
The Activation of GABAAR Alleviated Cerebral Ischemic Injury via the Suppression of Oxidative Stress, Autophagy, and Apoptosis Pathways


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cerebral Ischemia Model and Experimental Design
2.3. Cell Culture and OGD Exposure
2.4. Cell Viability Assay
2.5. Open-Field Test
2.6. Nissl Staining
2.7. Immunohistochemistry
2.8. ELISA Measurement
2.9. Western Blot Analysis
2.10. Immunofluorescence Assays
2.11. Measurement of Oxidative Stress Indexes
2.12. Transmission Electron Microscopy
2.13. Measurement of NO and NOS Contents
2.14. Statistical Analysis
3. Results
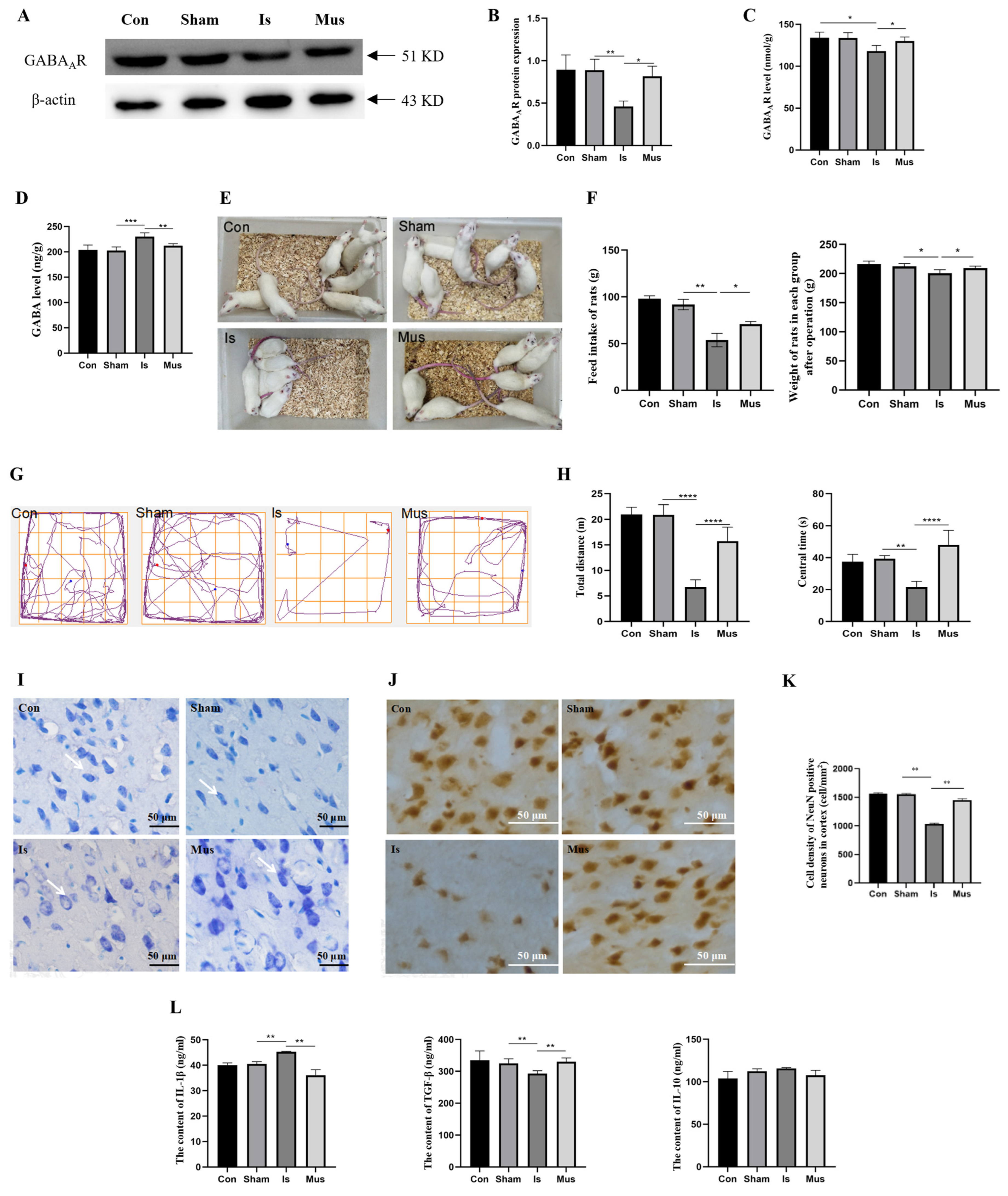
3.1. The Activation of GABAAR Regulated GABAergic Signaling and Attenuated Neuronal Damage Caused by Cerebral Ischemia
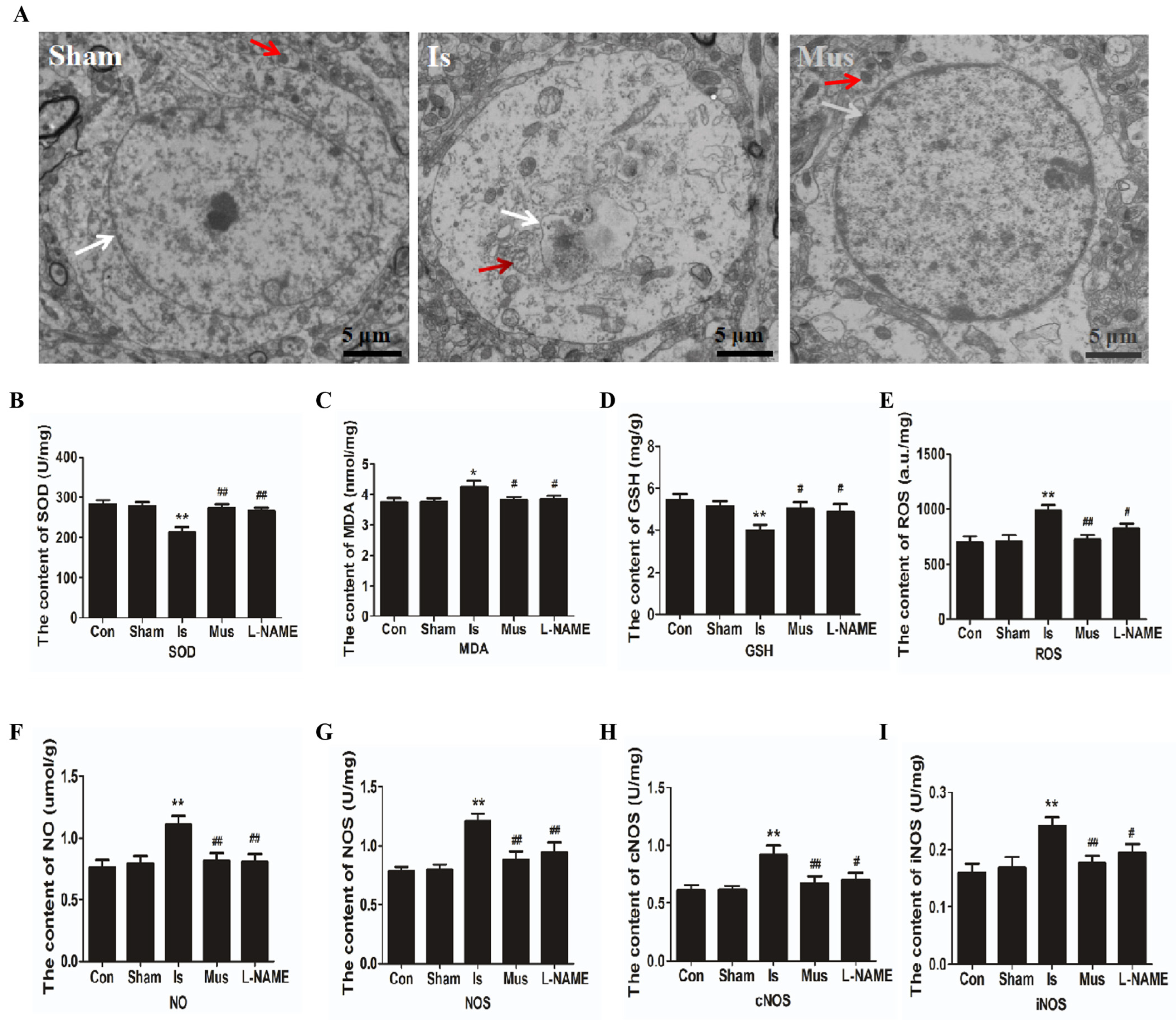
3.2. The Activation of GABAAR Alleviated Cerebral Ischemia-Induced Oxidative Stress via Inhibiting the NO/NOS Pathway
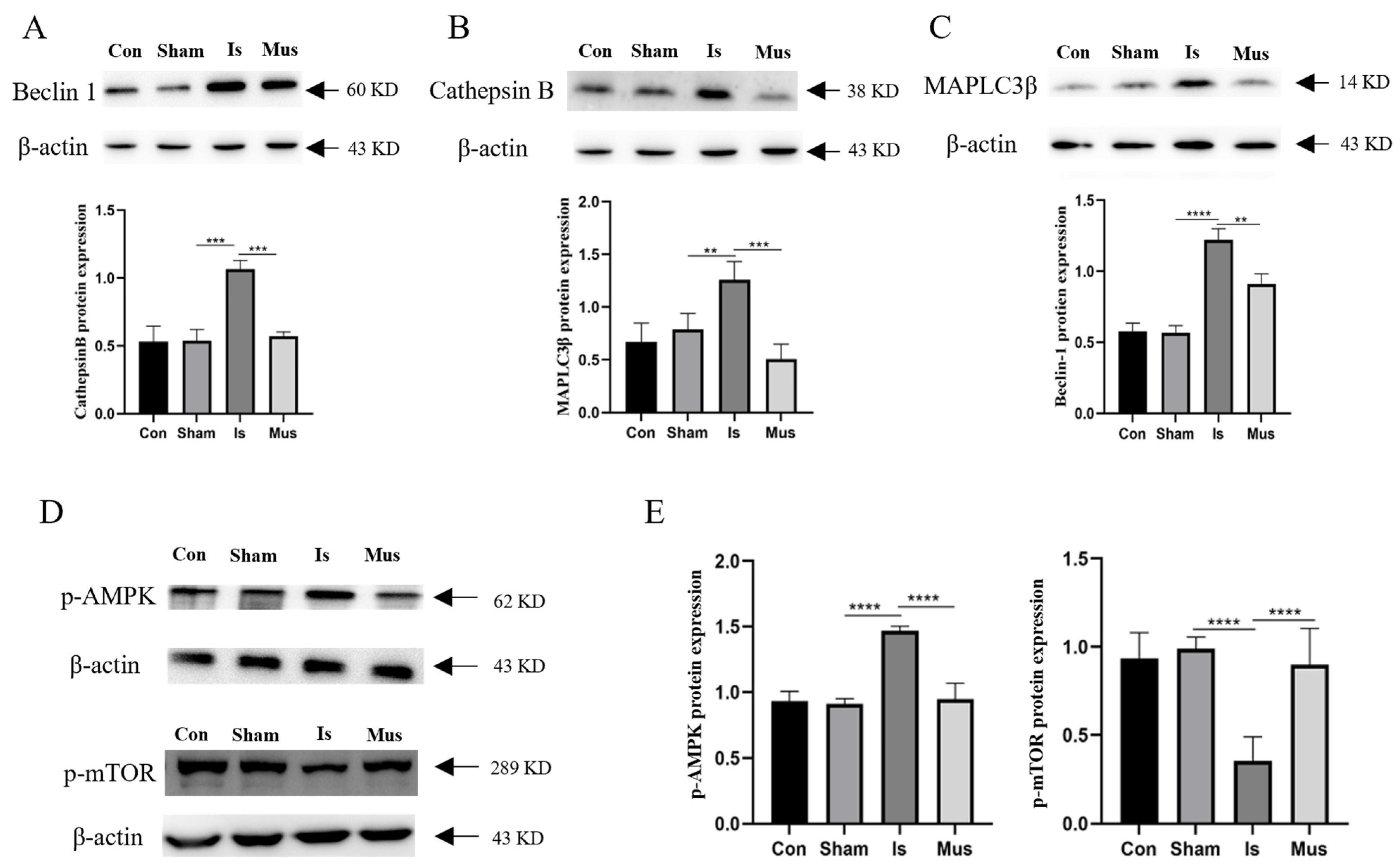
3.3. Inhibition of Autophagy-Alleviated Cerebral Ischemic Injury in Rats
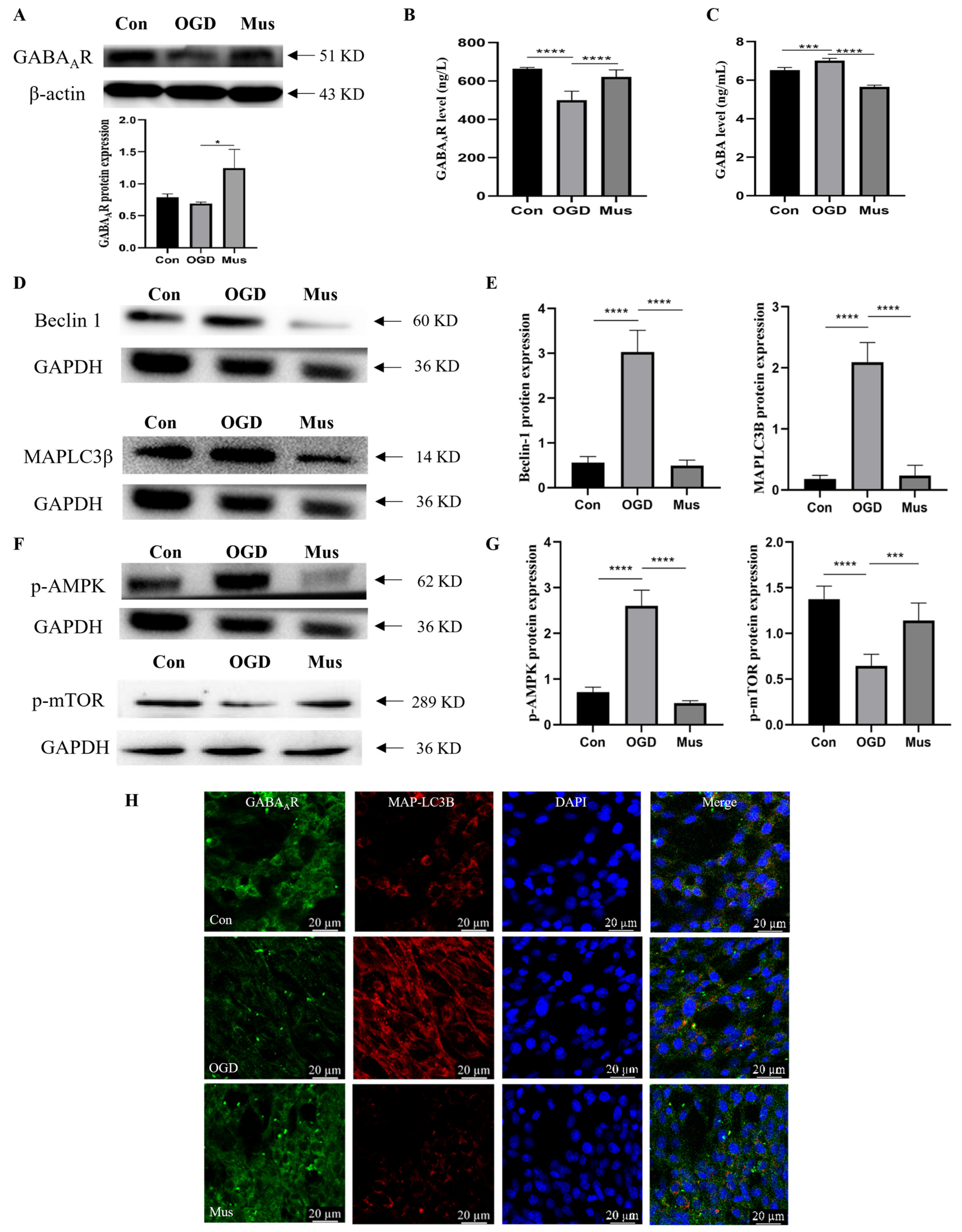
3.4. The Activation of GABAAR Inhibited Ischemia-Induced Autophagy through the AMPK/mTOR Signaling Pathway
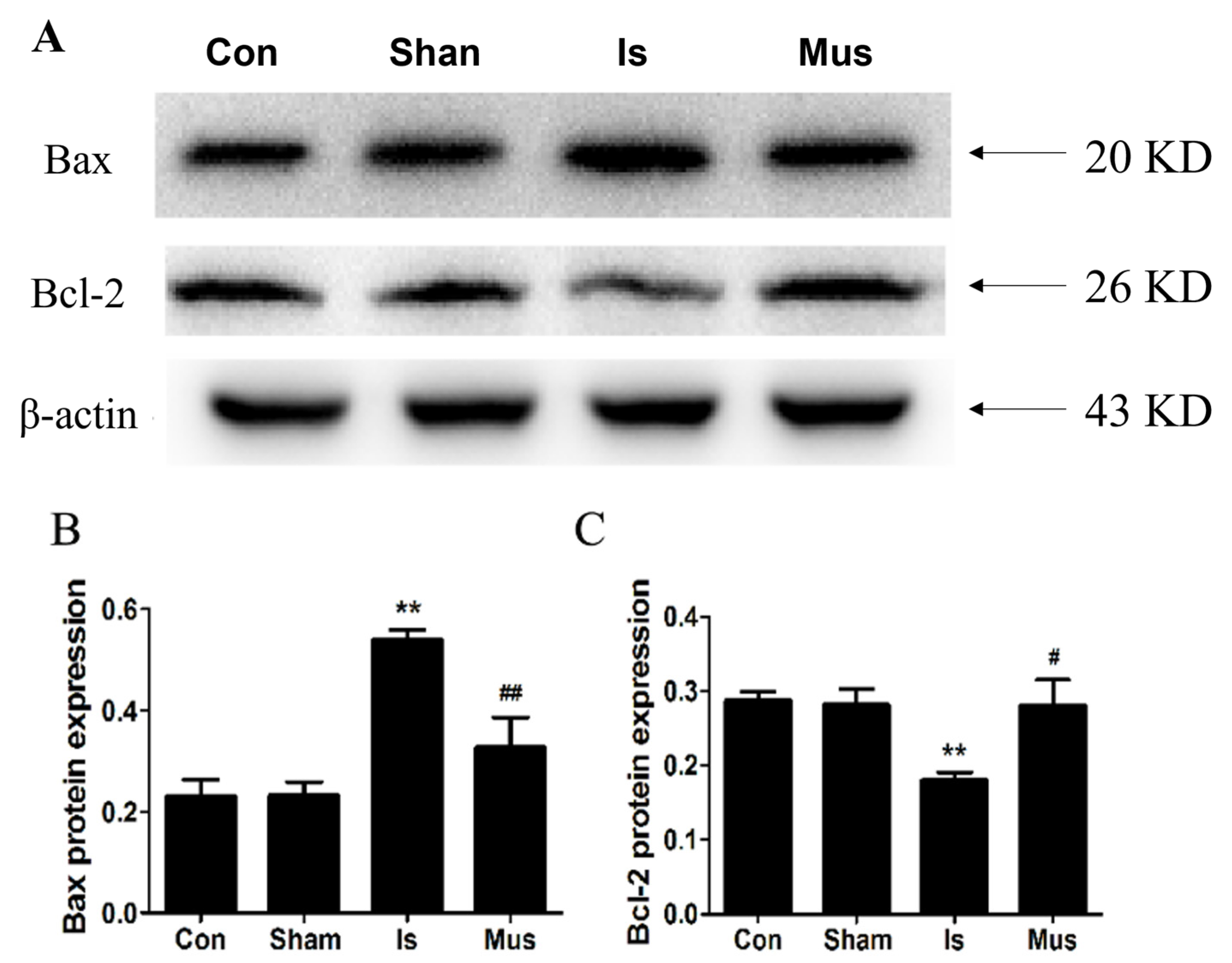
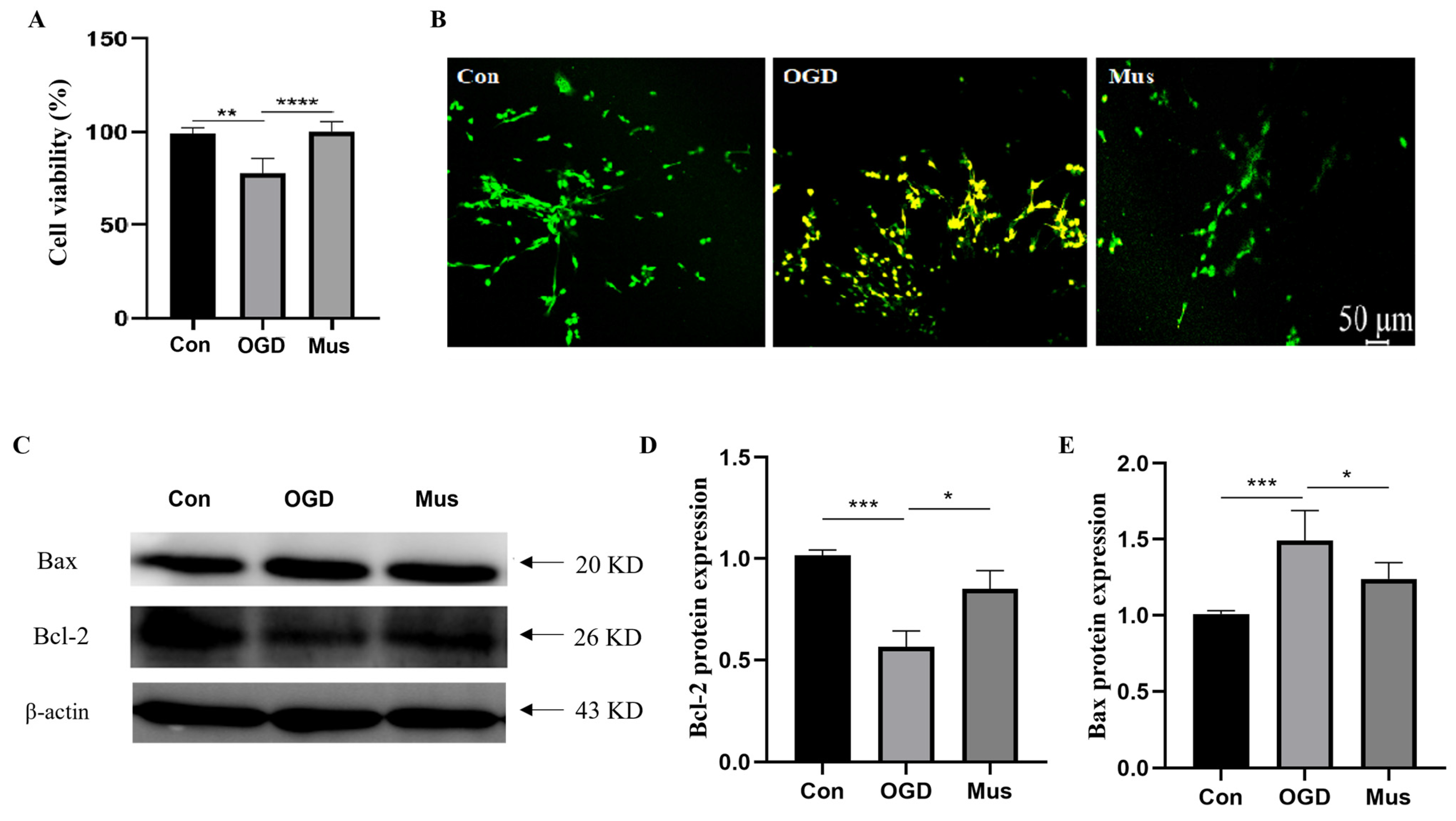
3.5. The Activation of GABAAR Inhibited Ischemia-Induced Apoptosis through Modulating the Bcl-2/Bax Signaling Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mukherjee, D.; Patil, C.G. Epidemiology and the global burden of stroke. World Neurosurg. 2011, 76, S85–S90. [Google Scholar] [CrossRef]
- Mao, R.; Zong, N.; Hu, Y.; Chen, Y.; Xu, Y. Neuronal Death Mechanisms and Therapeutic Strategy in Ischemic Stroke. Neurosci. Bull. 2022, 38, 1229–1247. [Google Scholar] [CrossRef]
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 2009, 4, 461–470. [Google Scholar] [CrossRef]
- Chen, H.S.; Chen, X.; Li, W.T.; Shen, J.G. Targeting RNS/caveolin-1/MMP signaling cascades to protect against cerebral ischemia-reperfusion injuries: Potential application for drug discovery. Acta Pharmacol. Sin. 2018, 39, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yin, Y.; Liu, S.; Dong, K.; Wang, J.; Guo, C. Fucoidan-proanthocyanidins nanoparticles protect against cisplatin-induced acute kidney injury by activating mitophagy and inhibiting mtDNA-cGAS/STING signaling pathway. Int. J. Biol. Macromol. 2023, 245, 125541. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Rosenbaum, Z.; Melamed, E.; Offen, D. Antioxidant therapy in acute central nervous system injury: Current state. Pharmacol. Rev. 2002, 54, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhao, X.Y.; Cheng, J.W.; Li, L.T.; Jiang, Q.; Zhang, Y.X.; Han, F. Signaling pathways in brain ischemia: Mechanisms and therapeutic implications. Pharmacol. Ther. 2023, 251, 108541. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Hernández, R.; Franchelli, G.; Ramos-Álvarez, M.M.; Peinado, M. Melatonin influences NO/NOS pathway and reduces oxidative and nitrosative stress in a model of hypoxic-ischemic brain damage. Nitric Oxide-Biol. Chem. 2017, 62, 32–43. [Google Scholar] [CrossRef]
- Guo, C.; Xia, Y.; Niu, P.; Jiang, L.; Duan, J.; Yu, Y.; Zhou, X.; Li, Y.; Sun, Z. Silica nanoparticles induce oxidative stress, inflammation, and endothelial dysfunction in vitro via activation of the MAPK/Nrf2 pathway and nuclear factor-κB signaling. Int. J. Nanomed. 2015, 10, 1463–1477. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef]
- Lin, W.J.; Kuang, H.Y. Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy 2014, 10, 1692–1701. [Google Scholar] [CrossRef]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhang, J.; Ding, Y.; Wu, J.; Chen, G. Novel Therapeutic Strategies for Ischemic Stroke: Recent Insights into Autophagy. Oxid. Med. Cell. Longev. 2022, 2022, 3450207. [Google Scholar] [CrossRef]
- Sheng, R.; Zhang, L.S.; Han, R.; Liu, X.Q.; Gao, B.; Qin, Z.H. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 2010, 6, 482–494. [Google Scholar] [CrossRef]
- Yan, W.; Zhang, H.; Bai, X.; Lu, Y.; Dong, H.; Xiong, L. Autophagy activation is involved in neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Brain Res. 2011, 1402, 109–121. [Google Scholar] [CrossRef]
- Shi, R.; Weng, J.; Zhao, L.; Li, X.M.; Gao, T.M.; Kong, J. Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci. Ther. 2012, 18, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.D.; Sheng, R.; Zhang, L.S.; Han, R.; Zhang, X.; Zhang, X.D.; Han, F.; Fukunaga, K.; Qin, Z.H. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 2008, 4, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Hao, Y.; Sun, L.; Zhao, Y.; Zheng, X.; Song, L. The dual roles of autophagy and the GPCRs-mediating autophagy signaling pathway after cerebral ischemic stroke. Mol. Brain 2022, 15, 14. [Google Scholar] [CrossRef]
- Montemurro, N.; Pahwa, B.; Tayal, A.; Shukla, A.; De Jesus Encarnacion, M.; Ramirez, I.; Nurmukhametov, R.; Chavda, V.; De Carlo, A. Macrophages in Recurrent Glioblastoma as a Prognostic Factor in the Synergistic System of the Tumor Microenvironment. Neurol. Int. 2023, 15, 595–608. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, C.; Jing, Y.; Cao, H.; Wang, X.; Zhao, J.; Gong, Q.; Chen, S. Deferoxamine Induces Autophagy Following Traumatic Brain Injury via TREM2 on Microglia. Mol. Neurobiol. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Feng, D.; Wang, B.; Wang, L.; Abraham, N.; Tao, K.; Huang, L.; Shi, W.; Dong, Y.; Qu, Y. Pre-ischemia melatonin treatment alleviated acute neuronal injury after ischemic stroke by inhibiting endoplasmic reticulum stress-dependent autophagy via PERK and IRE1 signalings. J. Pineal Res. 2017, 62, e12395. [Google Scholar] [CrossRef]
- Xing, S.; Zhang, Y.; Li, J.; Zhang, J.; Li, Y.; Dang, C.; Li, C.; Fan, Y.; Yu, J.; Pei, Z.; et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 2012, 8, 63–76. [Google Scholar] [CrossRef]
- Zheng, Y.Q.; Liu, J.X.; Li, X.Z.; Xu, L.; Xu, Y.G. RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacol. Sin. 2009, 30, 919–927. [Google Scholar] [CrossRef]
- Choi, D.W.; Koh, J.Y.; Peters, S. Pharmacology of glutamate neurotoxicity in cortical cell culture: Attenuation by NMDA antagonists. J. Neurosci. 1988, 8, 185–196. [Google Scholar] [CrossRef]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol. Brain 2018, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, B. Protection against ischaemic neuronal damage by drugs acting on excitatory neurotransmission. Cerebrovasc. Brain Metab. Rev. 1990, 2, 27–57. [Google Scholar] [PubMed]
- Chen, C.; Zhou, X.; He, J.; Xie, Z.; Xia, S.; Lu, G. The Roles of GABA in Ischemia-Reperfusion Injury in the Central Nervous System and Peripheral Organs. Oxidative Med. Cell. Longev. 2019, 2019, 4028394. [Google Scholar] [CrossRef] [PubMed]
- Green, A.R.; Hainsworth, A.H.; Jackson, D.M. GABA potentiation: A logical pharmacological approach for the treatment of acute ischaemic stroke. Neuropharmacology 2000, 39, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.A. Muscimol as an ionotropic GABA receptor agonist. Neurochem. Res. 2014, 39, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Winkelman, M.J.; Szabo, A.; Frecska, E. The potential of psychedelics for the treatment of Alzheimer’s disease and related dementias. Eur. Neuropsychopharmacol. 2023, 76, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, L.; Zhang, Q.; Chang, N.; Wang, D.; Shan, Y.; Li, L.; Wang, H.; Feng, H.; Zhang, L.; et al. Preservation of GABAA receptor function by PTEN inhibition protects against neuronal death in ischemic stroke. Stroke 2010, 41, 1018–1026. [Google Scholar] [CrossRef]
- Shuaib, A.; Mazagri, R.; Ijaz, S. GABA agonist “muscimol” is neuroprotective in repetitive transient forebrain ischemia in gerbils. Exp. Neurol. 1993, 123, 284–288. [Google Scholar] [CrossRef]
- Zhang, F.; Li, C.; Wang, R.; Han, D.; Zhang, Q.G.; Zhou, C.; Yu, H.M.; Zhang, G.Y. Activation of GABA receptors attenuates neuronal apoptosis through inhibiting the tyrosine phosphorylation of NR2A by Src after cerebral ischemia and reperfusion. Neuroscience 2007, 150, 938–949. [Google Scholar] [CrossRef]
- Ding, Y.; Xie, L.; Chang, C.Q.; Chen, Z.M.; Ai, H. Activation of γ-aminobutyric Acid (A) Receptor Protects Hippocampus from Intense Exercise-induced Synapses Damage and Apoptosis in Rats. Chin. Med. J. 2015, 128, 2330–2339. [Google Scholar] [CrossRef]
- Ohkuma, S.; Chen, S.H.; Katsura, M.; Chen, D.Z.; Kuriyama, K. Muscimol prevents neuronal injury induced by NMDA. Jpn. J. Pharmacol. 1994, 64, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Li, J.; Tang, X.; Li, D.; Wang, Y.; Wu, S.; Fan, K.; Ma, Y. Leonurine Reduces Oxidative Stress and Provides Neuroprotection against Ischemic Injury via Modulating Oxidative and NO/NOS Pathway. Int. J. Mol. Sci. 2022, 23, 10188. [Google Scholar] [CrossRef]
- Watson, B.D.; Dietrich, W.D.; Busto, R.; Wachtel, M.S.; Ginsberg, M.D. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann. Neurol. 1985, 17, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, C.; Yin, X.H.; Zhang, G.Y. Additive neuroprotection of GABA A and GABA B receptor agonists in cerebral ischemic injury via PI-3K/Akt pathway inhibiting the ASK1-JNK cascade. Neuropharmacology 2008, 54, 1029–1040. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, H.; Kai, J.; Zhu, F.; Dong, J.; Xu, Z.; Wong, M.; Zeng, L.H. Rapamycin prevents cerebral stroke by modulating apoptosis and autophagy in penumbra in rats. Ann. Clin. Transl. Neurol. 2018, 5, 138–146. [Google Scholar] [CrossRef]
- Yang, S.S.; Liu, Y.B.; Yu, J.B.; Fan, Y.; Tang, S.Y.; Duan, W.T.; Wang, Z.; Gan, R.T.; Yu, B. Rapamycin protects heart from ischemia/reperfusion injury independent of autophagy by activating PI3 kinase-Akt pathway and mitochondria K(ATP) channel. Pharmazie 2010, 65, 760–765. [Google Scholar]
- Gu, W.W.; Ao, G.Z.; Zhu, Y.M.; Sun, S.C.; Zhou, Q.; Fan, J.H.; Nobuhiko, K.; Ishidoh, K.; Zhang, H.L.; Gao, X.M. Autophagy and cathepsin L are involved in the antinociceptive effect of DMBC in a mouse acetic acid-writhing model. Acta Pharmacol. 2013, 34, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, D.; Hu, H.; Zhang, P.; Xie, R.; Cui, W. HIF-1α/BNIP3 signaling pathway-induced-autophagy plays protective role during myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 2019, 120, 109464. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Liu, X.; Han, D.; Xu, J.; Ma, Y. Neuroprotective effects of leonurine against oxygen-glucose deprivation by targeting Cx36/CaMKII in PC12 cells. PLoS ONE 2018, 13, e0200705. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, G.; Wang, H.; He, J.; Zhang, N.; Wu, Z.; Xiao, L.; Yang, C. Sex-dependent impact of different degrees of maternal separation experience on OFT behavioral performances after adult chronic unpredictable mild stress exposure in rats. Physiol. Behav. 2018, 194, 153–161. [Google Scholar] [CrossRef]
- Aktas, O.; Ullrich, O.; Infante-Duarte, C.; Nitsch, R.; Zipp, F. Neuronal damage in brain inflammation. Arch. Neurol. 2007, 64, 185–189. [Google Scholar] [CrossRef]
- Waldbaum, S.; Patel, M. Mitochondrial dysfunction and oxidative stress: A contributing link to acquired epilepsy? J. Bioenerg. Biomembr. 2010, 42, 449–455. [Google Scholar] [CrossRef]
- Orellana-Urzúa, S.; Rojas, I.; Líbano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. Curr. Pharm. Des. 2020, 26, 4246–4260. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhang, Y.; Chen, Q.; He, Y.; Zhou, H.; Wan, H.; Yang, J. Formononetin protects against inflammation associated with cerebral ischemia-reperfusion injury in rats by targeting the JAK2/STAT3 signaling pathway. Biomed. Pharmacother. 2022, 149, 112836. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.; Chaurasia, B.; Fiorindi, A.; Umana, G.E.; Lu, B.; Montemurro, N. Ischemic Stroke and SARS-CoV-2 Infection: The Bidirectional Pathology and Risk Morbidities. Neurol. Int. 2022, 14, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, H.; Drejer, J.; Schousboe, A.; Diemer, N.H. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem. 1984, 43, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef]
- Kondo, H.M.; Lin, I.F. Excitation-inhibition balance and auditory multistable perception are correlated with autistic traits and schizotypy in a non-clinical population. Sci. Rep. 2020, 10, 8171. [Google Scholar] [CrossRef] [PubMed]
- Lyden, P.D.; Lonzo, L. Combination therapy protects ischemic brain in rats. A glutamate antagonist plus a gamma-aminobutyric acid agonist. Stroke 1994, 25, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Pilipenko, V.; Narbute, K.; Beitnere, U.; Rumaks, J.; Pupure, J.; Jansone, B.; Klusa, V. Very low doses of muscimol and baclofen ameliorate cognitive deficits and regulate protein expression in the brain of a rat model of streptozocin-induced Alzheimer’s disease. Eur. J. Pharmacol. 2018, 818, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Guo, H.; Wang, X.; Jiao, H.; Li, L.; Zheng, J. c-myc protects mice from ischemia stroke through elevating microRNA-200b-5p-regulated SIRT1 expression. Brain Res. Bull. 2021, 176, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Fraunfelder, M.; Fujise, K.; Chen, S.Y. ADAR1 exacerbates ischemic brain injury via astrocyte-mediated neuron apoptosis. Redox Biol. 2023, 67, 102903. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, J.; Sun, S.; Zhao, J.; Dong, X.; Wang, J. Effects of Estradiol on Autophagy and Nrf-2/ARE Signals after Cerebral Ischemia. Cell. Physiol. Biochem. 2017, 41, 2027–2036. [Google Scholar] [CrossRef]
- Lyden, P.D.; Hedges, B. Protective effect of synaptic inhibition during cerebral ischemia in rats and rabbits. Stroke 1992, 23, 1463–1469, discussion 1469–1470. [Google Scholar] [CrossRef]
- Sah, R.; Galeffi, F.; Ahrens, R.; Jordan, G.; Schwartz-Bloom, R.D. Modulation of the GABA(A)-gated chloride channel by reactive oxygen species. J. Neurochem. 2002, 80, 383–391. [Google Scholar] [CrossRef]
- Ozcelik, D.; Uzun, H. Copper intoxication; antioxidant defenses and oxidative damage in rat brain. Biol. Trace Elem. Res. 2009, 127, 45–52. [Google Scholar] [CrossRef]
- Moro, M.A.; Cárdenas, A.; Hurtado, O.; Leza, J.C.; Lizasoain, I. Role of nitric oxide after brain ischaemia. Cell Calcium 2004, 36, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Shao, B.Z.; Deng, Z.; Chen, S.; Yue, Z.; Miao, C.Y. Autophagy in ischemic stroke. Prog. Neurobiol. 2018, 163–164, 98–117. [Google Scholar] [CrossRef]
- Baehrecke, E.H. Autophagic programmed cell death in Drosophila. Cell Death Differ. 2003, 10, 940–945. [Google Scholar] [CrossRef]
- Clarke, P.G.; Puyal, J. Autophagic cell death exists. Autophagy 2012, 8, 867–869. [Google Scholar] [CrossRef] [PubMed]
- Pi, Q.Z.; Wang, X.W.; Jian, Z.L.; Chen, D.; Zhang, C.; Wu, Q.C. Melatonin Alleviates Cardiac Dysfunction Via Increasing Sirt1-Mediated Beclin-1 Deacetylation and Autophagy During Sepsis. Inflammation 2021, 44, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Li, X.; Li, W.; Fu, Q.; Mu, Y.; Chen, X. Molecular characterization and role in virus infection of Beclin-1 in large yellow croaker (Larimichthys crocea). Fish Shellfish Immunol. 2021, 116, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Kay, A.; Jiang, Z.; Arkin, M.R. LC3B Binds to the Autophagy Protease ATG4b with High Affinity Using a Bipartite Interface. Biochemistry 2022, 61, 2295–2302. [Google Scholar] [CrossRef] [PubMed]
- Puyal, J.; Vaslin, A.; Mottier, V.; Clarke, P.G. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann. Neurol. 2009, 66, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Al-Bari, M.A.A.; Xu, P. Molecular regulation of autophagy machinery by mTOR-dependent and -independent pathways. Ann. N. Y. Acad. Sci. 2020, 1467, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.J.; Zhang, M.L.; Hou, Y.M.; Zhang, K.; Yao, D.H.; Li, G.Y.; Kou, W.B.; Wang, H.Y.; Wang, J.H. The Amomum tsao-ko Essential Oils Inhibited Inflammation and Apoptosis through p38/JNK MAPK Signaling Pathway and Alleviated Gentamicin-Induced Acute Kidney Injury. Molecules 2022, 27, 7121. [Google Scholar] [CrossRef] [PubMed]
- Pervin, S.; Singh, R.; Chaudhuri, G. Nitric-oxide-induced Bax integration into the mitochondrial membrane commits MDA-MB-468 cells to apoptosis: Essential role of Akt. Cancer Res. 2003, 63, 5470–5479. [Google Scholar]
- Ushmorov, A.; Ratter, F.; Lehmann, V.; Dröge, W.; Schirrmacher, V.; Umansky, V. Nitric-oxide-induced apoptosis in human leukemic lines requires mitochondrial lipid degradation and cytochrome C release. Blood 1999, 93, 2342–2352. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McCullough, L.D. Effects of AMP-activated protein kinase in cerebral ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 480–492. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, J.; Wang, J.; Wang, S.; Wang, J.; Huang, S.; Wang, Y.; Ma, Y. The Activation of GABAAR Alleviated Cerebral Ischemic Injury via the Suppression of Oxidative Stress, Autophagy, and Apoptosis Pathways. Antioxidants 2024, 13, 194. https://doi.org/10.3390/antiox13020194
Lan J, Wang J, Wang S, Wang J, Huang S, Wang Y, Ma Y. The Activation of GABAAR Alleviated Cerebral Ischemic Injury via the Suppression of Oxidative Stress, Autophagy, and Apoptosis Pathways. Antioxidants. 2024; 13(2):194. https://doi.org/10.3390/antiox13020194
Chicago/Turabian StyleLan, Jing, Jiaqi Wang, Shujing Wang, Jia Wang, Sijuan Huang, Yazhou Wang, and Yunfei Ma. 2024. "The Activation of GABAAR Alleviated Cerebral Ischemic Injury via the Suppression of Oxidative Stress, Autophagy, and Apoptosis Pathways" Antioxidants 13, no. 2: 194. https://doi.org/10.3390/antiox13020194
APA StyleLan, J., Wang, J., Wang, S., Wang, J., Huang, S., Wang, Y., & Ma, Y. (2024). The Activation of GABAAR Alleviated Cerebral Ischemic Injury via the Suppression of Oxidative Stress, Autophagy, and Apoptosis Pathways. Antioxidants, 13(2), 194. https://doi.org/10.3390/antiox13020194