
Colchicine Protects against Ethanol-Induced Senescence and Senescence-Associated Secretory Phenotype in Endothelial Cells

 ,
,  , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Culture
2.2. β-Gal Staining
2.3. Immunofluorescence Staining
2.4. Western Blot (WB)
2.5. Quantitative Polymerase Chain Reaction (qPCR)
2.6. Statistical Analysis
3. Results
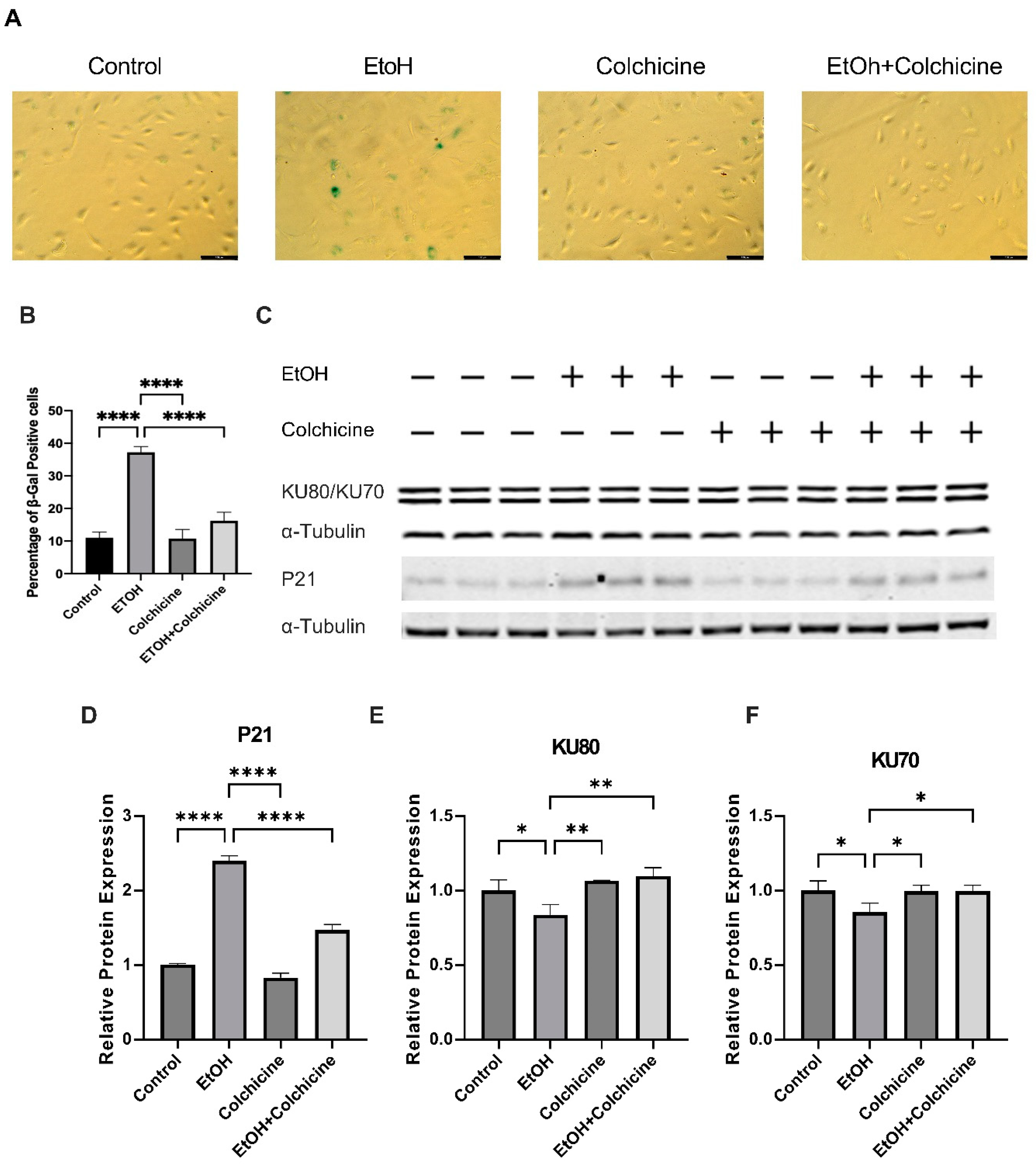
3.1. Colchicine Inhibited Ethanol-Induced Senescence
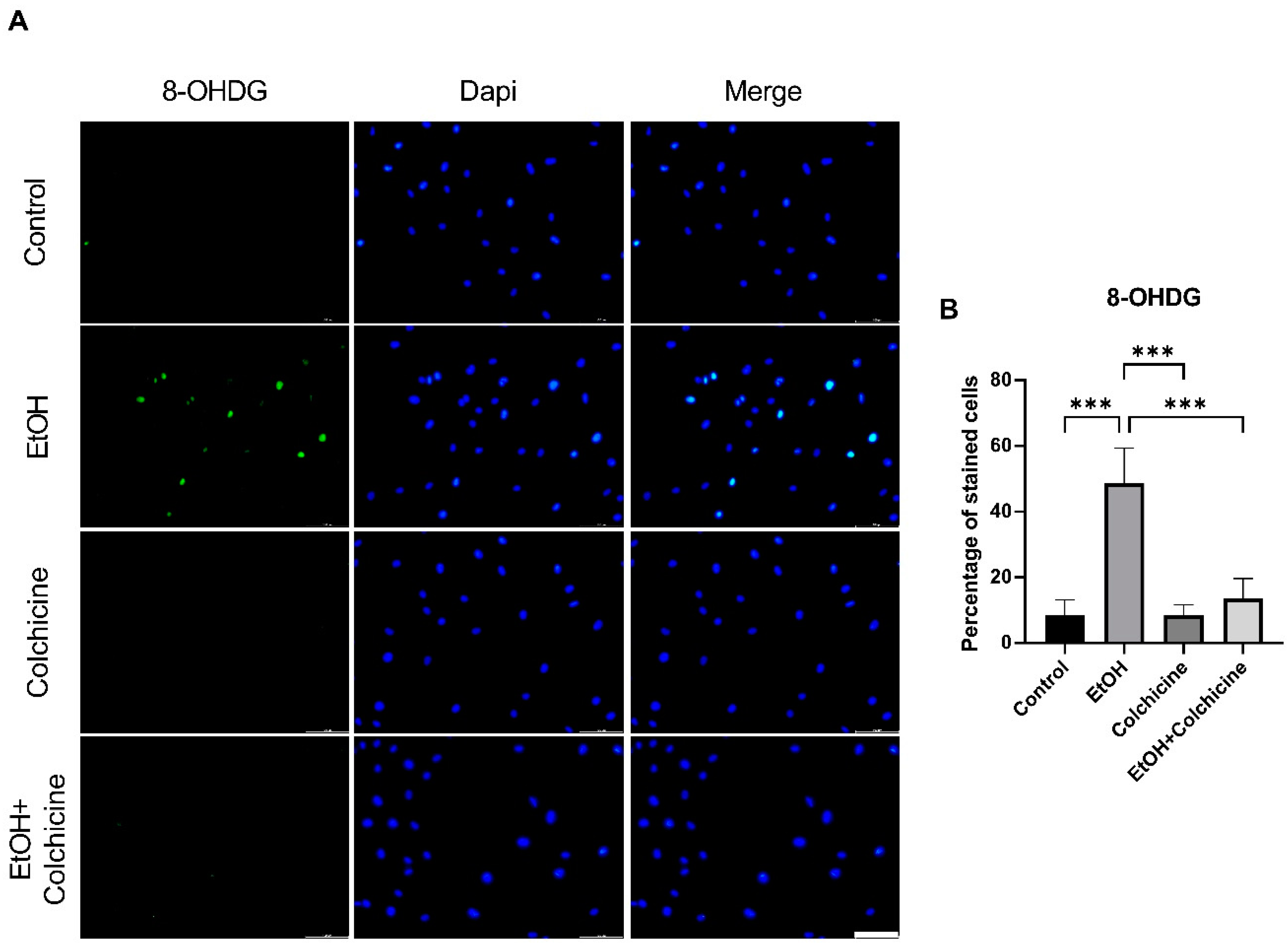
3.2. Colchicine Averted Ethanol-Induced Oxidative Stress in Endothelial Cells
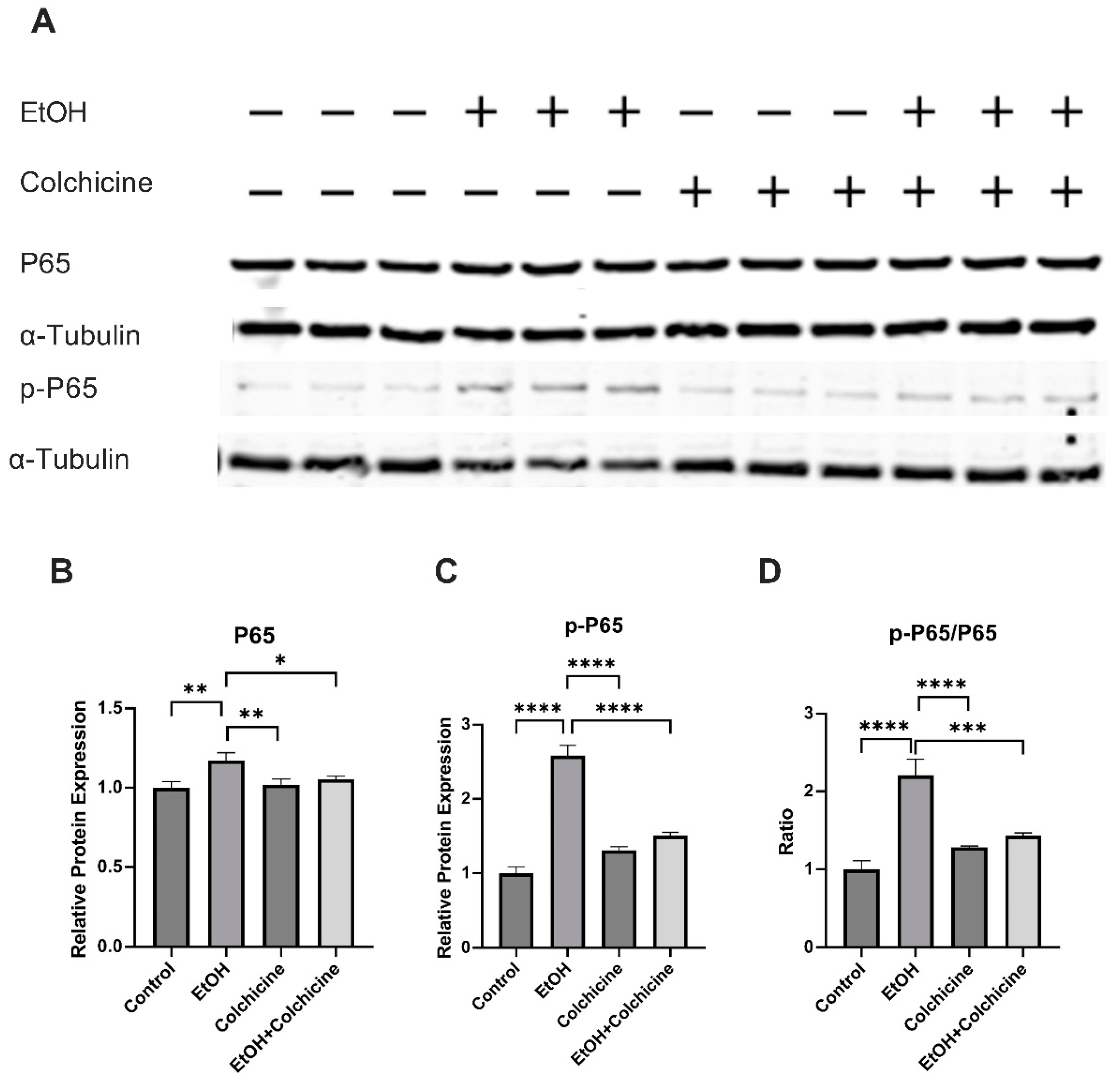
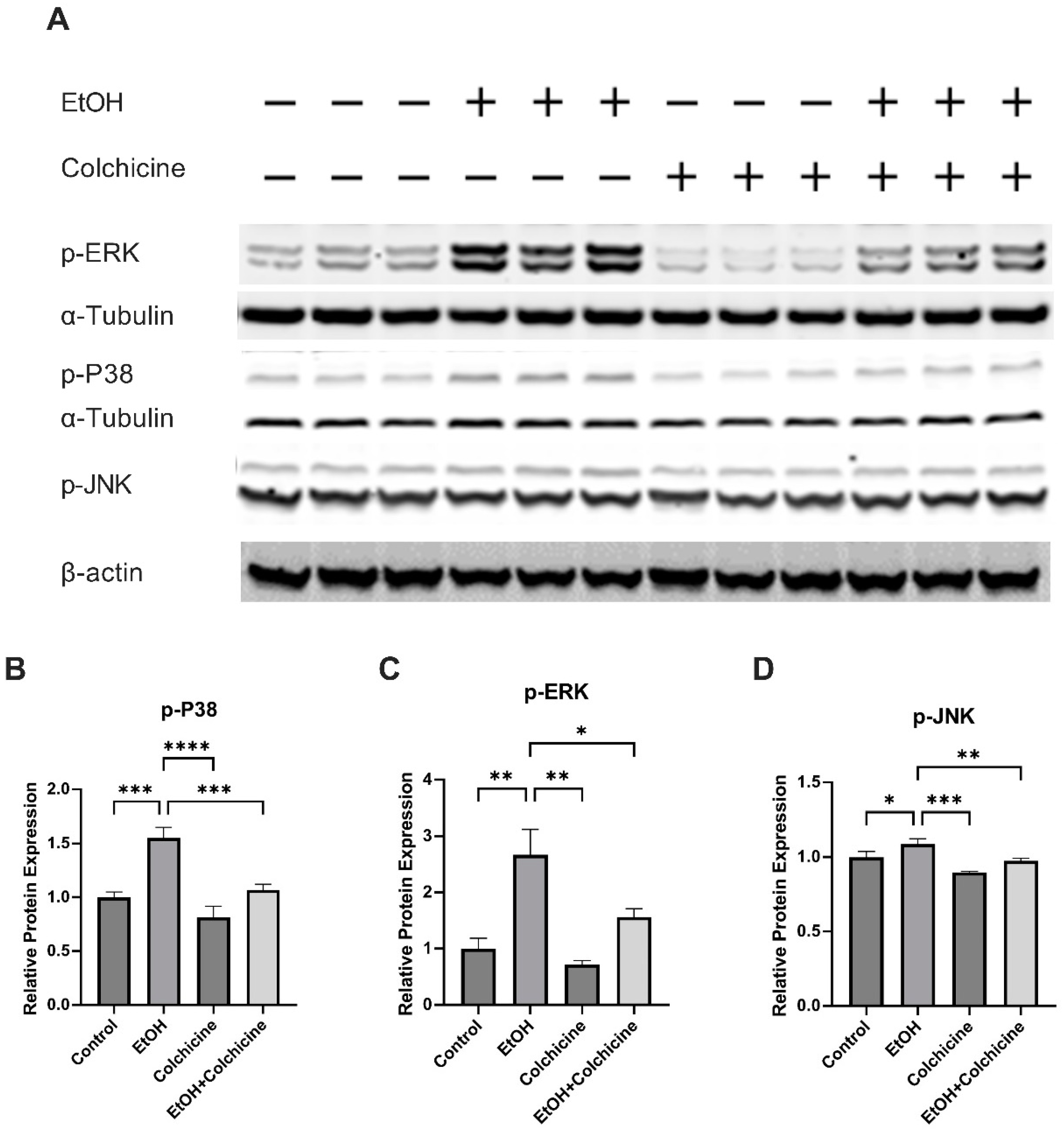
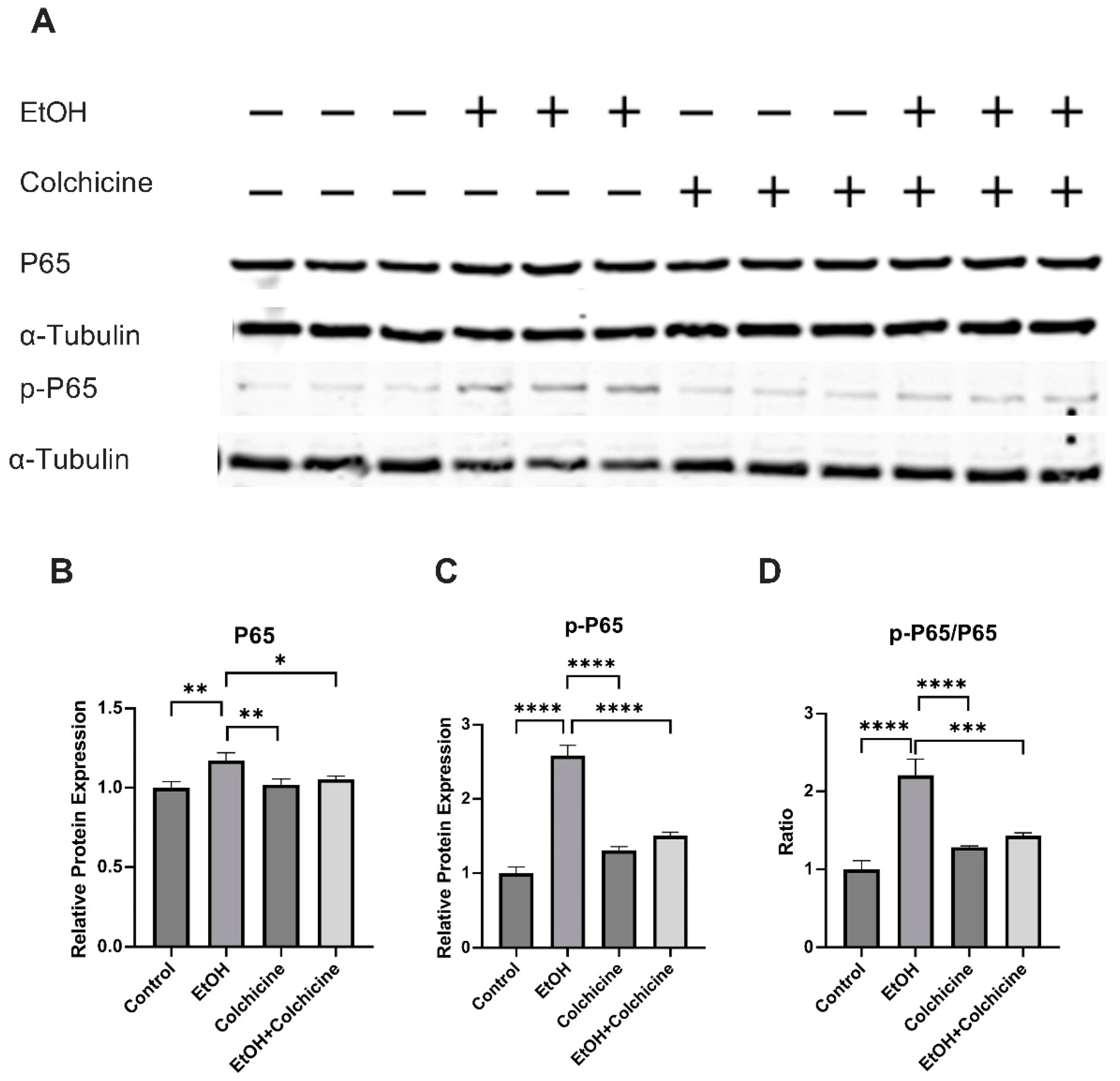
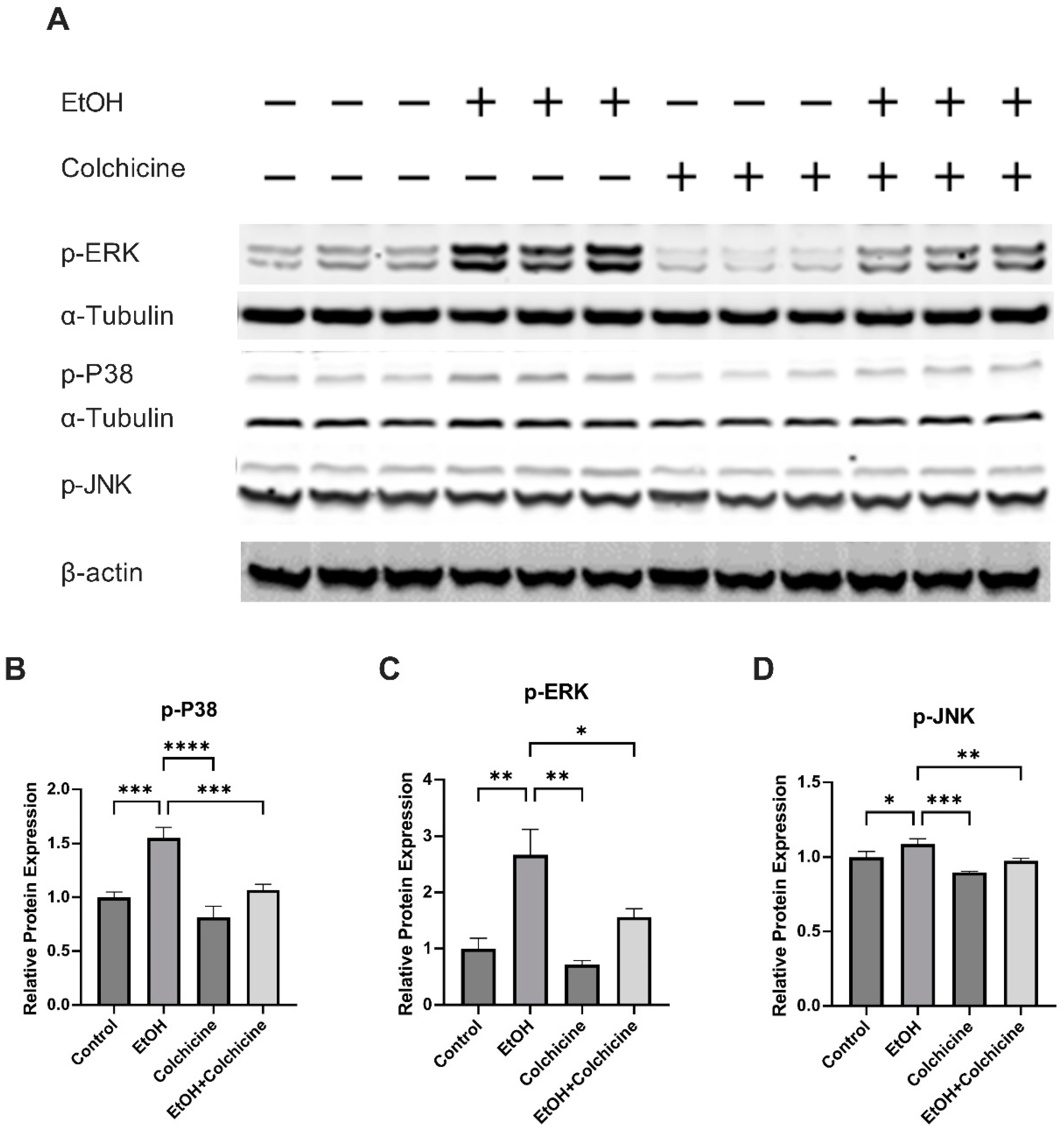
3.3. Colchicine Suppressed the Activation of NF-κB and MAPKs in Ethanol-Treated Endothelial Cells
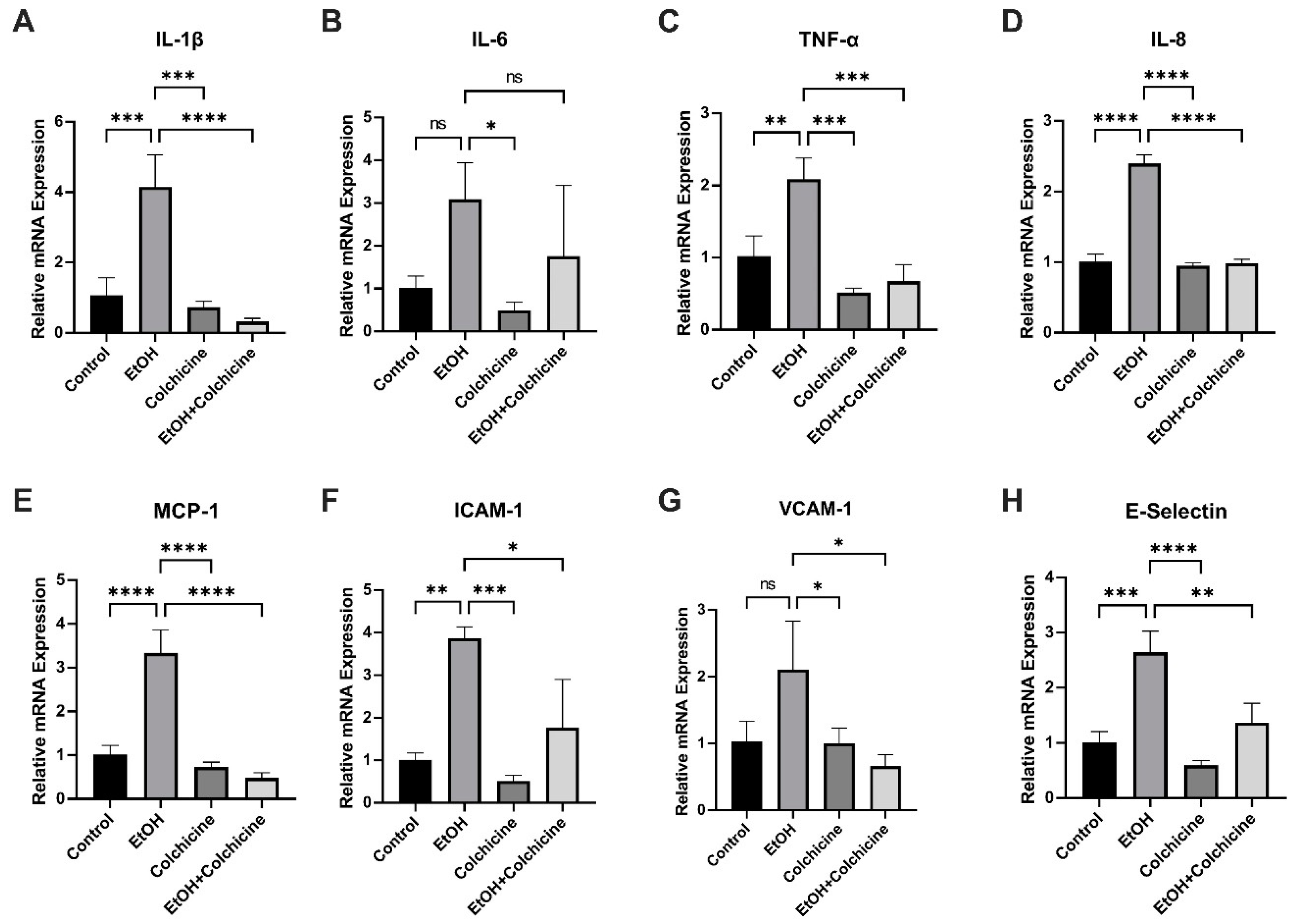
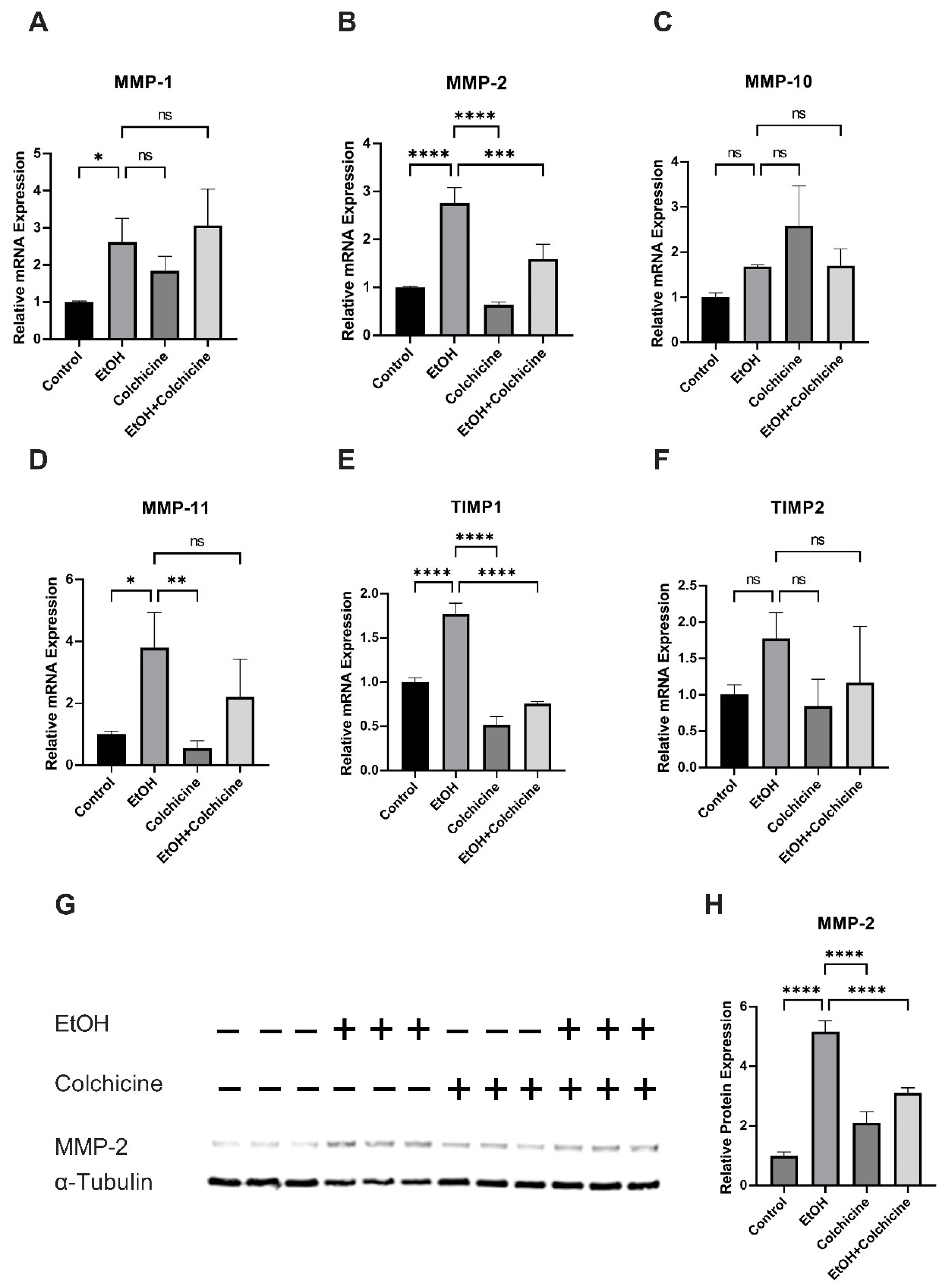
3.4. Colchicine Ameliorated Ethanol-Induced SASP in Endothelial Cells
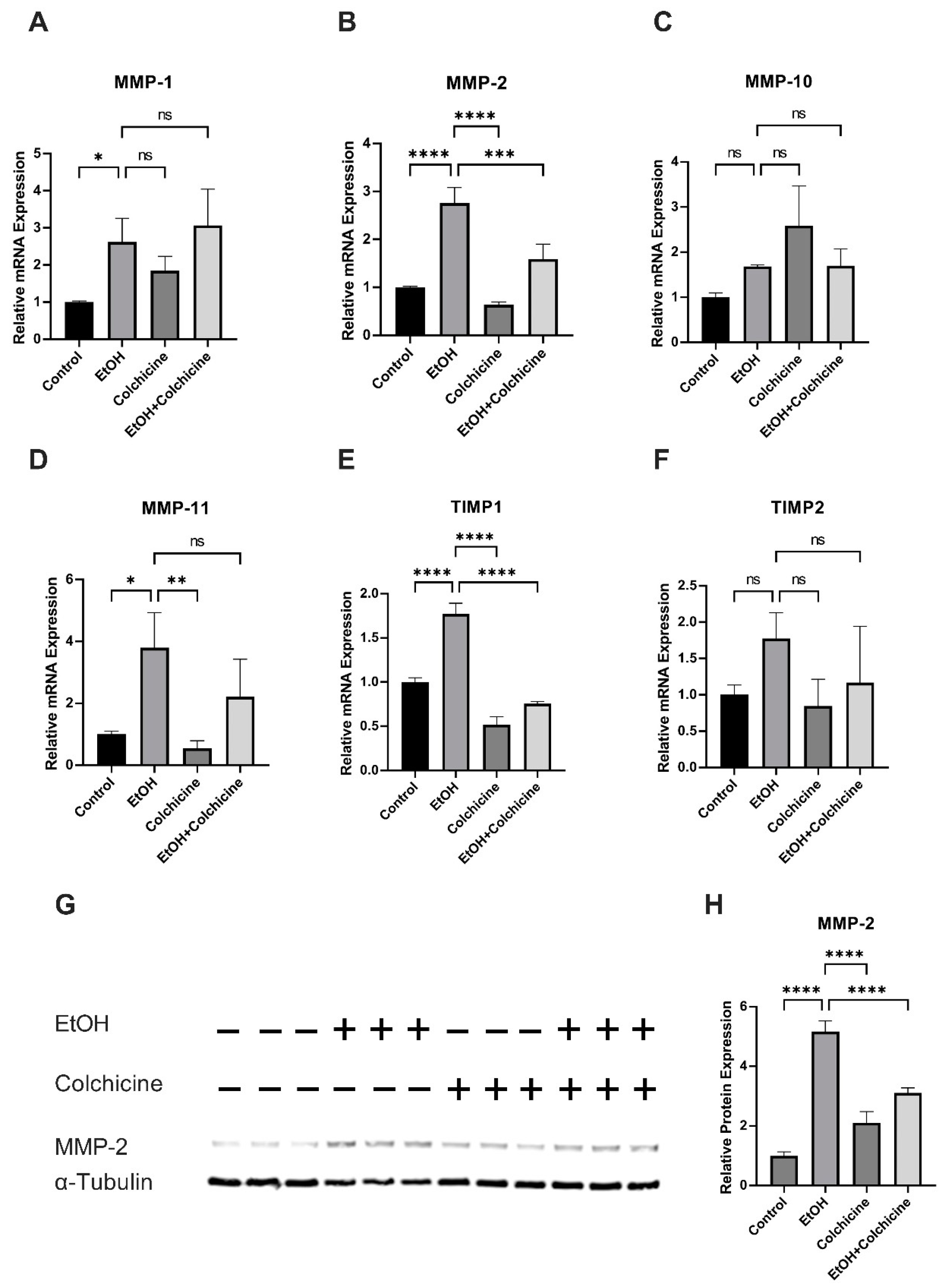
3.5. Colchicine Mitigated Ethanol-Induced Relative mRNA and Relative Protein Expression of MMP-2
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, X.; Liu, T.; Zhu, X.; Pan, X. The multifaceted role of the SASP in atherosclerosis: From mechanisms to therapeutic opportunities. Cell Biosci. 2022, 12, 74. [Google Scholar] [CrossRef]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5, 18. [Google Scholar] [CrossRef]
- Owens, W.A.; Walaszczyk, A.; Spyridopoulos, I.; Dookun, E.; Richardson, G.D. Senescence and senolytics in cardiovascular disease: Promise and potential pitfalls. Mech. Ageing Dev. 2021, 198, 111540. [Google Scholar] [CrossRef]
- Song, S.; Tchkonia, T.; Jiang, J.; Kirkland, J.L.; Sun, Y. Targeting Senescent Cells for a Healthier Aging: Challenges and Opportunities. Adv. Sci. Weinh. 2020, 7, 2002611. [Google Scholar] [CrossRef]
- Frej, F.; Peter, M.N. Chapter 20—Telomere Biology and Vascular Aging. In Early Vascular Aging (EVA); Nilsson, P.M., Olsen, M.H., Laurent, S., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 201–211. [Google Scholar]
- Haga, M.; Okada, M. Systems approaches to investigate the role of NF-kappaB signaling in aging. Biochem. J. 2022, 479, 161–183. [Google Scholar] [CrossRef]
- Aoki, T.; Fukuda, M.; Nishimura, M.; Nozaki, K.; Narumiya, S. Critical role of TNF-alpha-TNFR1 signaling in intracranial aneurysm formation. Acta Neuropathol. Commun. 2014, 2, 34. [Google Scholar] [CrossRef]
- Aoki, T.; Kataoka, H.; Ishibashi, R.; Nozaki, K.; Egashira, K.; Hashimoto, N. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke 2009, 40, 942–951. [Google Scholar] [CrossRef]
- Bot, I.; Ortiz Zacarias, N.V.; de Witte, W.E.; de Vries, H.; van Santbrink, P.J.; van der Velden, D.; Kroner, M.J.; van der Berg, D.J.; Stamos, D.; de Lange, E.C.; et al. A novel CCR2 antagonist inhibits atherogenesis in apoE deficient mice by achieving high receptor occupancy. Sci. Rep. 2017, 7, 52. [Google Scholar] [CrossRef]
- Branen, L.; Hovgaard, L.; Nitulescu, M.; Bengtsson, E.; Nilsson, J.; Jovinge, S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2137–2142. [Google Scholar] [CrossRef]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell 1998, 2, 275–281. [Google Scholar] [CrossRef]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef]
- Starke, R.M.; Chalouhi, N.; Jabbour, P.M.; Tjoumakaris, S.I.; Gonzalez, L.F.; Rosenwasser, R.H.; Wada, K.; Shimada, K.; Hasan, D.M.; Greig, N.H.; et al. Critical role of TNF-alpha in cerebral aneurysm formation and progression to rupture. J. Neuroinflamm. 2014, 11, 77. [Google Scholar] [CrossRef]
- Saha, R.N.; Jana, M.; Pahan, K. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J. Immunol. 2007, 179, 7101–7109. [Google Scholar] [CrossRef]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; St Croix, C.M.; Usas, A.; Vo, N.; et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Investig. 2012, 122, 2601–2612. [Google Scholar] [CrossRef]
- Garcia-Garcia, V.A.; Alameda, J.P.; Page, A.; Casanova, M.L. Role of NF-kappaB in Ageing and Age-Related Diseases: Lessons from Genetically Modified Mouse Models. Cells 2021, 10, 1906. [Google Scholar] [CrossRef]
- Hongo, A.; Okumura, N.; Nakahara, M.; Kay, E.P.; Koizumi, N. The Effect of a p38 Mitogen-Activated Protein Kinase Inhibitor on Cellular Senescence of Cultivated Human Corneal Endothelial Cells. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3325–3334. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Li, X.; Khan, D.; Rana, M.; Hänggi, D.; Muhammad, S. Doxycycline Attenuated Ethanol-Induced Inflammaging in Endothelial Cells: Implications in Alcohol-Mediated Vascular Diseases. Antioxidants 2022, 11, 2413. [Google Scholar] [CrossRef]
- Chen, X.; Li, M.; Yan, J.; Liu, T.; Pan, G.; Yang, H.; Pei, M.; He, F. Alcohol Induces Cellular Senescence and Impairs Osteogenic Potential in Bone Marrow-Derived Mesenchymal Stem Cells. Alcohol Alcohol. 2017, 52, 289–297. [Google Scholar] [CrossRef]
- Chen, J.R.; Lazarenko, O.P.; Haley, R.L.; Blackburn, M.L.; Badger, T.M.; Ronis, M.J. Ethanol impairs estrogen receptor signaling resulting in accelerated activation of senescence pathways, whereas estradiol attenuates the effects of ethanol in osteoblasts. J. Bone Miner. Res. 2009, 24, 221–230. [Google Scholar] [CrossRef]
- Blanco, A.M.; Valles, S.L.; Pascual, M.; Guerri, C. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J. Immunol. 2005, 175, 6893–6899. [Google Scholar] [CrossRef]
- Ku, B.M.; Lee, Y.K.; Jeong, J.Y.; Mun, J.; Han, J.Y.; Roh, G.S.; Kim, H.J.; Cho, G.J.; Choi, W.S.; Yi, G.S.; et al. Ethanol-induced oxidative stress is mediated by p38 MAPK pathway in mouse hippocampal cells. Neurosci. Lett. 2007, 419, 64–67. [Google Scholar] [CrossRef]
- Zhang, F.S.; He, Q.Z.; Qin, C.H.; Little, P.J.; Weng, J.P.; Xu, S.W. Therapeutic potential of colchicine in cardiovascular medicine: A pharmacological review. Acta Pharmacol. Sin. 2022, 43, 2173–2190. [Google Scholar] [CrossRef]
- Deftereos, S.G.; Beerkens, F.J.; Shah, B.; Giannopoulos, G.; Vrachatis, D.A.; Giotaki, S.G.; Siasos, G.; Nicolas, J.; Arnott, C.; Patel, S.; et al. Colchicine in Cardiovascular Disease: In-Depth Review. Circulation 2022, 145, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P. Colchicine, Biologic Agents and More for the Treatment of Familial Mediterranean Fever. The Old, the New, and the Rare. Curr. Med. Chem. 2016, 23, 60–86. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Seluanov, A.; Danek, J.; Hause, N.; Gorbunova, V. Changes in the level and distribution of Ku proteins during cellular senescence. DNA Repair. Amst. 2007, 6, 1740–1748. [Google Scholar] [CrossRef]
- Liang, F.; Romanienko, P.J.; Weaver, D.T.; Jeggo, P.A.; Jasin, M. Chromosomal double-strand break repair in Ku80-deficient cells. Proc. Natl. Acad. Sci. USA 1996, 93, 8929–8933. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Hande, M.P.; Tong, W.M.; Roth, D.; Lansdorp, P.M.; Wang, Z.Q.; Jackson, S.P. Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Curr. Biol. 2001, 11, 1192–1196. [Google Scholar] [CrossRef]
- Smith, A.J.; Ball, S.S.; Manzar, K.; Bowater, R.P.; Wormstone, I.M. Ku80 Counters Oxidative Stress-Induced DNA Damage and Cataract Formation in the Human Lens. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7868–7874. [Google Scholar] [CrossRef]
- Das, S.K.; Vasudevan, D.M. Alcohol-induced oxidative stress. Life Sci. 2007, 81, 177–187. [Google Scholar] [CrossRef]
- Zalar, D.M.; Pop, C.; Buzdugan, E.; Kiss, B.; Stefan, M.G.; Ghibu, S.; Crisan, D.; Buruiana-Simic, A.; Grozav, A.; Borda, I.M.; et al. Effects of Colchicine in a Rat Model of Diet-Induced Hyperlipidemia. Antioxidants 2022, 11, 230. [Google Scholar] [CrossRef]
- Pennings, G.J.; Reddel, C.J.; Traini, M.; Campbell, H.; Chen, V.; Kritharides, L. Colchicine inhibits ROS generation in response to glycoprotein VI stimulation. Sci. Rep. 2021, 11, 11965. [Google Scholar] [CrossRef]
- Zhang, B.; Huang, R.; Yang, D.; Chen, G.; Chen, Y.; Han, J.; Zhang, S.; Ma, L.; Yang, X. Combination of Colchicine and Ticagrelor Inhibits Carrageenan-Induced Thrombi in Mice. Oxid. Med. Cell. Longev. 2022, 2022, 3087198. [Google Scholar] [CrossRef]
- Anerillas, C.; Abdelmohsen, K.; Gorospe, M. Regulation of senescence traits by MAPKs. Geroscience 2020, 42, 397–408. [Google Scholar] [CrossRef]
- Chen, Z.; Yao, L.; Liu, Y.; Pan, Z.; Peng, S.; Wan, G.; Cheng, J.; Wang, J.; Cao, W. Astragaloside IV regulates NF-kappaB-mediated cellular senescence and apoptosis of hepatic stellate cells to suppress PDGF-BB-induced activation. Exp. Ther. Med. 2019, 18, 3741–3750. [Google Scholar] [CrossRef]
- Rovillain, E.; Mansfield, L.; Caetano, C.; Alvarez-Fernandez, M.; Caballero, O.L.; Medema, R.H.; Hummerich, H.; Jat, P.S. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene 2011, 30, 2356–2366. [Google Scholar] [CrossRef]
- Nicolae, C.M.; O’Connor, M.J.; Constantin, D.; Moldovan, G.L. NFkappaB regulates p21 expression and controls DNA damage-induced leukemic differentiation. Oncogene 2018, 37, 3647–3656. [Google Scholar] [CrossRef]
- Saha, K.; Adhikary, G.; Kanade, S.R.; Rorke, E.A.; Eckert, R.L. p38delta regulates p53 to control p21Cip1 expression in human epidermal keratinocytes. J. Biol. Chem. 2014, 289, 11443–11453. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Saito, S.; Hollander, M.C.; Sakaguchi, K.; Anderson, C.W.; Appella, E.; Fornace, A.J., Jr. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999, 18, 6845–6854. [Google Scholar] [CrossRef]
- Lafarga, V.; Cuadrado, A.; Lopez de Silanes, I.; Bengoechea, R.; Fernandez-Capetillo, O.; Nebreda, A.R. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol. Cell. Biol. 2009, 29, 4341–4351. [Google Scholar] [CrossRef]
- Shin, S.Y.; Kim, C.G.; Lim, Y.; Lee, Y.H. The ETS family transcription factor ELK-1 regulates induction of the cell cycle-regulatory gene p21(Waf1/Cip1) and the BAX gene in sodium arsenite-exposed human keratinocyte HaCaT cells. J. Biol. Chem. 2011, 286, 26860–26872. [Google Scholar] [CrossRef]
- Meyer-Lindemann, U.; Mauersberger, C.; Schmidt, A.C.; Moggio, A.; Hinterdobler, J.; Li, X.; Khangholi, D.; Hettwer, J.; Grasser, C.; Dutsch, A.; et al. Colchicine Impacts Leukocyte Trafficking in Atherosclerosis and Reduces Vascular Inflammation. Front. Immunol. 2022, 13, 898690. [Google Scholar] [CrossRef]
- Li, J.J.; Lee, S.H.; Kim, D.K.; Jin, R.; Jung, D.S.; Kwak, S.J.; Kim, S.H.; Han, S.H.; Lee, J.E.; Moon, S.J.; et al. Colchicine attenuates inflammatory cell infiltration and extracellular matrix accumulation in diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2009, 297, F200–F209. [Google Scholar] [CrossRef]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, regulation, and involvement in disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef]
- Muller, W.A. Getting leukocytes to the site of inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef]
- Choi, S.; Park, M.; Kim, J.; Park, W.; Kim, S.; Lee, D.K.; Hwang, J.Y.; Choe, J.; Won, M.H.; Ryoo, S.; et al. TNF-alpha elicits phenotypic and functional alterations of vascular smooth muscle cells by miR-155-5p-dependent down-regulation of cGMP-dependent kinase 1. J. Biol. Chem. 2018, 293, 14812–14822. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, X.; Zhu, G.; Liu, H.; Chen, J.; Wang, Y.; He, X. Quercetin inhibits TNF-alpha induced HUVECs apoptosis and inflammation via downregulating NF-kB and AP-1 signaling pathway in vitro. Med. Baltim. 2020, 99, e22241. [Google Scholar] [CrossRef] [PubMed]
- Siwik, D.A.; Chang, D.L.; Colucci, W.S. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ. Res. 2000, 86, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuna, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- Young, D.; Das, N.; Anowai, A.; Dufour, A. Matrix Metalloproteases as Influencers of the Cells’ Social Media. Int. J. Mol. Sci. 2019, 20, 3847. [Google Scholar] [CrossRef]
- Rastogi, S.; Rizwani, W.; Joshi, B.; Kunigal, S.; Chellappan, S.P. TNF-alpha response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2012, 19, 274–283. [Google Scholar] [CrossRef]
- Kjaergaard, A.G.; Dige, A.; Krog, J.; Tonnesen, E.; Wogensen, L. Soluble adhesion molecules correlate with surface expression in an in vitro model of endothelial activation. Basic Clin. Pharmacol. Toxicol. 2013, 113, 273–279. [Google Scholar] [CrossRef]
- Wang, L.; Tang, C. Targeting Platelet in Atherosclerosis Plaque Formation: Current Knowledge and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 9760. [Google Scholar] [CrossRef]
- Shah, B.; Allen, N.; Harchandani, B.; Pillinger, M.; Katz, S.; Sedlis, S.P.; Echagarruga, C.; Samuels, S.K.; Morina, P.; Singh, P.; et al. Effect of Colchicine on Platelet-Platelet and Platelet-Leukocyte Interactions: A Pilot Study in Healthy Subjects. Inflammation 2016, 39, 182–189. [Google Scholar] [CrossRef]
- Vaidya, K.; Tucker, B.; Kurup, R.; Khandkar, C.; Pandzic, E.; Barraclough, J.; Machet, J.; Misra, A.; Kavurma, M.; Martinez, G.; et al. Colchicine Inhibits Neutrophil Extracellular Trap Formation in Patients With Acute Coronary Syndrome After Percutaneous Coronary Intervention. J. Am. Heart Assoc. 2021, 10, e018993. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Z.; Liu, Z. Impact of Neutrophil Extracellular Traps on Thrombosis Formation: New Findings and Future Perspective. Front. Cell. Infect. Microbiol. 2022, 12, 910908. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Nepal, S.; Lee, E.S.; Jeong, T.C.; Kim, S.H.; Park, P.H. Ethanol increases matrix metalloproteinase-12 expression via NADPH oxidase-dependent ROS production in macrophages. Toxicol. Appl. Pharmacol. 2013, 273, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Sillanaukee, P.; Kalela, A.; Seppa, K.; Hoyhtya, M.; Nikkari, S.T. Matrix metalloproteinase-9 is elevated in serum of alcohol abusers. Eur. J. Clin. Investig. 2002, 32, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Koken, T.; Gursoy, F.; Kahraman, A. Long-term alcohol consumption increases pro-matrix metalloproteinase-9 levels via oxidative stress. J. Med. Toxicol. 2010, 6, 126–130. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Zhang, L.; Ji, J.; Wang, B.; Jin, W.; Zhang, C.; Chu, H. Effects of increased matrix metalloproteinase-9 expression on skeletal muscle fibrosis in prolonged alcoholic myopathies of rats. Mol. Med. Rep. 2012, 5, 60–65. [Google Scholar] [CrossRef]
- Yin, L.; Li, F.; Li, J.; Yang, X.; Xie, X.; Xue, L.; Li, Y.; Zhang, C. Chronic Intermittent Ethanol Exposure Induces Upregulation of Matrix Metalloproteinase-9 in the Rat Medial Prefrontal Cortex and Hippocampus. Neurochem. Res. 2019, 44, 1593–1601. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Khan, D.; Gerdes, N.; Hagenbeck, C.; Rana, M.; Cornelius, J.F.; Muhammad, S. Colchicine Protects against Ethanol-Induced Senescence and Senescence-Associated Secretory Phenotype in Endothelial Cells. Antioxidants 2023, 12, 960. https://doi.org/10.3390/antiox12040960
Zhou H, Khan D, Gerdes N, Hagenbeck C, Rana M, Cornelius JF, Muhammad S. Colchicine Protects against Ethanol-Induced Senescence and Senescence-Associated Secretory Phenotype in Endothelial Cells. Antioxidants. 2023; 12(4):960. https://doi.org/10.3390/antiox12040960
Chicago/Turabian StyleZhou, Huakang, Dilaware Khan, Norbert Gerdes, Carsten Hagenbeck, Majeed Rana, Jan Frederick Cornelius, and Sajjad Muhammad. 2023. "Colchicine Protects against Ethanol-Induced Senescence and Senescence-Associated Secretory Phenotype in Endothelial Cells" Antioxidants 12, no. 4: 960. https://doi.org/10.3390/antiox12040960
APA StyleZhou, H., Khan, D., Gerdes, N., Hagenbeck, C., Rana, M., Cornelius, J. F., & Muhammad, S. (2023). Colchicine Protects against Ethanol-Induced Senescence and Senescence-Associated Secretory Phenotype in Endothelial Cells. Antioxidants, 12(4), 960. https://doi.org/10.3390/antiox12040960