
Korean Red Ginseng Attenuates Particulate Matter-Induced Senescence of Skin Keratinocytes

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. KRG Preparation
2.3. PM2.5 Preparation
2.4. Keratinocyte Culture
2.5. Cell Viability
2.6. Detection of Intracellular Reactive Oxygen Species (ROS)
2.7. Colony Formation Detection
2.8. SAHF Detection
2.9. Immunofluorescence
2.10. β-Galactosidase Activity Detection
2.11. Western Blot Analysis
2.12. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.13. Quantitative Methylation-Specific PCR (qMSP) and Bisulfite Sequencing
2.14. Chromatin Immunoprecipitation (ChIP) Assay
2.15. Transfection of Small Interfering RNA (siRNA)
2.16. Statistical Analysis
3. Results
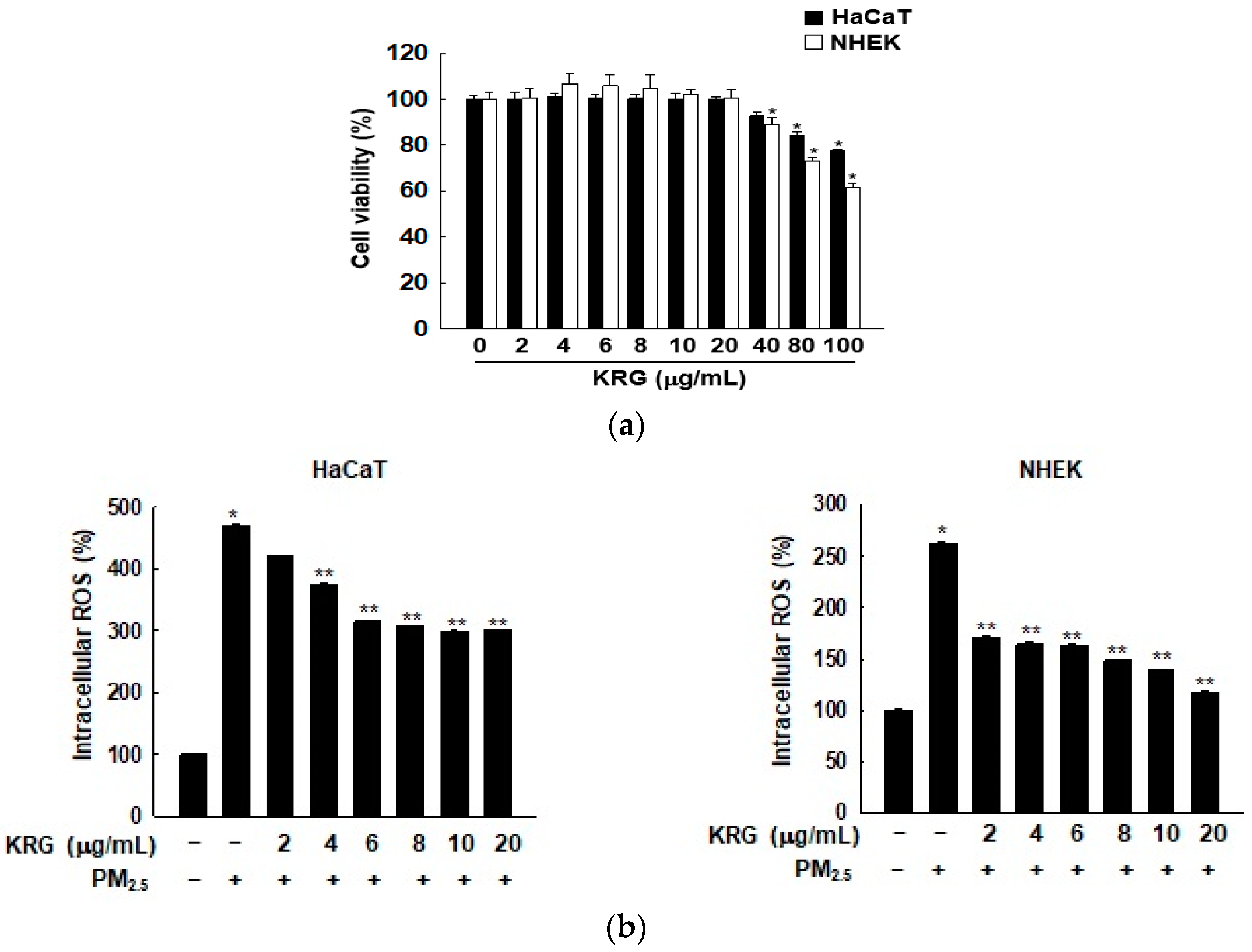
3.1. KRG Decreased ROS Generation Induced by PM2.5
3.2. KRG Attenuated Cellular Senescence Induced by PM2.5
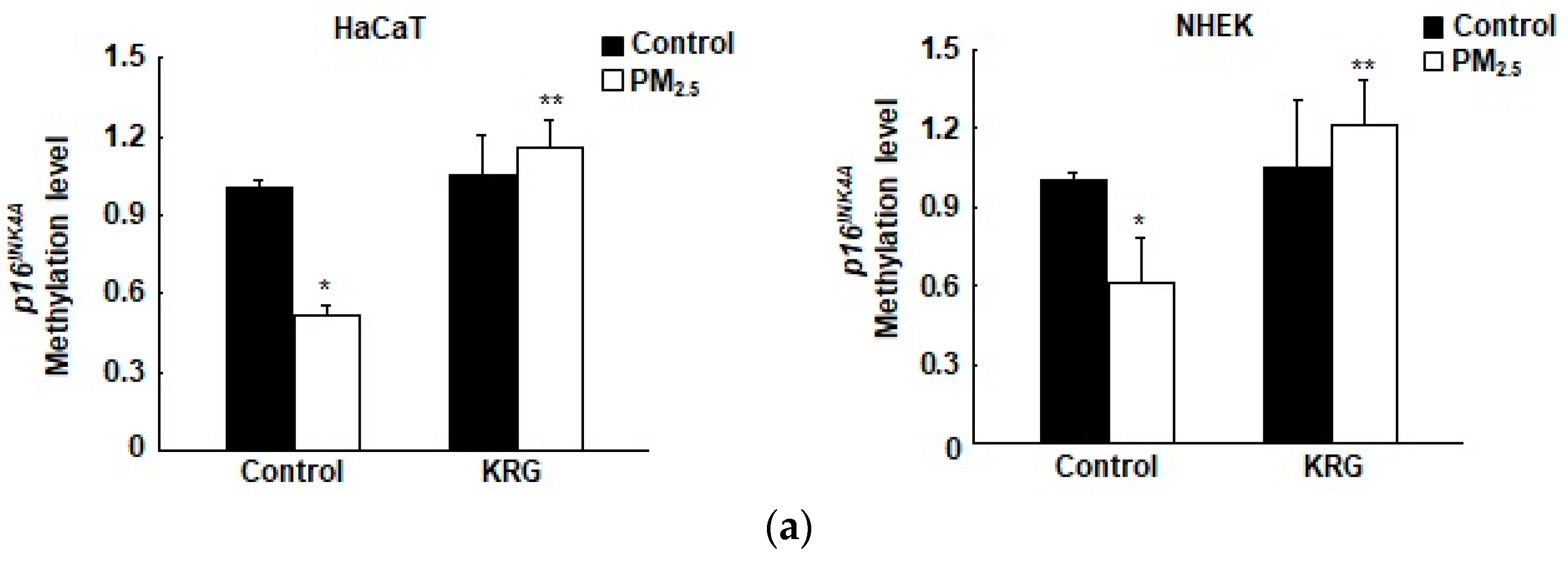
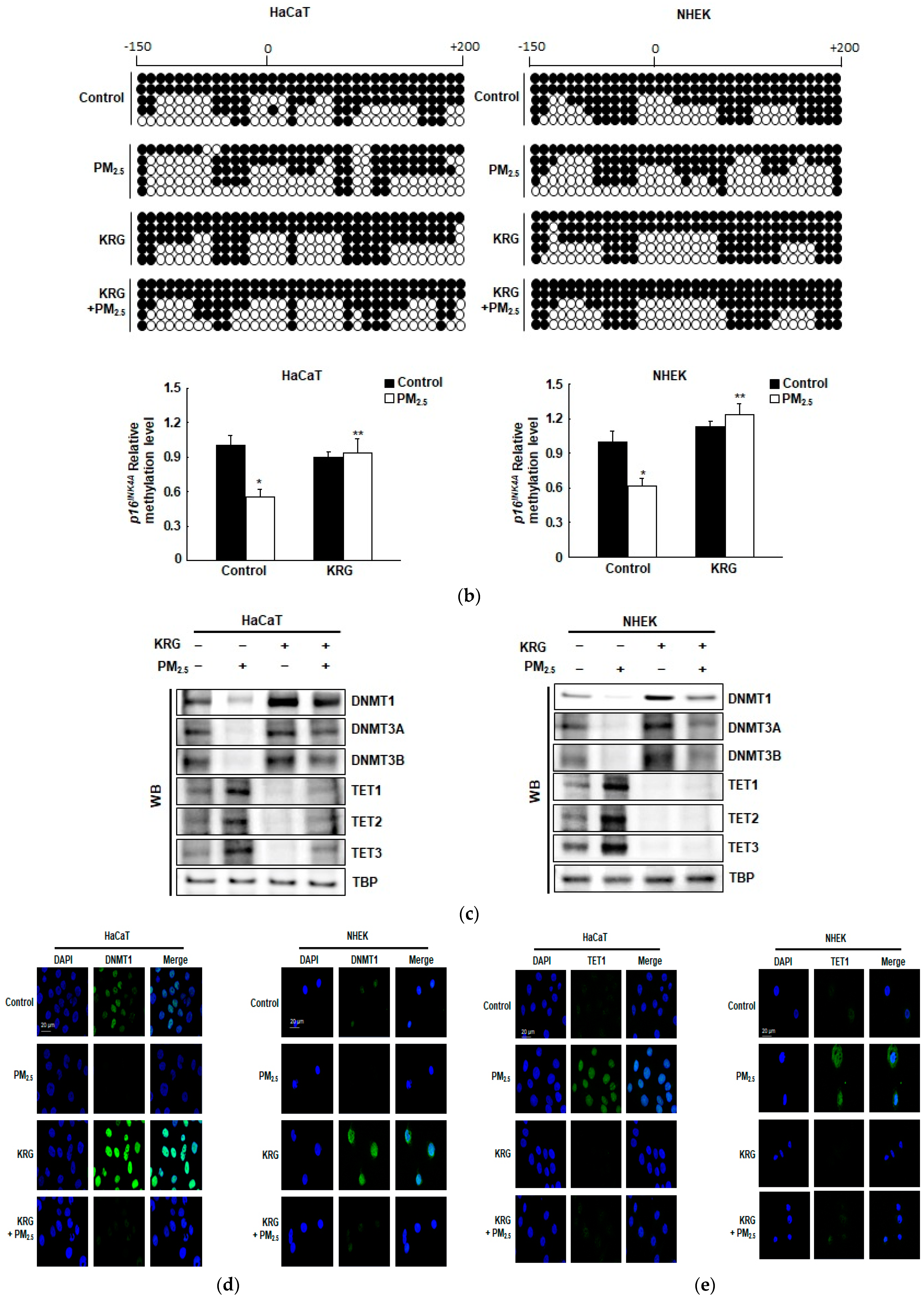
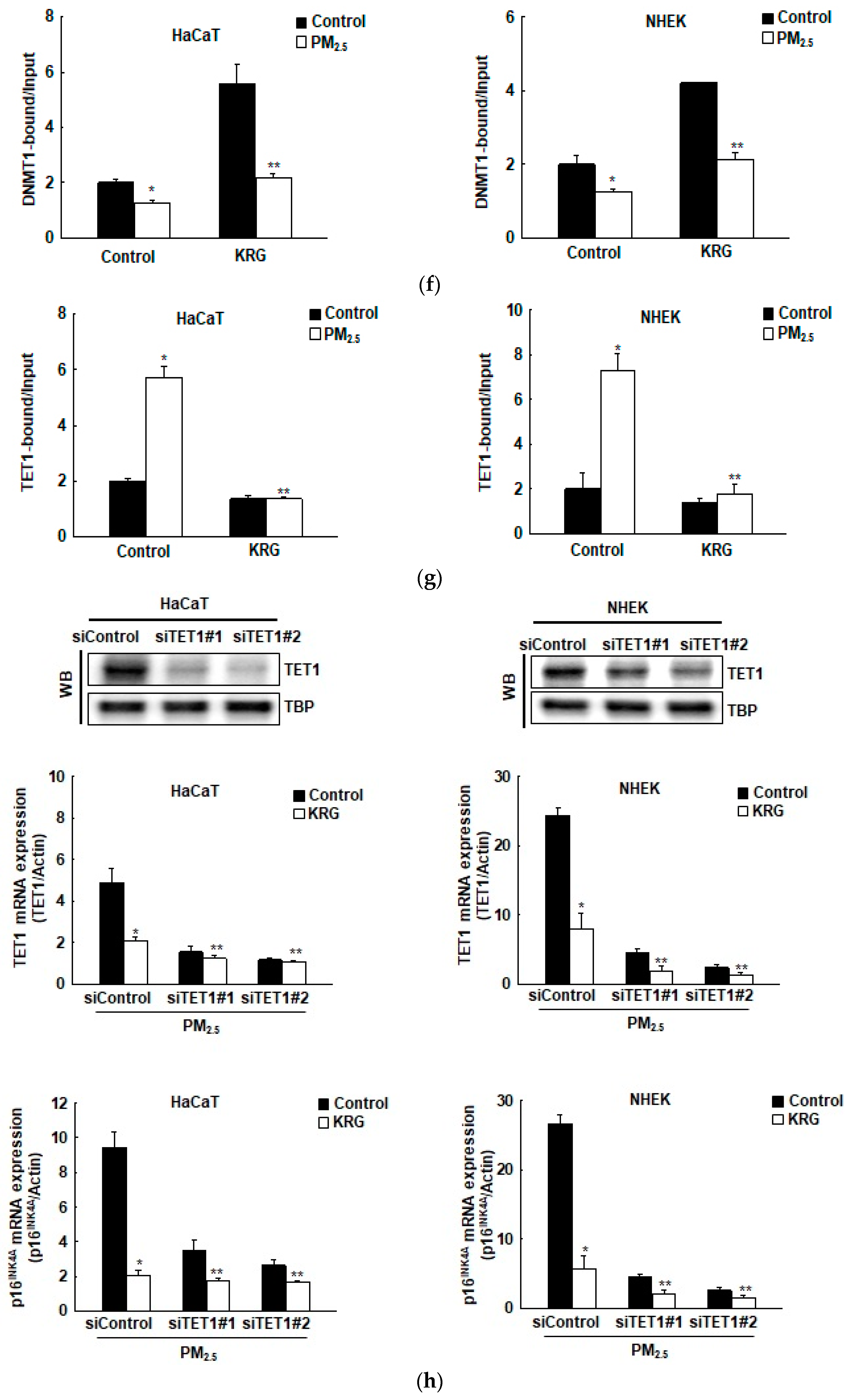
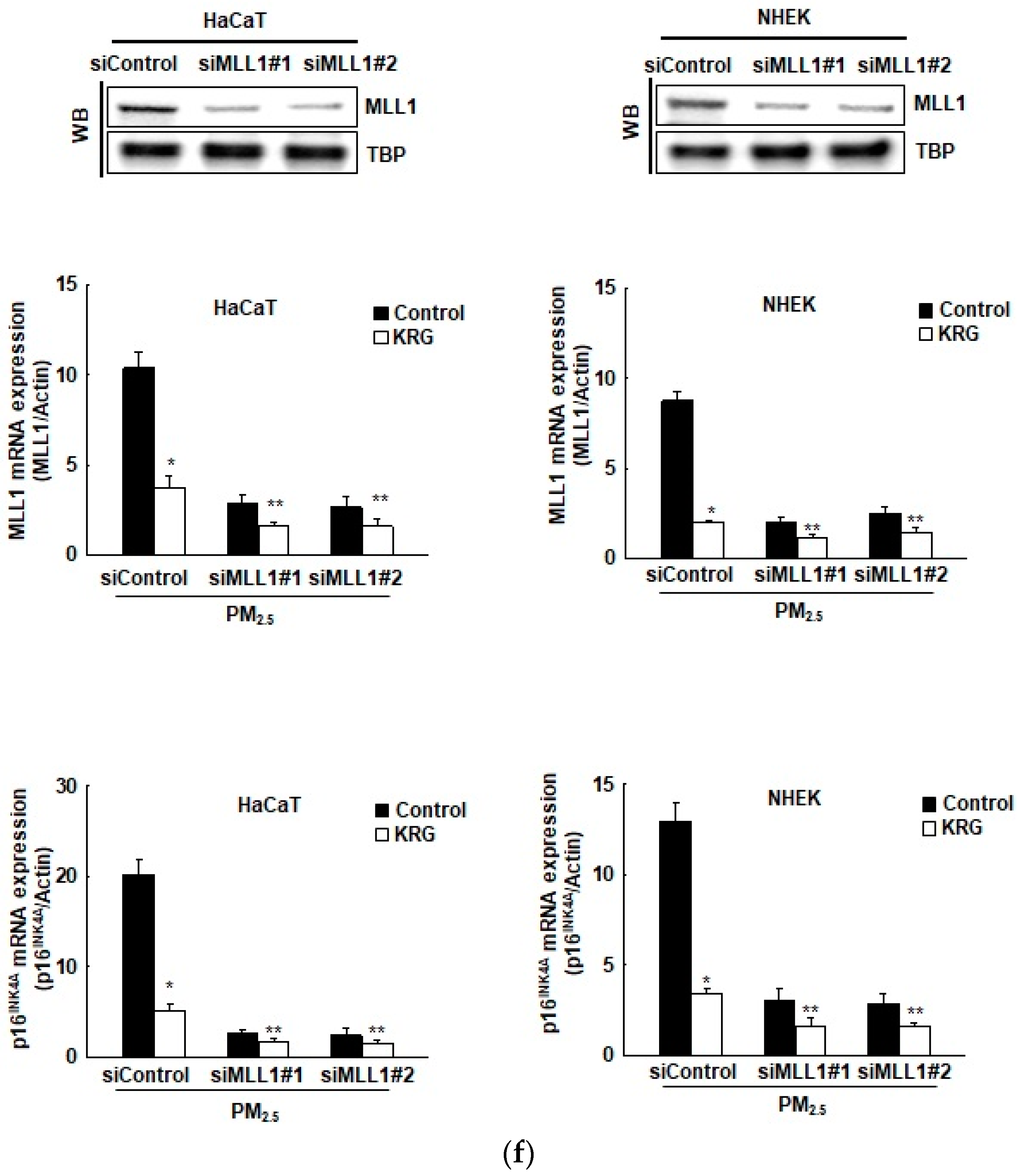
3.3. p16INK4A Expression in KRG-Treated Cells Was Attenuated through Decreased DNA Demethylation in Cellular Senescence Induced by PM2.5
3.4. p16INK4A Expression in KRG-Treated Cells Was Attenuated via Changes in Histone Methylation in Cellular Senescence Induced by PM2.5
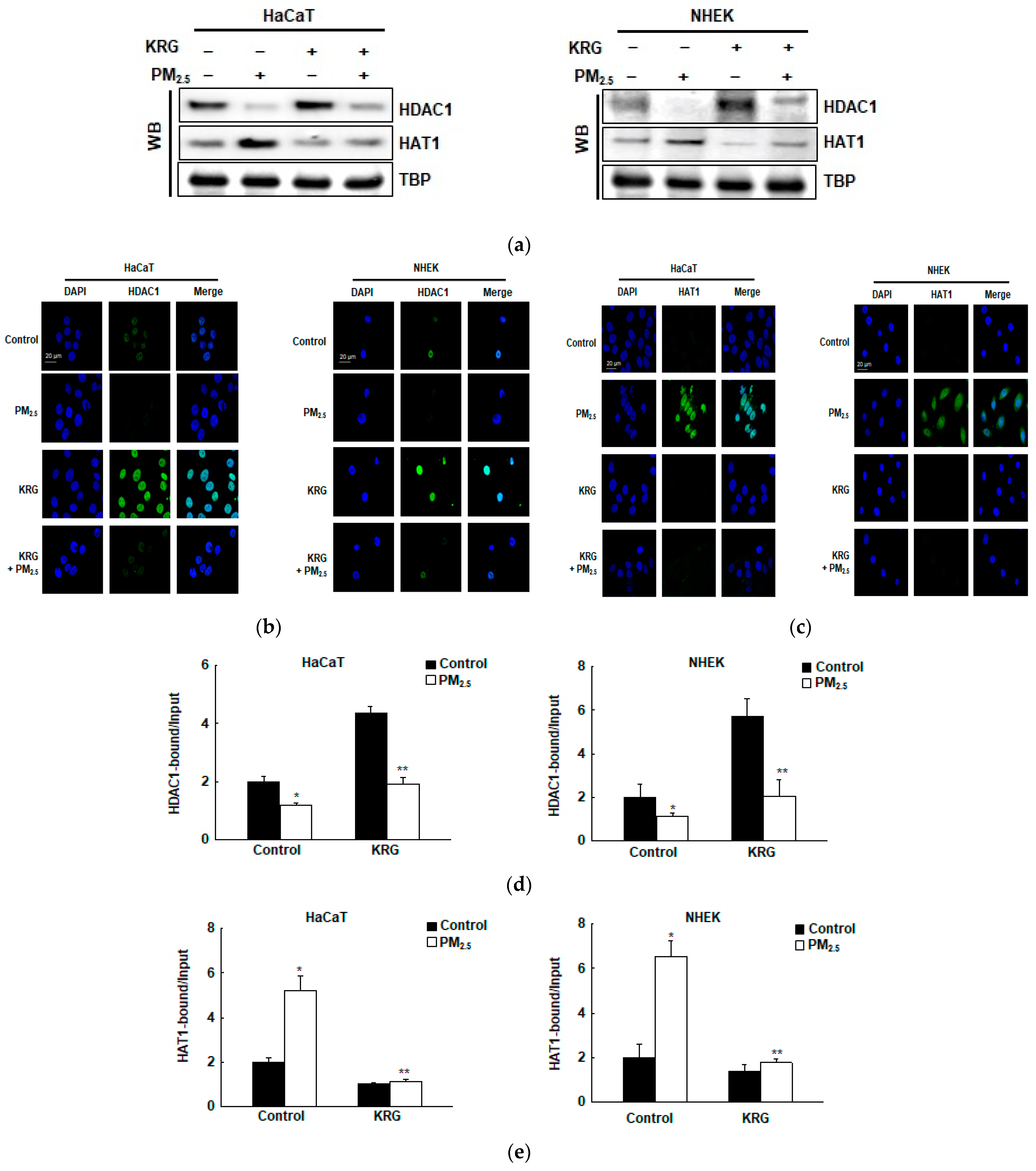
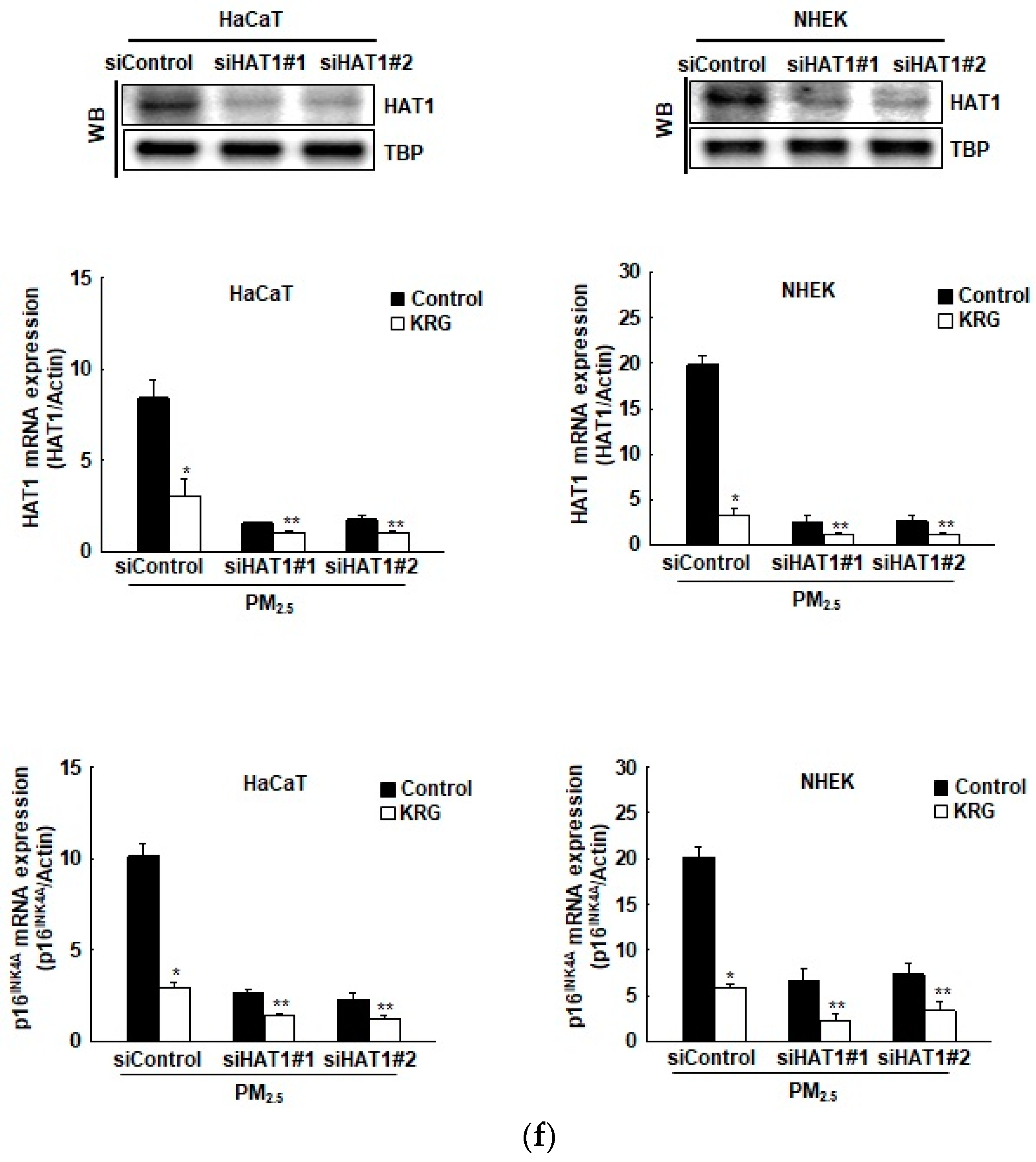
3.5. p16INK4A Expression in KRG-Treated Cells Was Attenuated via Changes in Histone Acetylation and Deacetylation in Cellular Senescence Induced by PM2.5
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Son, J.Y.; Lee, J.T.; Kim, K.H.; Jung, K.; Bell, M.L. Characterization of fine particulate matter and associations between particulate chemical constituents and mortality in Seoul, Korea. Environ. Health Perspect. 2012, 120, 872–878. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Chen, Y.; Li, X.; Zhang, Z.; Chu, H. Ambient fine particulate matter (PM2.5) induces oxidative stress and pro-inflammatory response via up-regulating the expression of CYP1A1/1B1 in human bronchial epithelial cells in vitro. Mutat. Res. 2019, 839, 40–48. [Google Scholar] [CrossRef]
- Cao, J.; Qin, G.; Shi, R.; Bai, F.; Yang, G.; Zhang, M.; Lv, J. Overproduction of reactive oxygen species and activation of MAPKs are involved in apoptosis induced by PM2.5 in rat cardiac H9c2 cells. J. Appl. Toxicol. 2016, 36, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiong, L.; Tang, M. Toxicity of inhaled particulate matter on the central nervous system: Neuroinflammation, neuropsychological effects and neurodegenerative disease. J. Appl. Toxicol. 2017, 37, 644–667. [Google Scholar] [CrossRef]
- Li, N.; He, F.; Liao, B.; Zhou, Y.; Li, B.; Ran, P. Exposure to ambient particulate matter alters the microbial composition and induces immune changes in rat lung. Respir. Res. 2017, 18, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.L.; Wang, P.W.; Aljuffali, I.A.; Huang, C.T.; Lee, C.W.; Fang, J.Y. The impact of urban particulate pollution on skin barrier function and the subsequent drug absorption. J. Dermatol. Sci. 2015, 78, 51–60. [Google Scholar] [CrossRef]
- Kim, K.E.; Cho, D.; Park, H.J. Air pollution and skin diseases: Adverse effects of airborne particulate matter on various skin diseases. Life Sci. 2016, 152, 126–134. [Google Scholar] [CrossRef]
- Magnani, N.D.; Muresan, X.M.; Belmonte, G.; Cervellati, F.; Sticozzi, C.; Pecorelli, A.; Miracco, C.; Marchini, T.; Evelson, P.; Valacchi, G. Skin damage mechanisms related to airborne particulate matter exposure. Toxicol. Sci. 2016, 149, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Bossis, G.; Hyun, Y.M.; Park, C.O.; Hyun, J.W. Particulate matter-induced senescence of skin keratinocytes involves oxidative stress-dependent epigenetic modifications. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rider, C.F.; Carlsten, C. Air pollution and DNA methylation: Effects of exposure in humans. Clin. Epigenetics. 2019, 11, 131. [Google Scholar] [CrossRef] [Green Version]
- Pulliero, A.; Traversi, D.; Franchitti, E.; Barchitta, M.; Izzotti, A.; Agodi, A. The interaction among microbiota, epigenetic regulation, and air pollutants in disease prevention. J. Pers. Med. 2021, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.H.; Jung, S.H.; Hong, S.J.; Rhyu, M.G. DNA methylation as surrogate marker for gastric cancer. J. Cancer Prev. 2015, 20, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, Y.; Gao, Y.; Geng, P.; Lu, Y.; Liu, X.; Yao, R.; Hou, P.; Liu, D.; Lu, J.; et al. JMJD3 promotes SAHF formation in senescent WI38 cells by triggering an interplay between demethylation and phosphorylation of RB protein. Cell Death Differ. 2015, 22, 1630–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Sharpless, N.E. Senescence in health and disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [Green Version]
- Crouch, J.; Shvedova, M.; Thanapaul, R.J.R.S.; Botchkarev, V.; Roh, D. Epigenetic regulation of cellular senescence. Cells 2022, 11, 672. [Google Scholar] [CrossRef]
- Zhao, R.; Choi, B.Y.; Lee, M.H.; Bode, A.M.; Dong, Z. Implications of genetic and epigenetic alterations of CDKN2A (p16(INK4a)) in cancer. EBioMedicine 2016, 8, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Liu, Y.; Liu, Y.; Zhang, C.; Yuan, B.; Zhang, L.; Sun, S. Increased p16 DNA methylation in mouse thymic lymphoma induced by irradiation. PLoS ONE 2014, 9, e93850. [Google Scholar] [CrossRef]
- Zhu, B.; Gong, Y.; Yan, G.; Wang, D.; Wang, Q.; Qiao, Y.; Hou, J.; Liu, B.; Tang, C. Atorvastatin treatment modulates p16 promoter methylation to regulate p16 expression. FEBS J. 2017, 284, 1868–1881. [Google Scholar] [CrossRef] [Green Version]
- D’Arcangelo, D.; Tinaburri, L.; Dellambra, E. The role of p16INK4a pathway in human epidermal stem cell self-renewal, aging and cancer. Int. J. Mol. Sci. 2017, 18, 1591. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Leboeuf, M.; Liu, F.; Grachtchouk, M.; Seykora, J.T.; Morrisey, E.E.; Dlugosz, A.A.; Millar, S.E. HDAC1/2 control proliferation and survival in adult epidermis and pre-basal cell carcinoma through p16 and p53. J. Investig. Dermatol. 2022, 142, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Naemura, M.; Murasaki, C.; Inoue, Y.; Okamoto, H. Transcriptional regulation of the p16 tumor suppressor gene. Anticancer Res. 2015, 35, 4397–4401. [Google Scholar] [PubMed]
- Wang, W.; Pan, K.; Chen, Y.; Huang, C.; Zhang, X. The acetylation of transcription factor HBP1 by p300/CBP enhances p16INK4A expression. Nucleic Acids Res. 2012, 40, 981–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, S.; Lee, J.W.; Kim, Y.; Hyun, S.H.; Han, C. Red ginseng monograph. J. Ginseng Res. 2018, 42, 549–561. [Google Scholar] [CrossRef]
- Kim, E.J.; Kwon, K.A.; Lee, Y.E.; Kim, J.H.; Kim, S.H.; Kim, J.H. Korean red ginseng extract reduces hypoxia-induced epithelial-mesenchymal transition by repressing NF-kB and ERK1/2 pathways in colon cancer. J. Ginseng Res. 2018, 42, 288–297. [Google Scholar] [CrossRef]
- Sung, W.N.; Kwok, H.H.; Rhee, M.H.; Yue, P.Y.; Wong, R.N. Korean red ginseng extract induces angiogenesis through activation of glucocorticoid receptor. J. Ginseng Res. 2017, 41, 477–486. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kim, J.H.; Kwon, H.M.; Lee, D.H.; Won, M.H.; Kwon, Y.G.; Kim, Y.M. Korean red ginseng protects endothelial cells from serum-deprived apoptosis by regulating Bcl-2 family protein dynamics and caspase S-nitrosylation. J. Ginseng Res. 2013, 37, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Lee, S.E.; Jeong, S.I.; Park, C.S.; Jin, Y.H.; Park, Y.S. Up-regulation of heme oxygenase-1 by Korean red ginseng water extract as a cytoprotective effect in human endothelial cells. J. Ginseng Res. 2011, 35, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Park, J.M.; Jeong, M.; Han, Y.M.; Go, E.J.; Ko, W.J.; Cho, J.Y.; Kwon, C.I.; Hahm, K.B. Korean red ginseng ameliorated experimental pancreatitis through the inhibition of hydrogen sulfide in mice. Pancreatology 2016, 16, 326–336. [Google Scholar] [CrossRef]
- Schubert, P.; Schantz, M.M.; Sander, L.C.; Wise, S.A. Determination of polycyclic aromatic hydrocarbons with molecular weight 300 and 302 in environmental-matrix standard reference materials by gas chromatography/mass spectrometry. Anal. Chem. 2003, 75, 234–246. [Google Scholar] [CrossRef]
- Bezabeh, D.Z.; Bamford, H.A.; Schantz, M.M.; Wise, S.A. Determination of nitrated polycyclic aromatic hydrocarbons in diesel particulate-related standard reference materials by using gas chromatography/mass spectrometry with negative ion chemical ionization. Anal. Bioanal. Chem. 2003, 375, 381–388. [Google Scholar] [CrossRef]
- Piao, M.J.; Ahn, M.J.; Kang, K.A.; Ryu, Y.S.; Hyun, Y.J.; Shilnikova, K.; Zhen, A.X.; Jeong, J.W.; Choi, Y.H.; Kang, H.K.; et al. Particulate matter 2.5 damages skin cells by inducing oxidative stress, subcellular organelle dysfunction, and apoptosis. Arch. Toxicol. 2018, 92, 2077–2091. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef]
- McGarvey, K.M.; Greene, E.; Fahrner, J.A.; Jenuwein, T.; Baylin, S.B. DNA methylation and complete transcriptional silencing of cancer genes persist after depletion of EZH2. Cancer Res. 2007, 67, 5097–5102. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Bae, B.S.; Park, H.W.; Ahn, N.G.; Cho, B.G.; Cho, Y.L.; Kwak, Y.S. Characterization of Korean red ginseng (Panax ginseng Meyer): History, preparation method, and chemical composition. J. Ginseng Res. 2015, 39, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Ratan, Z.A.; Haidere, M.F.; Hong, Y.H.; Park, S.H.; Lee, J.O.; Lee, J.; Cho, J.Y. Pharmacological potential of ginseng and its major component ginsenosides. J. Ginseng Res. 2021, 45, 199–210. [Google Scholar] [CrossRef]
- Wahid, F.; Khan, T.; Subhan, F.; Khan, M.; Kim, Y. Ginseng pharmacology: Multiple molecular targets and recent clinical trials. Drugs Future 2010, 35, 399–407. [Google Scholar] [CrossRef]
- Lee, D.; Lee, D.S.; Jung, K.; Hwang, G.S.; Lee, H.L.; Yamabe, N.; Lee, H.J.; Eom, D.W.; Kim, K.H.; Kang, K.S. Protective effect of ginsenoside Rb1 against tacrolimus-induced apoptosis in renal proximal tubular LLC-PK1 cells. J. Ginseng Res. 2018, 42, 75–80. [Google Scholar] [CrossRef]
- Kim, M.K.; Kang, H.; Baek, C.W.; Jung, Y.H.; Woo, Y.C.; Choi, G.J.; Shin, H.Y.; Kim, K.S. Antinociceptive and anti-inflammatory effects of ginsenoside Rf in a rat model of incisional pain. J. Ginseng Res. 2018, 42, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Saw, C.L.; Yang, A.Y.; Cheng, D.C.; Boyanapalli, S.S.; Su, Z.Y.; Khor, T.O.; Gao, S.; Wang, J.; Jiang, Z.H.; Kong, A.N. Pharmacodynamics of ginsenosides: Antioxidant activities, activation of Nrf2, and potential synergistic effects of combinations. Chem. Res. Toxicol. 2012, 25, 1574–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.H.; Kim, J.Y.; Bae, J.; Kim, Y.M.; Won, M.H.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Korean red ginseng prevents endothelial senescence by downregulating the HO-1/NF-κB/miRNA-155-5p/eNOS pathway. J. Ginseng Res. 2021, 45, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.G.; Choi, S.Y.; Kim, H.; Choi, E.J.; Lee, E.J.; Park, P.J.; Ko, J.; Kim, K.P.; Baek, H.S. Panax ginseng-derived extracellular vesicles facilitate anti-senescence effects in human skin cells: An eco-friendly and sustainable way to use ginseng substances. Cells 2021, 10, 486. [Google Scholar] [CrossRef]
- Chung, T.H.; Kim, J.H.; Seol, S.Y.; Kim, Y.J.; Lee, Y.J. The effects of Korean red ginseng on biological aging and antioxidant capacity in postmenopausal women: A double-blind randomized controlled study. Nutrients 2021, 13, 3090. [Google Scholar] [CrossRef]
- Hwang, E.; Lee, T.H.; Park, S.Y.; Yi, T.H.; Kim, S.Y. Enzyme-modified Panax ginseng inhibits UVB-induced skin aging through the regulation of procollagen type I and MMP-1 expression. Food Funct. 2014, 5, 265–274. [Google Scholar] [CrossRef]
- Shi, G.; Liu, D.; Zhou, B.; Liu, Y.; Hao, B.; Yu, S.; Wu, L.; Wang, M.; Song, Z.; Wu, C.; et al. Ginsenoside Rb1 alleviates oxidative low-density lipoprotein-induced vascular endothelium senescence via the SIRT1/beclin-1/autophagy axis. J. Cardiovasc. Pharmacol. 2020, 75, 155–167. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, R.; Kong, L.; Shen, Y.; Xu, D.; Zheng, F.; Liu, J.; Wu, Q.; Jia, B.; Zhang, J. Ginsenoside Rg3 inhibits the senescence of prostate stromal cells through down-regulation of interleukin 8 expression. Oncotarget 2017, 8, 64779–64792. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, L.; Jiang, R.; Li, C.; Chen, X.; Xiao, H.; Hou, J.; Hu, L.; Huang, C.; Wang, Y. Ginsenoside Rg1 prevents bone marrow mesenchymal stem cell senescence via NRF2 and PI3K/Akt signaling. Free Radic. Biol. Med. 2021, 174, 182–194. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, S.; Yang, S.; Li, R.; Yang, Y.; Chen, Y.; Zhang, W. Inhibitory role of ginsenoside Rb2 in endothelial senescence and inflammation mediated by microRNA-216a. Mol. Med. Rep. 2021, 23, 415. [Google Scholar] [CrossRef]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolic, N.; Anicic, B.; Carkic, J.; Simonovic, J.; Toljic, B.; Tanic, N.; Tepavcevic, Z.; Vukadinovic, M.; Konstantinovic, V.S.; Milasin, J. High frequency of p16 and p14 promoter hypermethylation and marked telomere instability in salivary gland tumors. Arch. Oral. Biol. 2015, 60, 1662–1666. [Google Scholar] [CrossRef]
- Zhou, W.; Tian, D.; He, J.; Wang, Y.; Zhang, L.; Cui, L.; Jia, L.; Zhang, L.; Li, L.; Shu, Y.; et al. Repeated PM2.5 exposure inhibits BEAS-2B cell p53 expression through ROS-Akt-DNMT3B pathway-mediated promoter hypermethylation. Oncotarget 2016, 7, 20691–20703. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Kang, H.K.; Chang, W.Y.; Keum, Y.S.; Hyun, J.W. Interaction of DNA demethylase and histone methyltransferase upregulates Nrf2 in 5-fluorouracil-resistant colon cancer cells. Oncotarget 2016, 7, 40594–40620. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, A.C.; Cortesão, E.; Oliveiros, B.; Alves, V.; Espadana, A.I.; Rito, L.; Magalhães, E.; Pereira, S.; Pereira, A.; Costa, J.M.; et al. Oxidative stress levels are correlated with p15 and p16 gene promoter methylation in myelodysplastic syndrome patients. Clin. Exp. Med. 2016, 16, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lin, H.; Gan, Y.; Cui, C.; Zhang, B.; Gu, L.; Zhou, J.; Zhu, G.; Deng, D. p16 methylation leads to paclitaxel resistance of advanced non-small cell lung cancer. J. Cancer 2019, 10, 1726–1733. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, K.A.; Piao, M.J.; Fernando, P.D.S.M.; Herath, H.M.U.L.; Yi, J.M.; Hyun, J.W. Korean Red Ginseng Attenuates Particulate Matter-Induced Senescence of Skin Keratinocytes. Antioxidants 2023, 12, 1516. https://doi.org/10.3390/antiox12081516
Kang KA, Piao MJ, Fernando PDSM, Herath HMUL, Yi JM, Hyun JW. Korean Red Ginseng Attenuates Particulate Matter-Induced Senescence of Skin Keratinocytes. Antioxidants. 2023; 12(8):1516. https://doi.org/10.3390/antiox12081516
Chicago/Turabian StyleKang, Kyoung Ah, Mei Jing Piao, Pincha Devage Sameera Madushan Fernando, Herath Mudiyanselage Udari Lakmini Herath, Joo Mi Yi, and Jin Won Hyun. 2023. "Korean Red Ginseng Attenuates Particulate Matter-Induced Senescence of Skin Keratinocytes" Antioxidants 12, no. 8: 1516. https://doi.org/10.3390/antiox12081516