–2421. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Yuan, Q.; Cheng, Y.; Li, J.; Liu, Z.; Liu, Y.; Li, Y.; Su, T.; Wang, J.; Salvo, M.E.; et al. Loss of KDM4B exacerbates bone-fat imbalance and mesenchymal stromal cell exhaustion in skeletal aging. Cell Stem Cell 2021, 28, 1057–1073.e7. [Google Scholar] [CrossRef]
- Yi, S.J.; Jang, Y.J.; Kim, H.J.; Lee, K.; Lee, H.; Kim, Y.; Kim, J.; Hwang, S.Y.; Song, J.S.; Okada, H.; et al. The KDM4B-CCAR1-MED1 axis is a critical regulator of osteoclast differentiation and bone homeostasis. Bone Res. 2021, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Forgetta, V.; Zhou, S.; Richards, J.B.; Greenwood, C.M. Identifying rare genetic determinants for improved polygenic risk prediction of bone mineral density and fracture risk. J. Bone Miner. Res. 2023, 38, 1771–1781. [Google Scholar] [CrossRef]
- Wang, C.; Wang, J.; Li, J.; Hu, G.; Shan, S.; Li, Q.; Zhang, X. KDM5A controls bone morphogenic protein 2-induced osteogenic differentiation of bone mesenchymal stem cells during osteoporosis. Cell Death Dis. 2016, 7, e2335. [Google Scholar] [CrossRef]
- Liu, H.; Zhai, L.; Liu, Y.; Lu, D.; Vander Ark, A.; Yang, T.; Krawczyk, C.M. The histone demethylase KDM5C controls female bone mass by promoting energy metabolism in osteoclasts. Sci. Adv. 2023, 9, eadg0731. [Google Scholar] [CrossRef]
- Gao, C.W.; Lin, W.; Riddle, R.C.; Kushwaha, P.; Boukas, L.; Björnsson, H.T.; Hansen, K.D.; Fahrner, J.A. A mouse model of Weaver syndrome displays overgrowth and excess osteogenesis reversible with KDM6A/6B inhibition. JCI Insight 2024, 9, e173392. [Google Scholar] [CrossRef] [PubMed]
- Pribadi, C.; Cakouros, D.; Camp, E.; Anderson, P.; Gronthos, S. KDM6A-mediated regulation of cranial frontal bone suture fusion in mice is sex dependent. Stem Cells Dev. 2023, 32, 398–409. [Google Scholar] [CrossRef]
- Liu, Z.; Lee, H.L.; Suh, J.S.; Deng, P.; Lee, C.R.; Bezouglaia, O.; Mirnia, M.; Chen, V.; Zhou, M.; Cui, Z.K.; et al. The ERα/KDM6B regulatory axis modulates osteogenic differentiation in human mesenchymal stem cells. Bone Res. 2022, 10, 3. [Google Scholar] [CrossRef]
- Behera, J.; Ison, J.; Voor, M.J.; Tyagi, N. Probiotics stimulate bone formation in obese mice via histone methylations. Theranostics 2021, 11, 8605–8623. [Google Scholar] [CrossRef]
- Shan, L.; Yang, X.; Liao, X.; Yang, Z.; Zhou, J.; Li, X.; Wang, B. Histone demethylase KDM7A regulates bone homeostasis through balancing osteoblast and osteoclast differentiation. Cell Death Dis. 2024, 15, 136. [Google Scholar] [CrossRef] [PubMed]
- Assi, R.; Cherifi, C.; Cornelis, F.M.F.; Zhou, Q.; Storms, L.; Pazmino, S.; Coutinho de Almeida, R.; Meulenbelt, I.; Lories, R.J.; Monteagudo, S. Inhibition of KDM7A/B histone demethylases restores H3K79 methylation and protects against osteoarthritis. Ann. Rheum. Dis. 2023, 82, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Xing, Z.; Hu, Q.; Kong, N.; Liao, C.; Xu, S.; Zhang, J.; Kang, F.; Zhu, X. A bone-targeting near-infrared luminescence nanocarrier facilitates alpha-ketoglutarate efficacy enhancement for osteoporosis therapy. Acta Biomater. 2024, 173, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hu, J.; Faber, J.; Miszuk, J.; Sun, H. Locally delivered metabolite derivative promotes bone regeneration in aged mice. ACS Appl. Bio Mater. 2022, 5, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, C.; Cao, X.; Jia, X.; Chen, X.; Wang, Z.; Xu, W.; Dai, F.; Zhang, S. Mitochondria-targeted supramolecular coordination container encapsulated with exogenous itaconate for synergistic therapy of joint inflammation. Theranostics 2022, 12, 3251–3272. [Google Scholar] [CrossRef]
- Ni, L.; Lin, Z.; Hu, S.; Shi, Y.; Jiang, Z.; Zhao, J.; Zhou, Y.; Wu, Y.; Tian, N.; Sun, L.; et al. Itaconate attenuates osteoarthritis by inhibiting STING/NF-κB axis in chondrocytes and promoting M2 polarization in macrophages. Biochem. Pharmacol. 2022, 198, 114935. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Name | Gene–Gene Interaction | Biological Activity |
|---|---|---|
| ACOS | SOD2, SDHA, IDH3G, IDH3B, IDH3A, IDH2, IDH1, FXN, CS, ACLY | TCA cycle, pyruvate metabolism, amino acid metabolism, cytosine methylation, cellular energetics, butyrate-induced histone acetylation, mitochondrial complex assembly, glutathione metabolism, fatty acid biosynthesis, lipid metabolism |
| IDH | ACO1, ACO2, DLST, IDH1, IDH2, IDH3B, IDH3G, OGDH, OGDHL, SUCLA2 | TCA cycle, pyruvate metabolism, cellular energetics, glycolysis and glucogenesis, neuroinflammation and glutamatergic signaling, one-carbon metabolism, glutathione metabolism, histone methylation |
| OGDH | DLAT, DLD, DLST, IDH1, IDH2, IDH3A, IDH3B, IDH3G, PDHX, SUCLG1 | TCA cycle, pyruvate metabolism, cytosine metabolism, amino acid, VEGF–VEGFRs signaling, angiogenesis, iron metabolism in the placenta, glutathione metabolism, tryptophan metabolism |
| SCS | ACLY, DLAT, DLST, OGDH, SDHA, SDHB, SDHC, SDHD, SUCLA2 SUCLG2 | TCA cycle, pyruvate metabolism, mitochondrial complex assembly, electron transfer chain in OXPHOS, amino acid metabolism, fatty acid biosynthesis |
| SDH | FH, NDUFS2, NDUFS8, NDUFV1, SDHAF2, SDHB, SDHC, SDHD, SUCLG1, F5H5T6 | Mitochondrial complex assembly, TCA cycle, electron transfer chain in mitochondrial OXPHOS, TCA cycle in senescence, urea cycle and associated metabolism, cellular energetics |
| FH | CS GOT2 MDH2 ME1 ME2 ME3 SDHA SDHB SDHC SDHD | Mitochondrial complex II assembly, amino acid metabolism, TCA cycle, pentose phosphate pathway, NAD metabolism, cellular energetics, glycolysis and glucogenesis |
| MDH | ACLY, CS, FH, GOT1, GOT1L1, GOT2, IDH2, ME1, ME3, PC | Butyrate-mediated histone acetylation, TCA cycle in senescence, alanine and aspartate metabolism, urea cycle-associated metabolism, glycolysis and glucogenesis, fatty acid biosynthesis, glutamine metabolism, trans-sulfuration metabolism, pentose phosphate pathway |
| CS | ACLY, ACO1, ACO2, DLAT, FH, IDH1, IDH2, MDH2, PC, SDHB | TCA cycle, cellular energetics, cytosine methylation, amino acid metabolism, glycolysis and glucogenesis, urea cycle-associated metabolism, iron metabolism in the placenta |
| Types | Name | Gene–Gene Interaction | Biological Activity |
|---|---|---|---|
| RNA m6N demethylase | FTO | ZMAT3, CLUAP1, CTSA, IRX5, TERF2IP, TKFC, NDRG1, AMFR, PBX3, CKB, NBAS, BCCIP, EEF2, SF3B3, PEPD, LDOC1, GNE, MPPED2, LDHB, CSTF2T | AMP-activated protein kinase (AMPK) signaling, sterol regulatory element binding protein (SREBP) signaling, FTO obesity variant mechanism, pre-implantation embryo, intercellular component of the RIG receptor pathway, miR-209-3 alteration of the YAP/ECM axis, microRNA regulation of the p53 pathway, Hippo-YAP signaling, urea cycle metabolism |
| Histone lysine demethylase | KDM4A | ARID1B, CAPZA1, EPHB2, FBXL4, H3C1, H4-16, HARS2, IFNB1, IRF3 JADE1, KDM4B, KDM4C, KDM4D, KDM4F, NCOR1, PBRM1, RNF168, RNF8, UTP14A | NIPBL in DNA damage, nucleic acid metabolism, and innate immunity, STING pathway, innate response to dsRNA, TLR4 signaling, and tolerance |
| Histone lysine demethylase | KDM5A | ARID4A, ARNTL, BPTF, CLOCK, COX20, EMSY, EPB41L5, GATAD1, GLIS1, GPN3, HDAC1 JARID2, MORF4L1, MTF2, PHF12, RSF1, SPAG9, STK4, SUZ12, | Interactome of polycomb repressive complex (PRC2), circadian rhythm pathway, melatonin metabolism and effects, exercise-induced circadian regulation, valproic acid pathway, hedgehog signaling pathway, sumolyation by RanBP2-regulated transcription repression, transcription co-factor SKI and SKIL protein partners, energy metabolism |
| Histone lysine demethylase | KDM6A | ASH2L, ATM, DRG1, DYRK1A, FNTA, KLF4, MAP3K2, MSH6, PNPLA4, POU5F1, PROSER1, PUDP, RB1, RBBP5, SIX4, SOX2, SUPT6H, TRAPPC2, WDR5, | Cell cycle G1/S phase transition, cell cycle G2/M phase transition, cell differentiation, DNA damage response, GABPα/β pathway |
| Histone lysine demethylase | KDM7A | ALAS2, CDC27, GPCPD1, HSF2BP, KDM2B, KDM3A, KDM4C, KDM6B, KRR1, KRT2, MCL1, PHF13, PHF2, PHF8, PRKD1, RHD, RNF19A, RORA, TDGF1, UHRF1 | Vitamin D-sensitive calcium signaling, coregulation androgen receptor activity, HIF-1α transcription factor network, apoptosis modulation and signaling, microRNA network, heme biosynthesis, nuclear receptors, regulation of apoptosis by parathyroid hormone-related protein, development and heterogeneity of the ILC family, white fat cell differentiation, FBXL10 enhancement of MAP/ERK signaling |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lian, W.-S.; Wu, R.-W.; Lin, Y.-H.; Chen, Y.-S.; Jahr, H.; Wang, F.-S. Tricarboxylic Acid Cycle Regulation of Metabolic Program, Redox System, and Epigenetic Remodeling for Bone Health and Disease. Antioxidants 2024, 13, 470. https://doi.org/10.3390/antiox13040470
Lian W-S, Wu R-W, Lin Y-H, Chen Y-S, Jahr H, Wang F-S. Tricarboxylic Acid Cycle Regulation of Metabolic Program, Redox System, and Epigenetic Remodeling for Bone Health and Disease. Antioxidants. 2024; 13(4):470. https://doi.org/10.3390/antiox13040470
Chicago/Turabian StyleLian, Wei-Shiung, Re-Wen Wu, Yu-Han Lin, Yu-Shan Chen, Holger Jahr, and Feng-Sheng Wang. 2024. "Tricarboxylic Acid Cycle Regulation of Metabolic Program, Redox System, and Epigenetic Remodeling for Bone Health and Disease" Antioxidants 13, no. 4: 470. https://doi.org/10.3390/antiox13040470
APA StyleLian, W.-S., Wu, R.-W., Lin, Y.-H., Chen, Y.-S., Jahr, H., & Wang, F.-S. (2024). Tricarboxylic Acid Cycle Regulation of Metabolic Program, Redox System, and Epigenetic Remodeling for Bone Health and Disease. Antioxidants, 13(4), 470. https://doi.org/10.3390/antiox13040470