
Extracellular Superoxide Dismutase Attenuates Hepatic Oxidative Stress in Nonalcoholic Fatty Liver Disease through the Adenosine Monophosphate-Activated Protein Kinase Activation
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Recombinant EC-SOD
2.2. Animals and Treatment
2.3. Blood and Urine Parameters
2.4. Histological Assessment
2.5. Western Blot Analysis
2.6. In Vitro Study
2.7. Statistical Analysis
3. Results
3.1. Characteristics of Experimental Mice Groups
3.2. Effects of hEC-SOD on Intrahepatic Histologic Changes Associated with Fibrosis, Inflammation and Lipid Accumulation
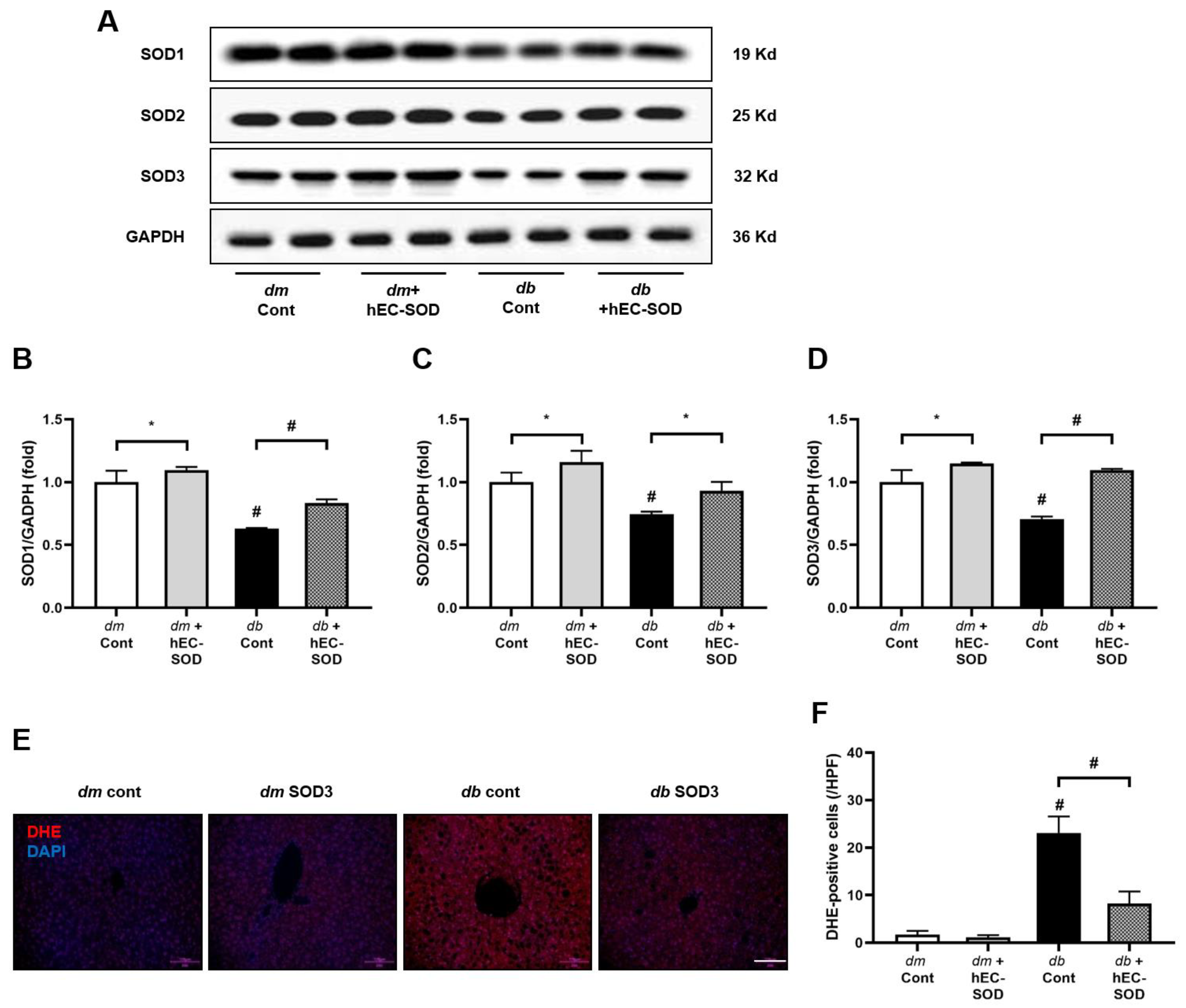
3.3. Intrahepatic SOD Isoforms and SOD3 Expression in Response to hEC-SOD Treatment
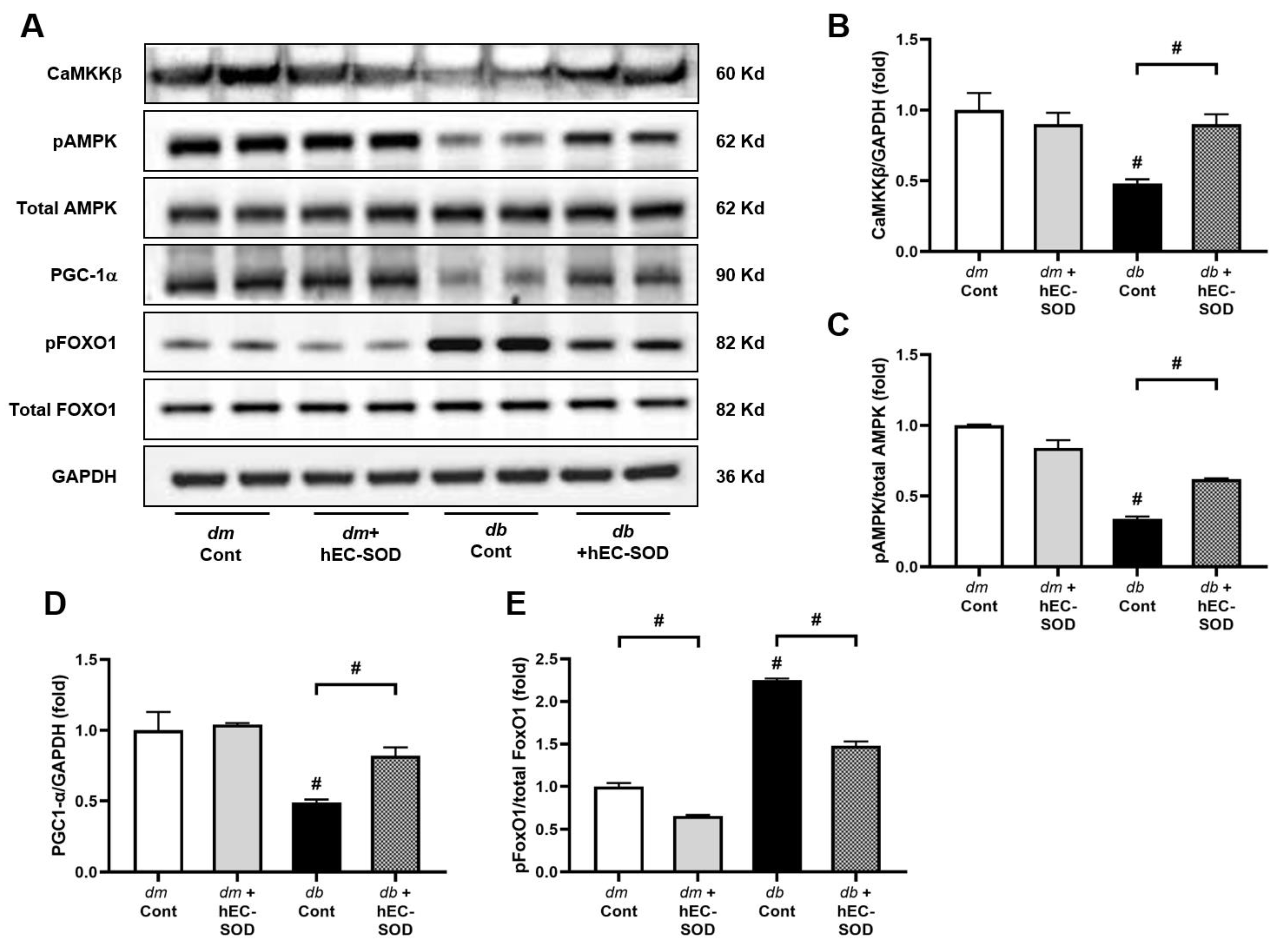
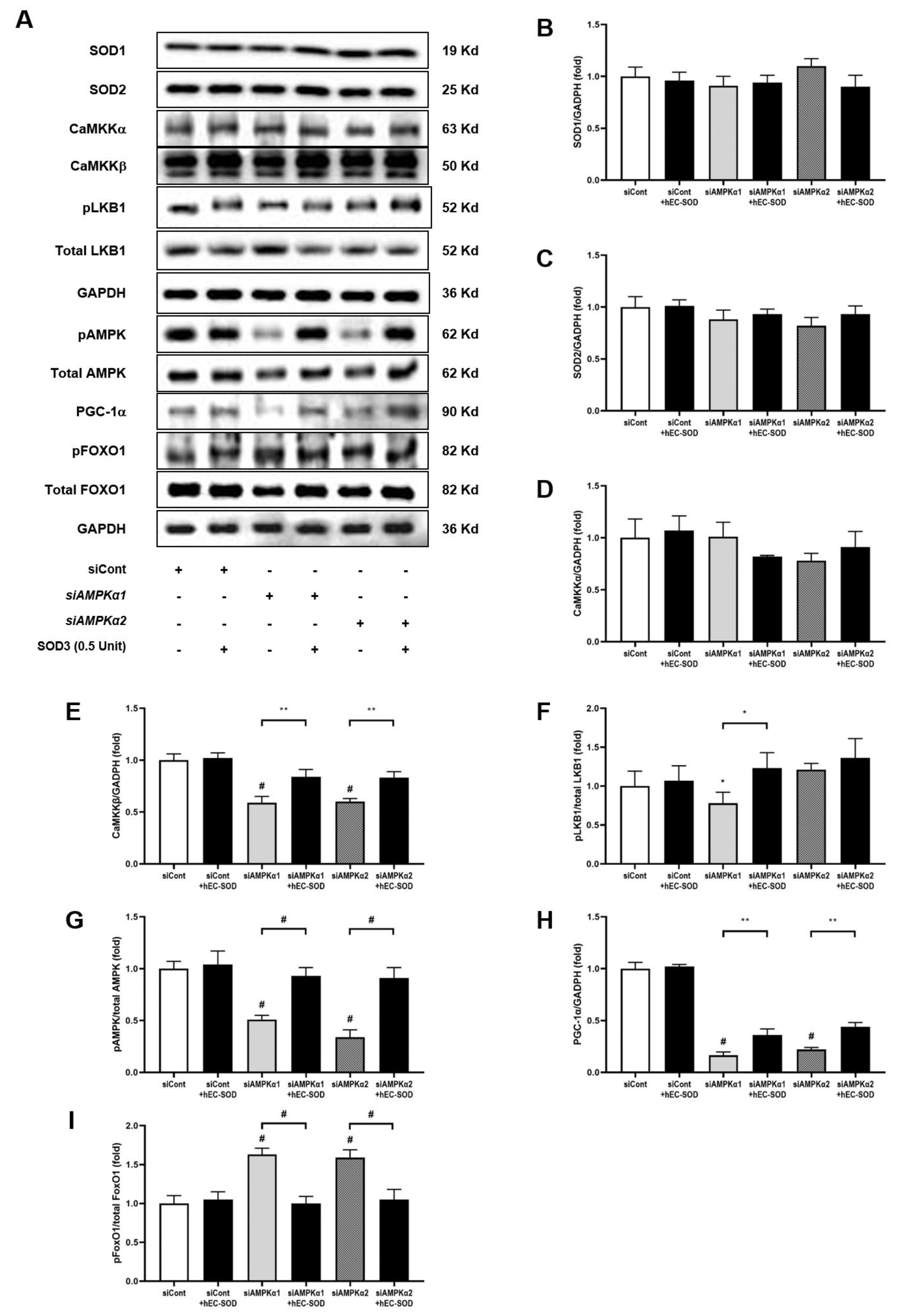
3.4. Effects of hEC-SOD on Intrahepatic Expression of Phospho/Total AMPK and Associated Downstream Signaling Pathways including Phospho-/Total FoxOs and PGC-1α
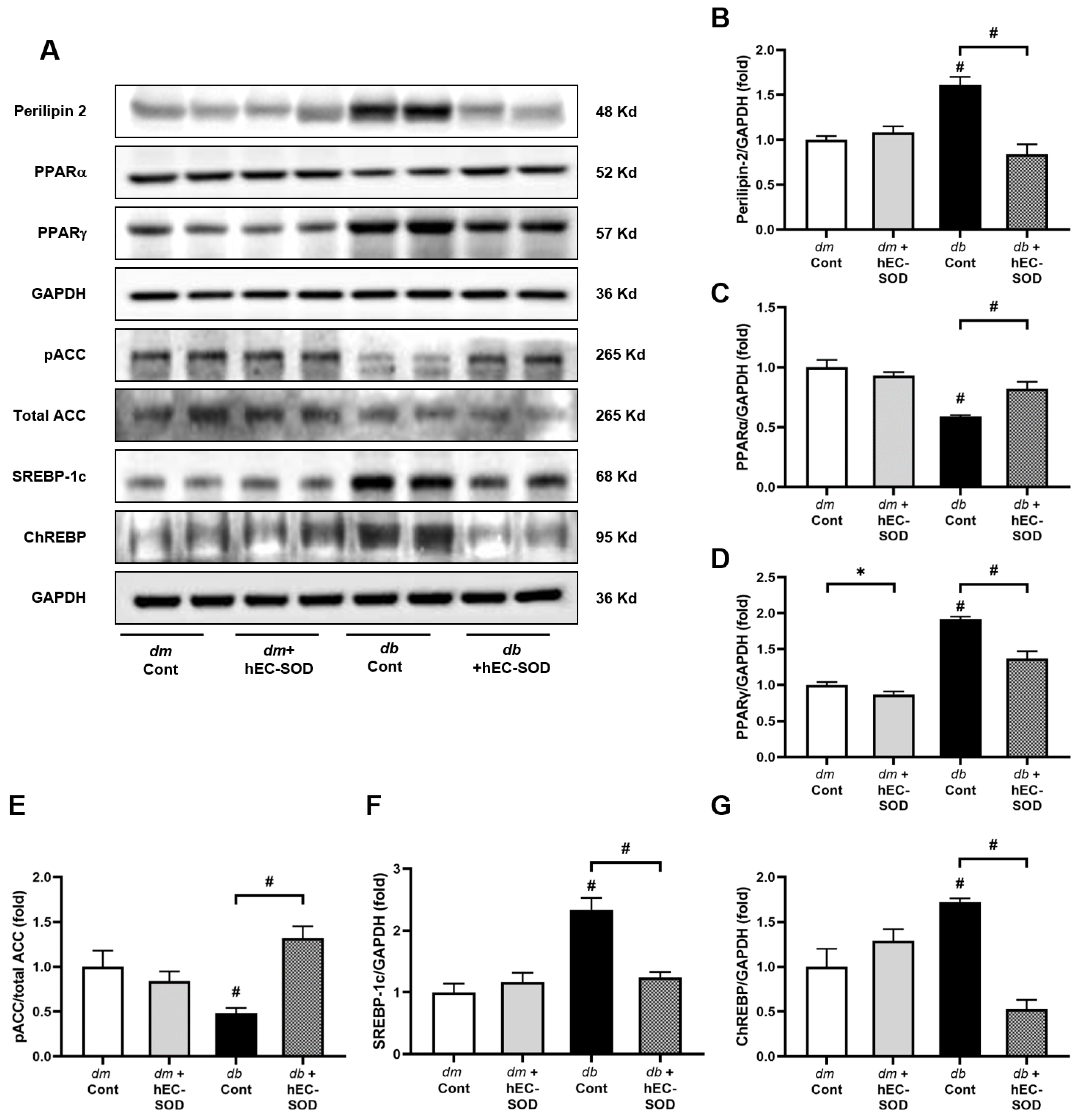
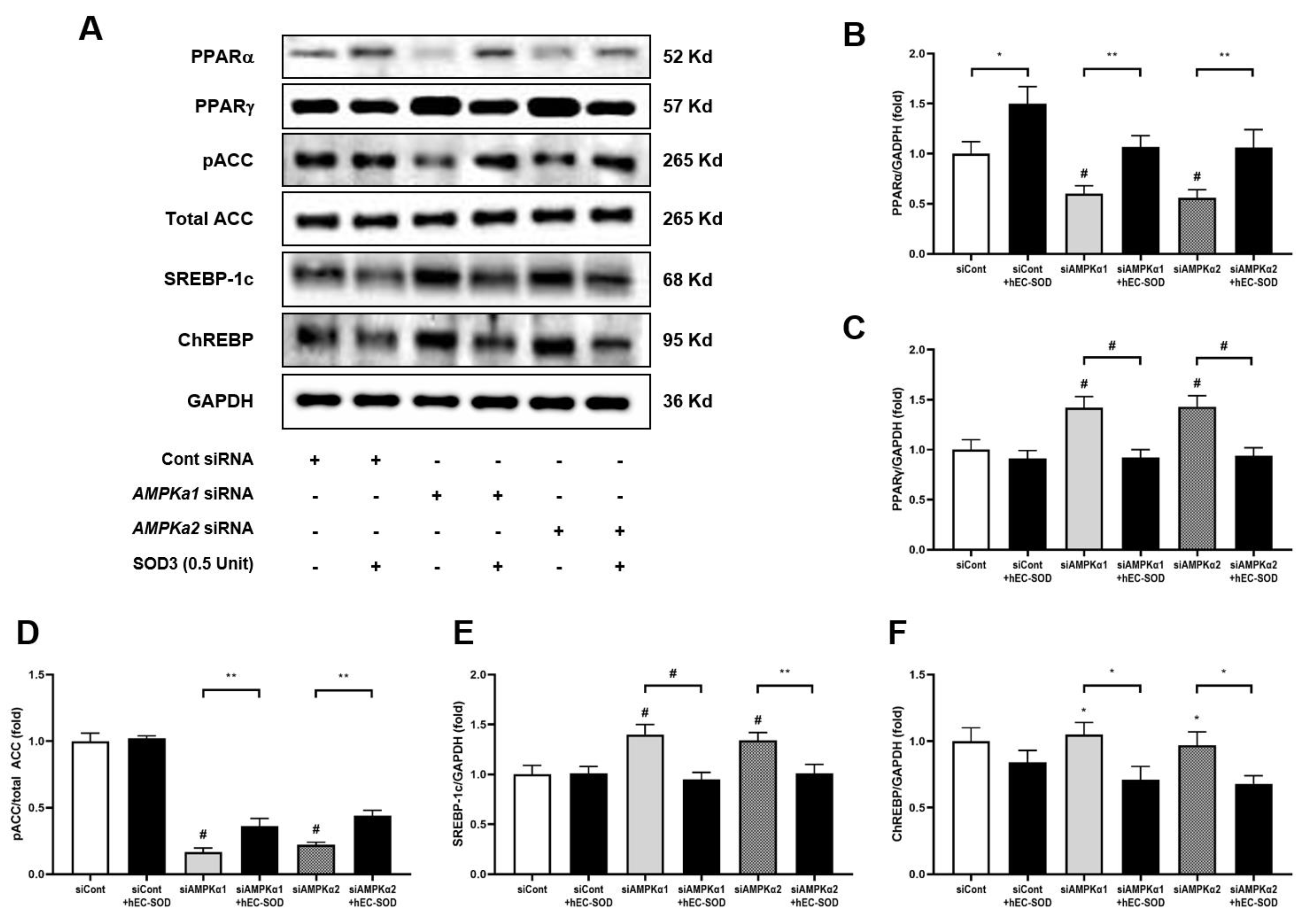
3.5. Effects of EC-SOD on Intrahepatic Expression of Perilipin-2, PPARα/γ, Phospho-ACC and SREBP-1c
3.6. Effect of EC-SOD on Intrahepatic Expression of Bcl-2, BAX, Beclin-1 and LC3
3.7. Inflammatory Cytokines and Cellular Alterations in the Liver Following EC-SOD Treatment
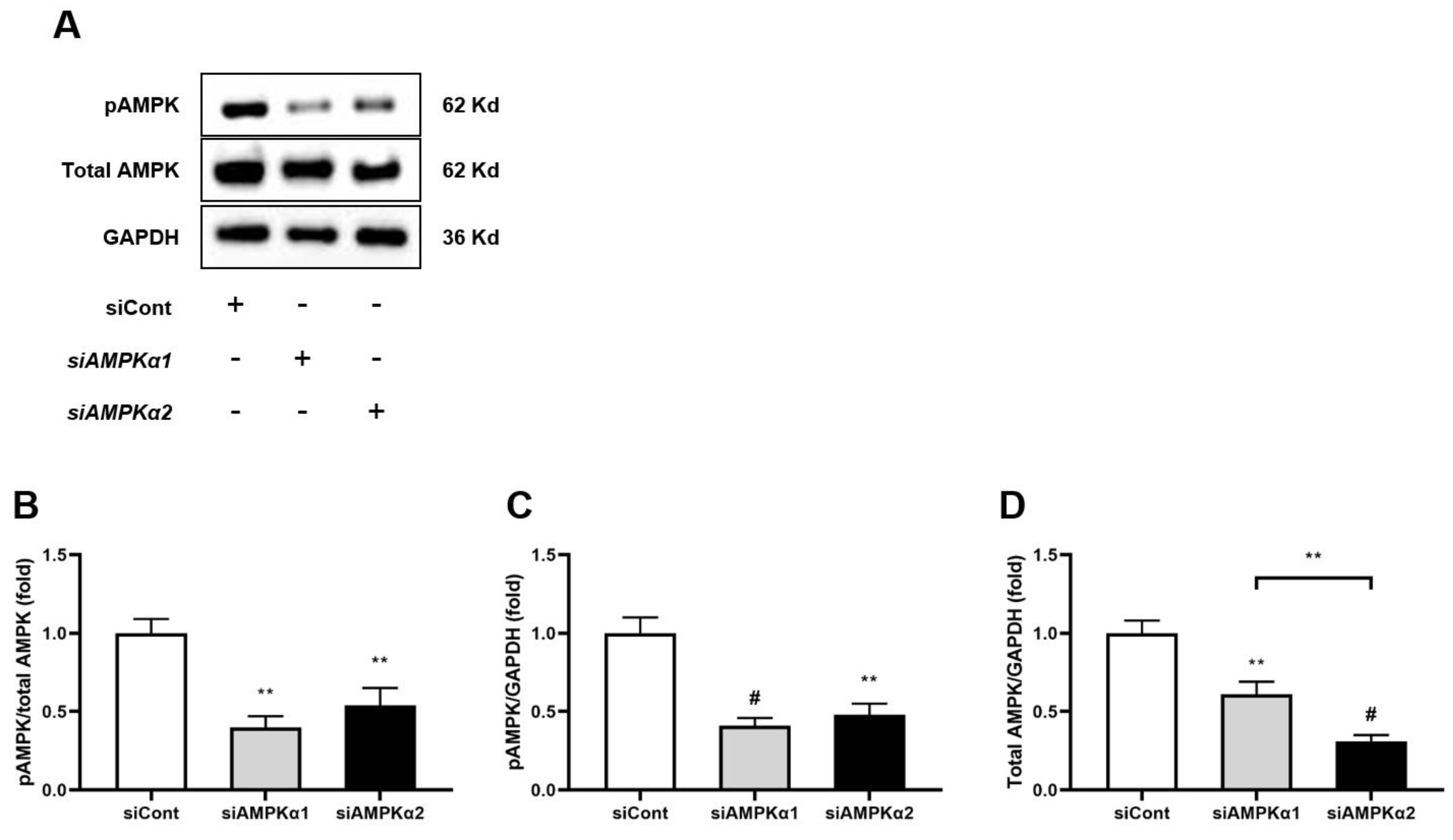
3.8. In Vitro Studies (HepG2)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Mitrofanova, A.; Merscher, S.; Fornoni, A. Kidney lipid dysmetabolism and lipid droplet accumulation in chronic kidney disease. Nat. Rev. Nephrol. 2023, 19, 629–645. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of beta-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- An, Y.; Xu, B.T.; Wan, S.R.; Ma, X.M.; Long, Y.; Xu, Y.; Jiang, Z.Z. The role of oxidative stress in diabetes mellitus-induced vascular endothelial dysfunction. Cardiovasc. Diabetol. 2023, 22, 237. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide anion radical (O2−·), superoxide dismutases, and related matters. J. Biol. Chem. 1997, 272, 18515–18517. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Spaulding, H.R. Extracellular superoxide dismutase, a molecular transducer of health benefits of exercise. Redox Biol. 2020, 32, 101508. [Google Scholar] [CrossRef] [PubMed]
- Fattman, C.L.; Schaefer, L.M.; Oury, T.D. Extracellular superoxide dismutase in biology and medicine. Free. Radic. Biol. Med. 2003, 35, 236–256. [Google Scholar] [CrossRef]
- Gao, D.; Hu, S.; Zheng, X.; Lin, W.; Gao, J.; Chang, K.; Zhao, D.; Wang, X.; Zhou, J.; Lu, S.; et al. SOD3 Is Secreted by Adipocytes and Mitigates High-Fat Diet-Induced Obesity, Inflammation, and Insulin Resistance. Antioxid. Redox Signal. 2020, 32, 193–212. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Gao, M.; Qu, S.; Liu, D. Overexpression of superoxide dismutase 3 gene blocks high-fat diet-induced obesity, fatty liver and insulin resistance. Gene Ther. 2014, 21, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, C.W. Adenosine monophosphate-activated protein kinase in diabetic nephropathy. Kidney Res. Clin. Pract. 2016, 35, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Pan, J.; Qu, N.; Lei, Y.; Han, J.; Zhang, J.; Han, D. The AMPK pathway in fatty liver disease. Front. Physiol. 2022, 13, 970292. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Root-McCaig, J.; Castellani, L.; Kemp, B.E.; Steinberg, G.R.; Wright, D.C. Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity 2014, 22, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, Y.; Suehiro, Y.; Mitani, S.; Xue, D. AMPK-FOXO-IP3R signaling pathway mediates neurological and developmental defects caused by mitochondrial DNA mutations. Proc. Natl. Acad. Sci. USA 2023, 120, e2302490120. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. PGC-1alpha buffers ROS-mediated removal of mitochondria during myogenesis. Cell Death Dis. 2014, 5, e1515. [Google Scholar] [CrossRef] [PubMed]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1alpha Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants 2023, 12, 1075. [Google Scholar] [CrossRef]
- Kim, Y.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Yoon, H.E.; Shin, S.J.; Choi, B.S.; Kim, Y.S.; Chang, Y.S.; Park, C.W. The Adiponectin Receptor Agonist AdipoRon Ameliorates Diabetic Nephropathy in a Model of Type 2 Diabetes. J. Am. Soc. Nephrol. 2018, 29, 1108–1127. [Google Scholar] [CrossRef]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, T.W.; Kim, Y.; Yang, K.S.; Park, H.S.; Choi, S.R.; Chung, S.; Kim, H.W.; et al. Fenofibrate improves renal lipotoxicity through activation of AMPK-PGC-1alpha in db/db mice. PLoS ONE 2014, 9, e96147. [Google Scholar] [CrossRef]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, Y.; Park, H.S.; Kim, H.W.; Choi, B.S.; Chang, Y.S.; Kim, H.W.; Kim, T.Y.; et al. Extracellular Superoxide Dismutase Attenuates Renal Oxidative Stress Through the Activation of Adenosine Monophosphate-Activated Protein Kinase in Diabetic Nephropathy. Antioxid. Redox Signal. 2018, 28, 1543–1561. [Google Scholar] [CrossRef]
- Kwon, M.J.; Han, J.; Kim, B.H.; Lee, Y.S.; Kim, T.Y. Superoxide dismutase 3 suppresses hyaluronic acid fragments mediated skin inflammation by inhibition of toll-like receptor 4 signaling pathway: Superoxide dismutase 3 inhibits reactive oxygen species-induced trafficking of toll-like receptor 4 to lipid rafts. Antioxid. Redox Signal. 2012, 16, 297–313. [Google Scholar] [CrossRef]
- Karlsson, K.; Sandstrom, J.; Edlund, A.; Edlund, T.; Marklund, S.L. Pharmacokinetics of extracellular-superoxide dismutase in the vascular system. Free Radic. Biol. Med. 1993, 14, 185–190. [Google Scholar] [CrossRef]
- Gorecki, M.; Beck, Y.; Hartman, J.R.; Fischer, M.; Weiss, L.; Tochner, Z.; Slavin, S.; Nimrod, A. Recombinant human superoxide dismutases: Production and potential therapeutical uses. Free Radic. Res. Commun. 1991, 12–13, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Arunachalam, G.; Hwang, J.W.; Chung, S.; Sundar, I.K.; Kinnula, V.L.; Crapo, J.D.; Rahman, I. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. USA 2010, 107, 15571–15576. [Google Scholar] [CrossRef]
- Agrahari, G.; Sah, S.K.; Nguyen, C.T.; Choi, S.S.; Kim, H.Y.; Kim, T.Y. Superoxide Dismutase 3 Inhibits LL-37/KLK-5-Mediated Skin Inflammation through Modulation of EGFR and Associated Inflammatory Cascades. J. Investig. Dermatol. 2020, 140, 656–665.e8. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, M.; Oh, S.B.; Kim, H.Y.; Kim, C.; Kim, T.Y.; Park, Y.H. Superoxide dismutase 3 prevents early stage diabetic retinopathy in streptozotocin-induced diabetic rat model. PLoS ONE 2022, 17, e0262396. [Google Scholar] [CrossRef]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef]
- Rinella, M.E.; Neuschwander-Tetri, B.A.; Siddiqui, M.S.; Abdelmalek, M.F.; Caldwell, S.; Barb, D.; Kleiner, D.E.; Loomba, R. AASLD Practice Guidance on the clinical assessment and management of nonalcoholic fatty liver disease. Hepatology 2023, 77, 1797–1835. [Google Scholar] [CrossRef] [PubMed]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Sohn, J.H. Pharmacological advances in the treatment of nonalcoholic fatty liver diseases: Focused on global results of randomized controlled trials. Clin. Mol. Hepatol. 2023, 29, S268–S275. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.C.; Corsi, D.; Cavi, N.; Bruni, N.; Dosio, F. Superoxide Dismutase Administration: A Review of Proposed Human Uses. Molecules 2021, 26, 1844. [Google Scholar] [CrossRef]
- Sun, Y.L.; Bai, T.; Zhou, L.; Zhu, R.T.; Wang, W.J.; Liang, R.P.; Li, J.; Zhang, C.X.; Gou, J.J. SOD3 deficiency induces liver fibrosis by promoting hepatic stellate cell activation and epithelial-mesenchymal transition. J. Cell. Physiol. 2021, 236, 4313–4329. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Y.; Sun, P.; Zhang, W.; Xin, Q.; Wang, N.; Niu, Y.; Chen, Y.; Luo, J.; Lu, J.; et al. Empagliflozin-Enhanced Antioxidant Defense Attenuates Lipotoxicity and Protects Hepatocytes by Promoting FoxO3a- and Nrf2-Mediated Nuclear Translocation via the CAMKK2/AMPK Pathway. Antioxidants 2022, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Barthel, A.; Schmoll, D.; Krüger, K.D.; Roth, R.A.; Joost, H.G. Regulation of the forkhead transcription factor FKHR (FOXO1a) by glucose starvation and AICAR, an activator of AMP-activated protein kinase. Endocrinology 2002, 143, 3183–3186. [Google Scholar] [CrossRef]
- Pettinelli, P.; Videla, L.A. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: An additional reinforcing lipogenic mechanism to SREBP-1c induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef]
- Schadinger, S.E.; Bucher, N.L.; Schreiber, B.M.; Farmer, S.R. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1195–E1205. [Google Scholar] [CrossRef]
- Panasiuk, A.; Dzieciol, J.; Panasiuk, B.; Prokopowicz, D. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J. Gastroenterol. 2006, 12, 6198–6202. [Google Scholar] [CrossRef]
- Park, H.; Hasegawa, G.; Obayashi, H.; Fujinami, A.; Ohta, M.; Hara, H.; Adachi, T.; Tamaki, S.; Nakajima, Y.; Kimura, F.; et al. Relationship between insulin resistance and inflammatory markers and anti-inflammatory effect of losartan in patients with type 2 diabetes and hypertension. Clin. Chim. Acta 2006, 374, 129–134. [Google Scholar] [CrossRef]
- Sasaki, T.; Abe, Y.; Takayama, M.; Adachi, T.; Okano, H.; Hirose, N.; Arai, Y. Association among extracellular superoxide dismutase genotype, plasma concentration, and comorbidity in the very old and centenarians. Sci. Rep. 2021, 11, 8539. [Google Scholar] [CrossRef] [PubMed]
- Mohammedi, K.; Bellili-Munoz, N.; Marklund, S.L.; Driss, F.; Le Nagard, H.; Patente, T.A.; Fumeron, F.; Roussel, R.; Hadjadj, S.; Marre, M.; et al. Plasma extracellular superoxide dismutase concentration, allelic variations in the SOD3 gene and risk of myocardial infarction and all-cause mortality in people with type 1 and type 2 diabetes. Cardiovasc. Diabetol. 2015, 14, 845. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Shankar, S.S.; Yates, K.P.; Bolognese, J.; Daly, E.; Dehn, C.A.; Neuschwander-Tetri, B.; Kowdley, K.; Vuppalanchi, R.; Behling, C.; et al. Diagnostic performance of circulating biomarkers for non-alcoholic steatohepatitis. Nat. Med. 2023, 29, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Tincopa, M.A.; Loomba, R. Non-invasive diagnosis and monitoring of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Lancet Gastroenterol. Hepatol. 2023, 8, 660–670. [Google Scholar] [CrossRef]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar]
- Kumar, A.; Sharma, A.; Duseja, A.; Das, A.; Dhiman, R.K.; Chawla, Y.K.; Kohli, K.K.; Bhansali, A. Patients with Nonalcoholic Fatty Liver Disease (NAFLD) have Higher Oxidative Stress in Comparison to Chronic Viral Hepatitis. J. Clin. Exp. Hepatol. 2013, 3, 12–18. [Google Scholar] [CrossRef]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef]
- Yao, N.; He, Y.; Wu, Y.; Wang, F.; Tian, Z. Prognostic value of plasma level of superoxide dismutase in HBV-related acute-on-chronic liver failure. BMC Gastroenterol. 2022, 22, 312. [Google Scholar] [CrossRef]
- He, Y.; Wang, F.; Yao, N.; Wu, Y.; Zhao, Y.; Tian, Z. Serum superoxide dismutase level is a potential biomarker of disease prognosis in patients with HEV-induced liver failure. BMC Gastroenterol. 2022, 22, 14. [Google Scholar] [CrossRef]
- Tian, Z.; Yao, N.; Wu, Y.; Wang, F.; Zhao, Y. Association between plasma level of superoxide dismutase and survival of patients with acute-on-chronic liver failure. BMC Gastroenterol. 2022, 22, 50. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | db/m Control | db/m hEC-SOD | db/db Control | db/db hEC-SOD |
|---|---|---|---|---|
| Body weight (g) | 30.9 ± 1.6 | 30.2 ± 0.8 | 55.6 ± 2.7 † | 40.5 ± 4.8 ‡ |
| Liver weight (g) | 1.6 ± 0.3 | 1.4 ± 0.1 | 3.4 ± 0.9 † | 2.0 ± 0.1 ** |
| Epididymal wt (g) | 0.7 ± 0.2 | 0.4 ± 0.1 | 2.5 ± 0.3 † | 1.7 ± 0.2 ‡ |
| Glucose (mg/dL) | 149.7 ± 37.9 | 137.3 ± 18.4 | 543.8 ± 34.1 † | 537.3 ± 25.0 † |
| HbA1c (%) | 4.1 ± 0.1 | 3.9 ± 0.1 | 12.7 ± 1.1 † | 12.0 ± 0.6 † |
| Insulin (ng/mL) | 0.36 ± 0.06 | 0.37 ± 0.14 | 4.75 ± 1.32 † | 1.81 ± 0.69 ‡ |
| HOMAIR | 1.30 ± 0.40 | 1.42 ± 0.49 | 7.09 ± 2.31 † | 3.13 ± 0.86 ‡ |
| Urinary 8-OH-dG (ng) | 30.8 ± 1.6 | 26.8 ± 7.7 | 224.5 ± 73.1 † | 42.0 ± 16.6 ‡ |
| Urinary isoprostane (ng/24 h) | 4.2 ± 1.5 | 3.5 ± 0.6 | 44.5 ± 13.8 † | 18.5 ± 3.8 ‡ |
| MCP-1 (pg/mL) | 141.5 ± 11.9 | 146.5 ± 18.6 | 343.1 ± 38.0 † | 187.9 ± 28.7 ‡ |
| TNF-α (pg/mL) | 2.2 ± 0.3 | 2.1 ± 0.3 | 2.9 ± 0.4 † | 2.0 ± 0.2 ‡ |
| ALT (U/L) | 23.7 ± 6.8 | 21.7 ± 2.8 | 52.2 ± 8.5 † | 31.5 ± 5.9 ‡ |
| AST (U/L) | 27.5 ± 3.3 | 24.3 ± 6.9 | 49.2 ± 4.1 † | 32.2 ± 2.9 ‡ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, H.; Lim, J.H.; Kim, T.W.; Kim, E.N.; Oum, S.-J.; Bae, S.H.; Park, C.W. Extracellular Superoxide Dismutase Attenuates Hepatic Oxidative Stress in Nonalcoholic Fatty Liver Disease through the Adenosine Monophosphate-Activated Protein Kinase Activation. Antioxidants 2023, 12, 2040. https://doi.org/10.3390/antiox12122040
Nam H, Lim JH, Kim TW, Kim EN, Oum S-J, Bae SH, Park CW. Extracellular Superoxide Dismutase Attenuates Hepatic Oxidative Stress in Nonalcoholic Fatty Liver Disease through the Adenosine Monophosphate-Activated Protein Kinase Activation. Antioxidants. 2023; 12(12):2040. https://doi.org/10.3390/antiox12122040
Chicago/Turabian StyleNam, Heechul, Ji Hee Lim, Tae Woo Kim, Eun Nim Kim, Sae-Jong Oum, Si Hyun Bae, and Cheol Whee Park. 2023. "Extracellular Superoxide Dismutase Attenuates Hepatic Oxidative Stress in Nonalcoholic Fatty Liver Disease through the Adenosine Monophosphate-Activated Protein Kinase Activation" Antioxidants 12, no. 12: 2040. https://doi.org/10.3390/antiox12122040