



7-O-tyrosyl Silybin Derivatives as a Novel Set of Anti-Prostate Cancer Compounds

 ,
,  , ,
, ,  ,
, 
Abstract
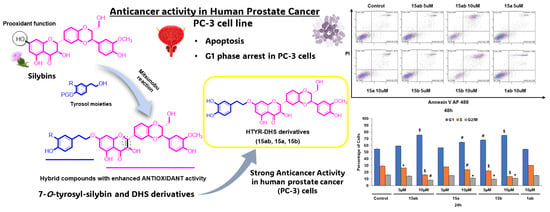
1. Introduction
2. Materials and Methods
2.1. General
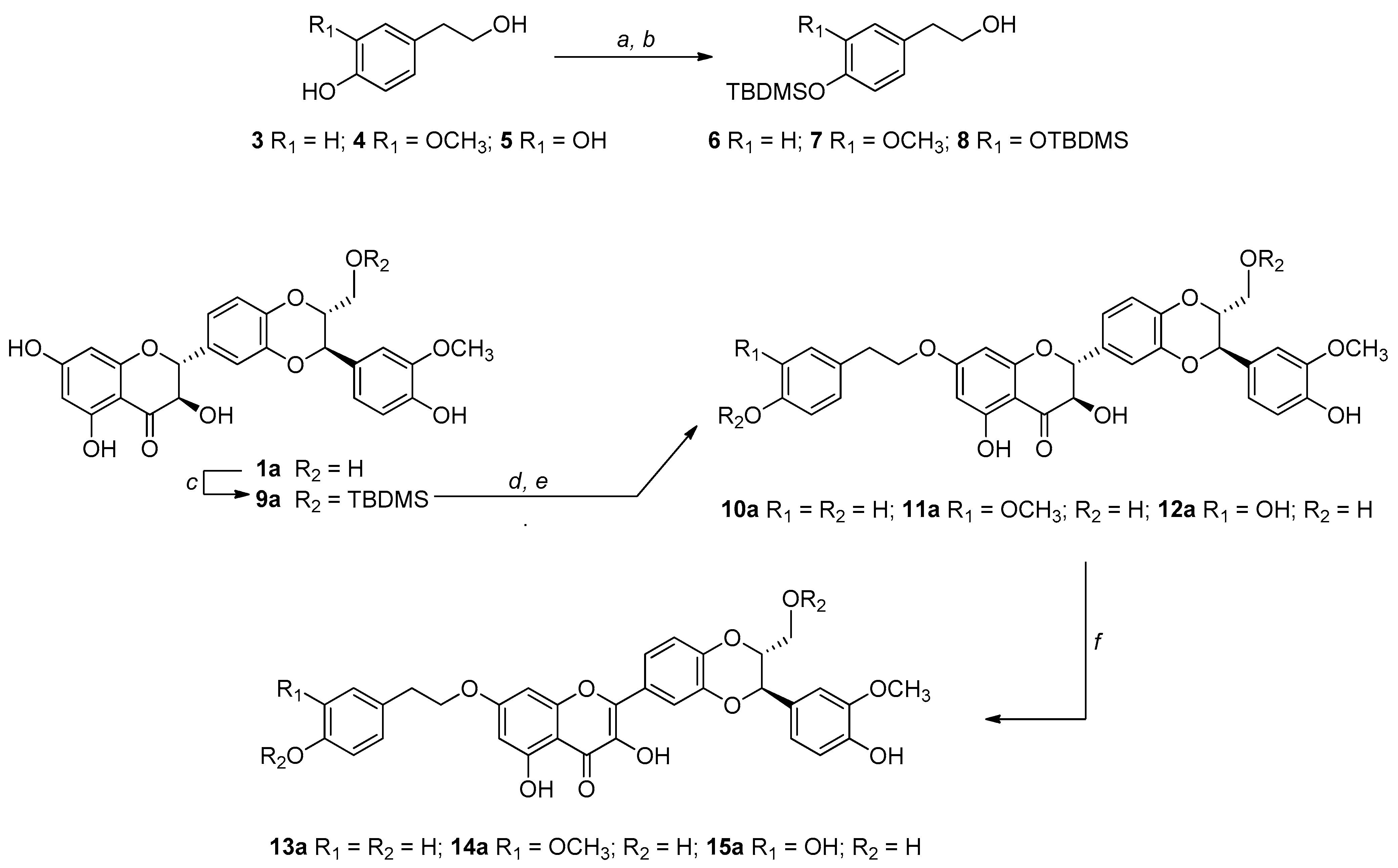
2.2. Synthesis of 9″-O-tert-butyldimethylsilyl-silybin A (9a) and 9″-O-tert-buthyldimethylsilyl-silybin B (9b)
2.3. Alkylation by Mitsunobu Reaction: General Procedure for the Synthesis of 10a–12a
2.4. Synthesis of 7-O-tyrosyl-2,3-dehydro Silybin Derivatives (13a–15a): General Procedure
2.5. Medium and Chemical Stability
2.6. DPPH Assay
2.7. ORAC Assay
2.8. Cell Culture, Reagents, and Treatments
2.9. Cell Growth and Death Assay
2.10. Flow Cytometry for Apoptosis and Cell Cycle Determination
2.11. Lysate Preparation and Immunoblot Analysis
2.12. Statistical Analysis
3. Results
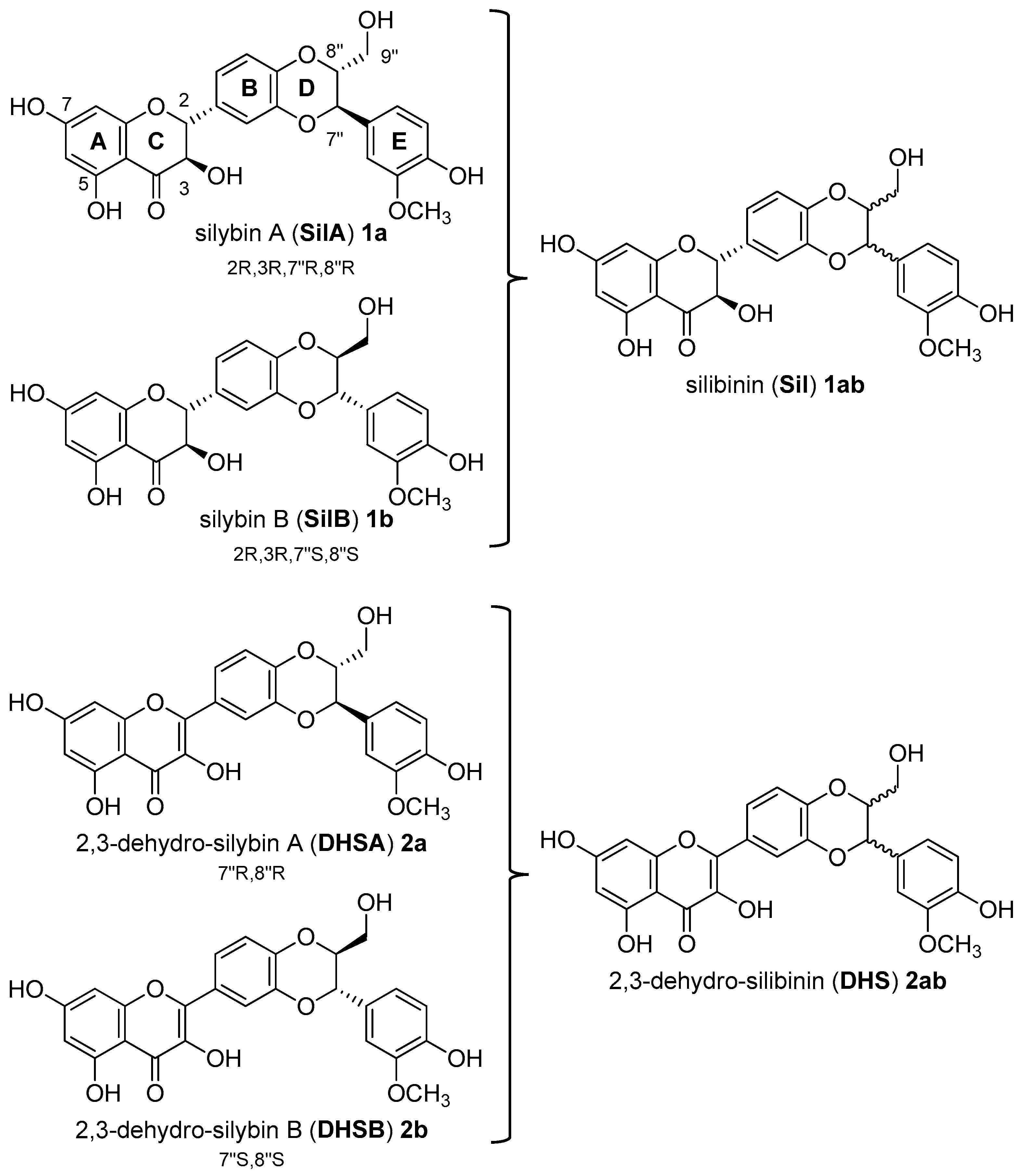
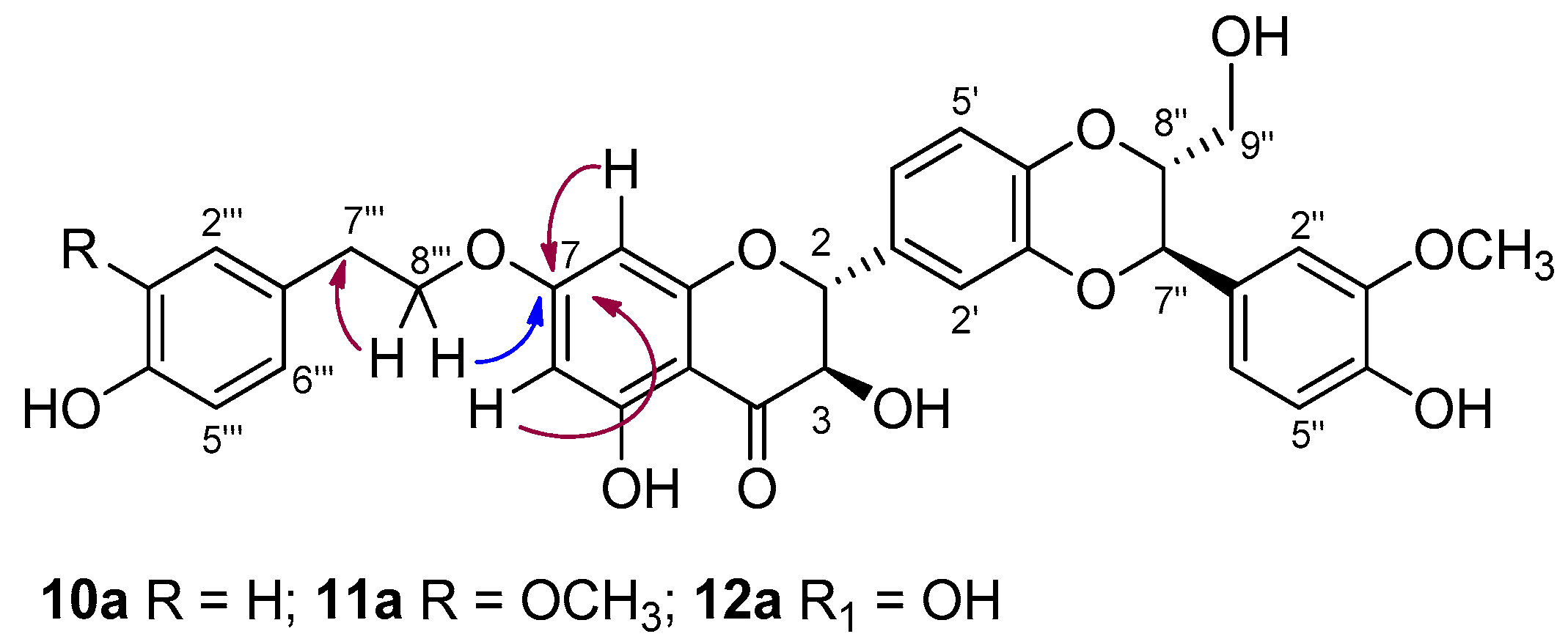
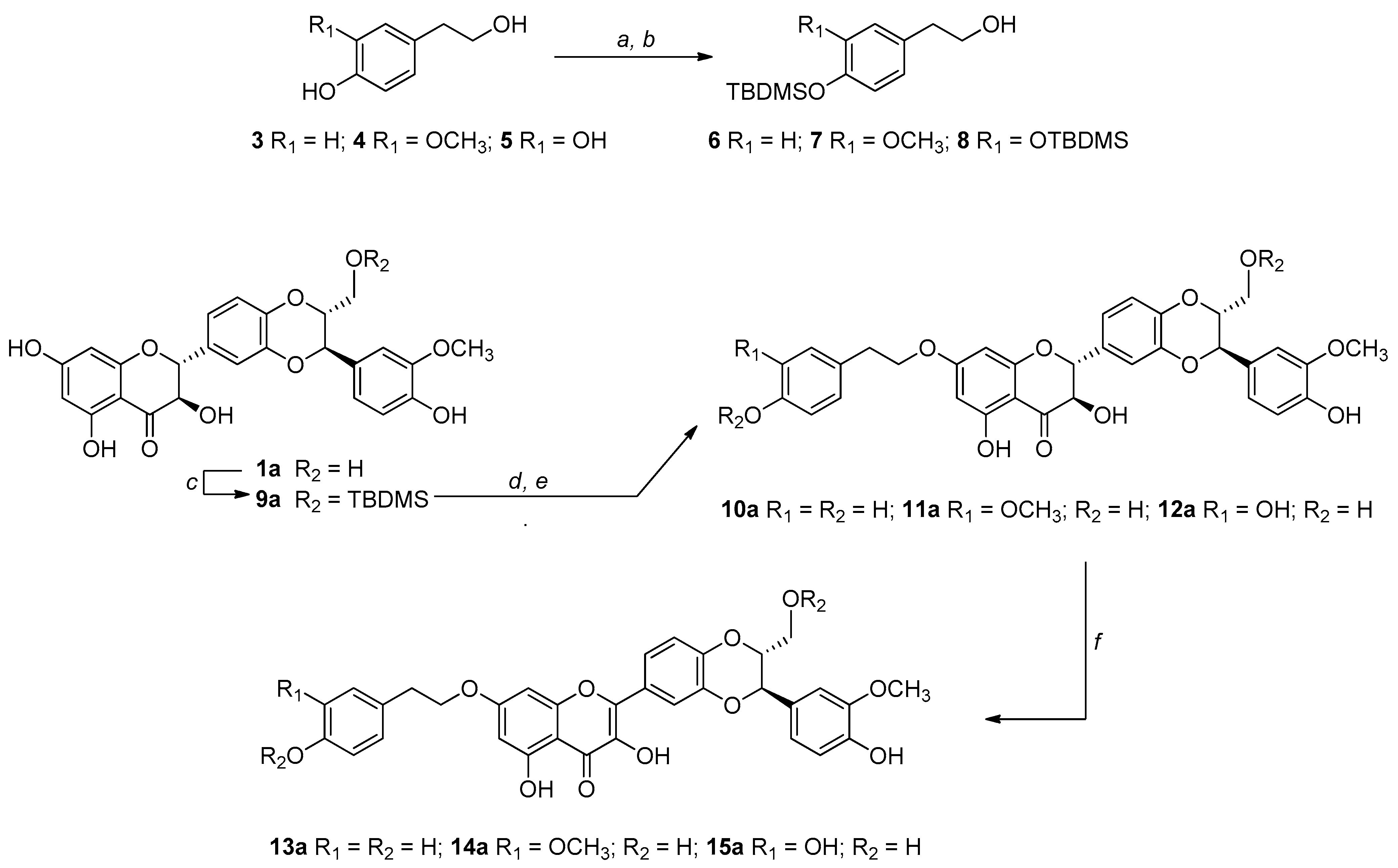
3.1. Synthesis, NMR Characterization, and Chemical Stability
3.2. Radical Scavenger Properties (DPPH and ORAC Assays)
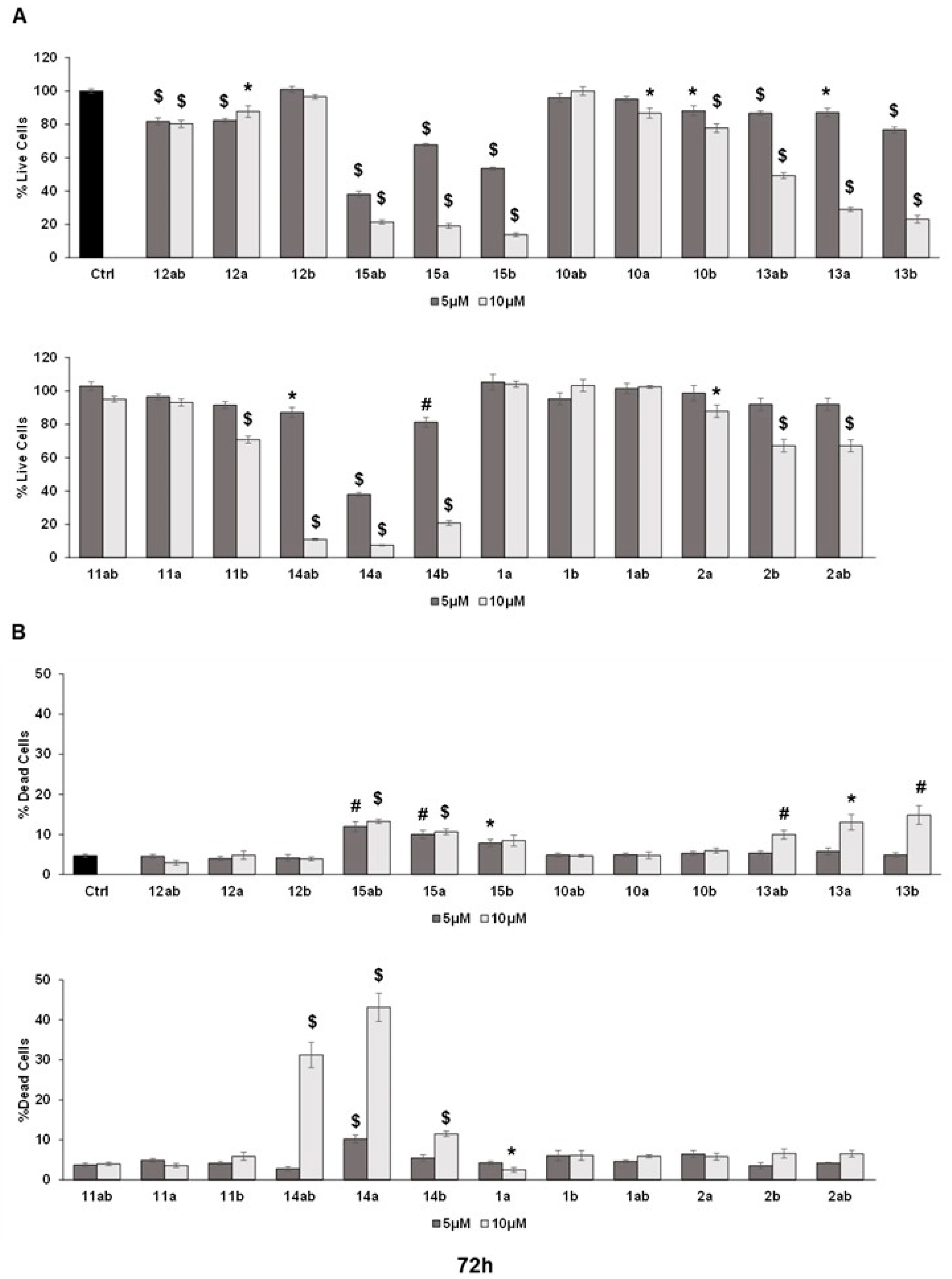
3.3. Anti-Proliferative Effects towards Prostate Cancer Cell Lines
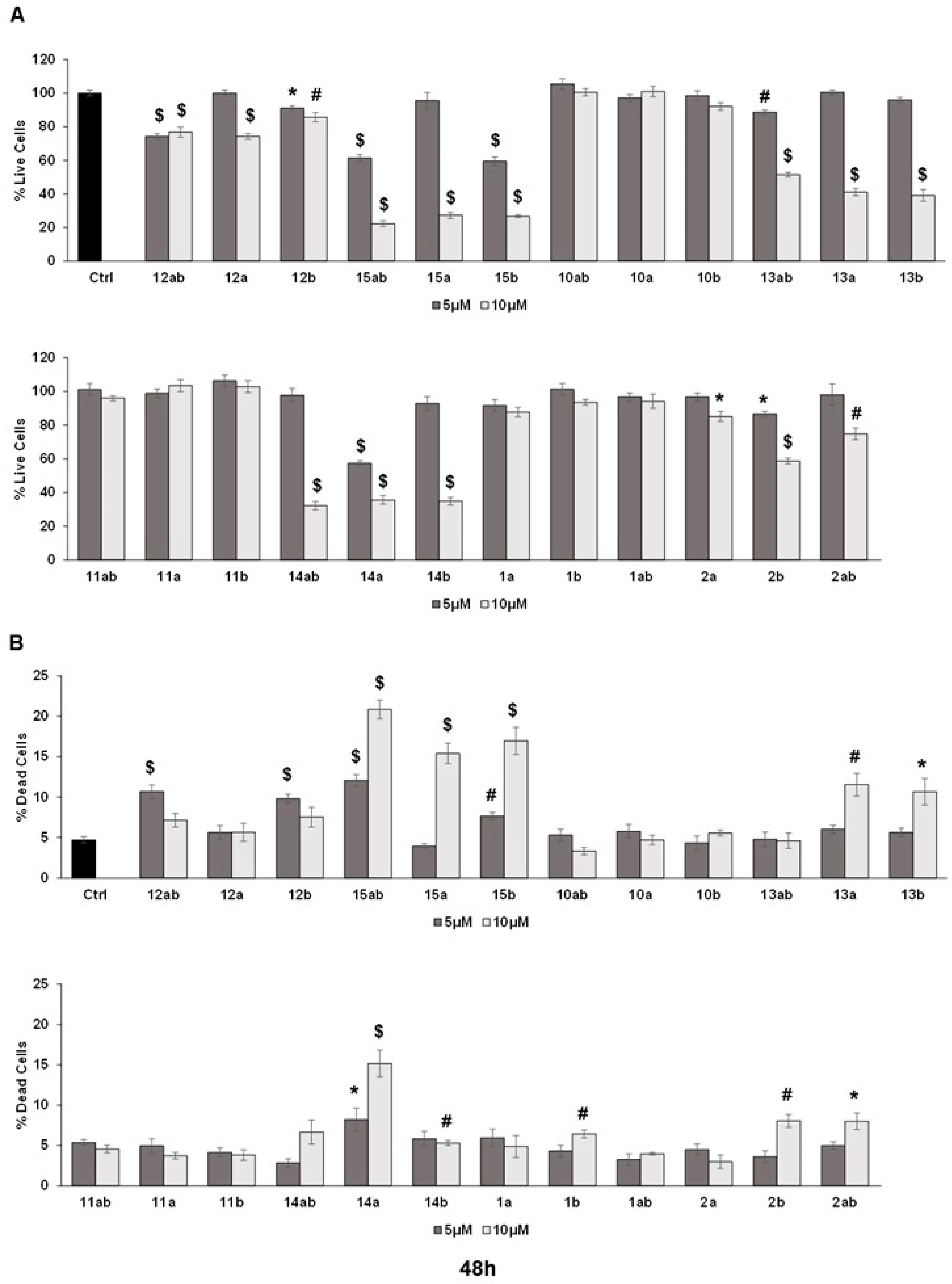
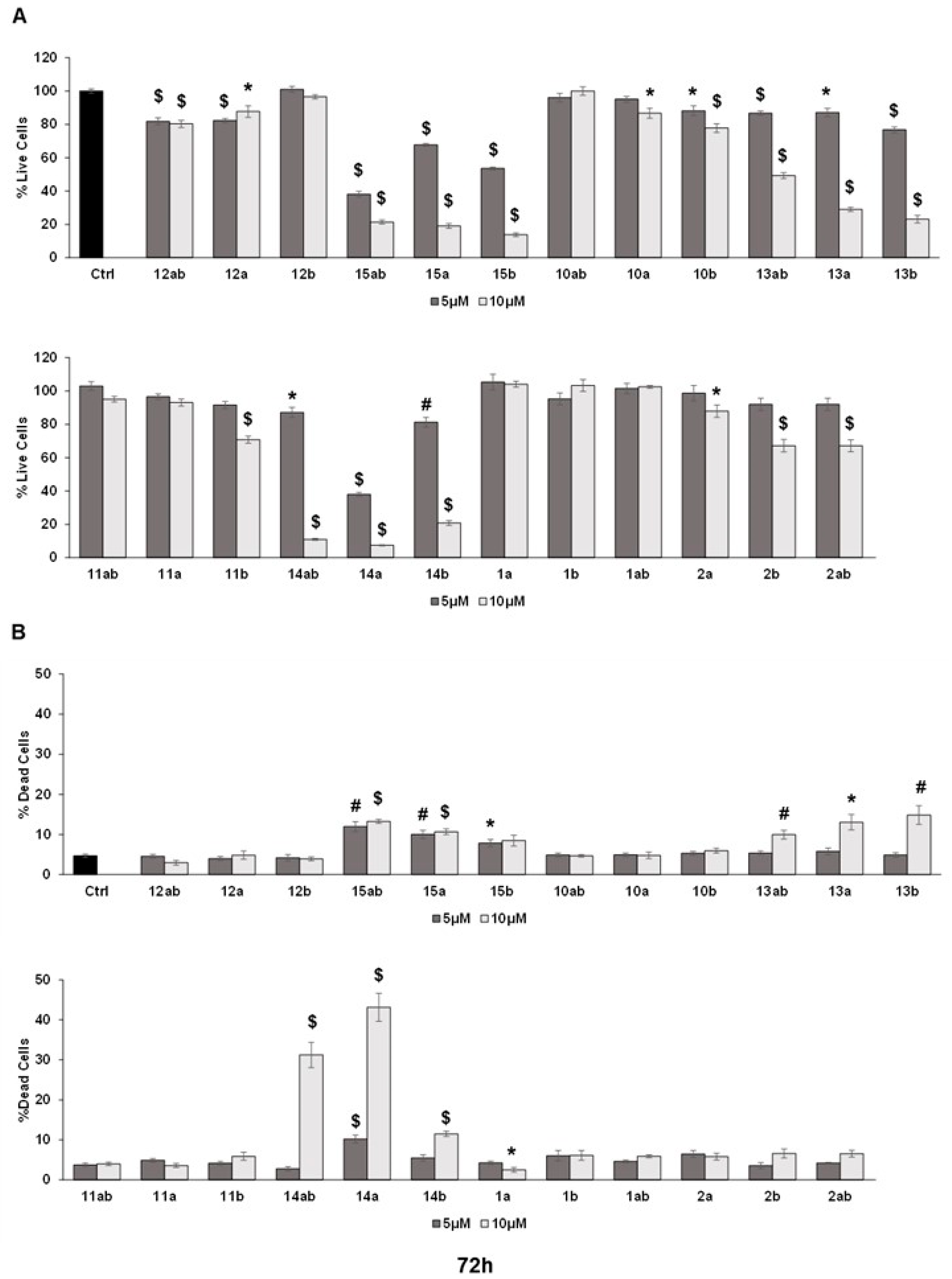
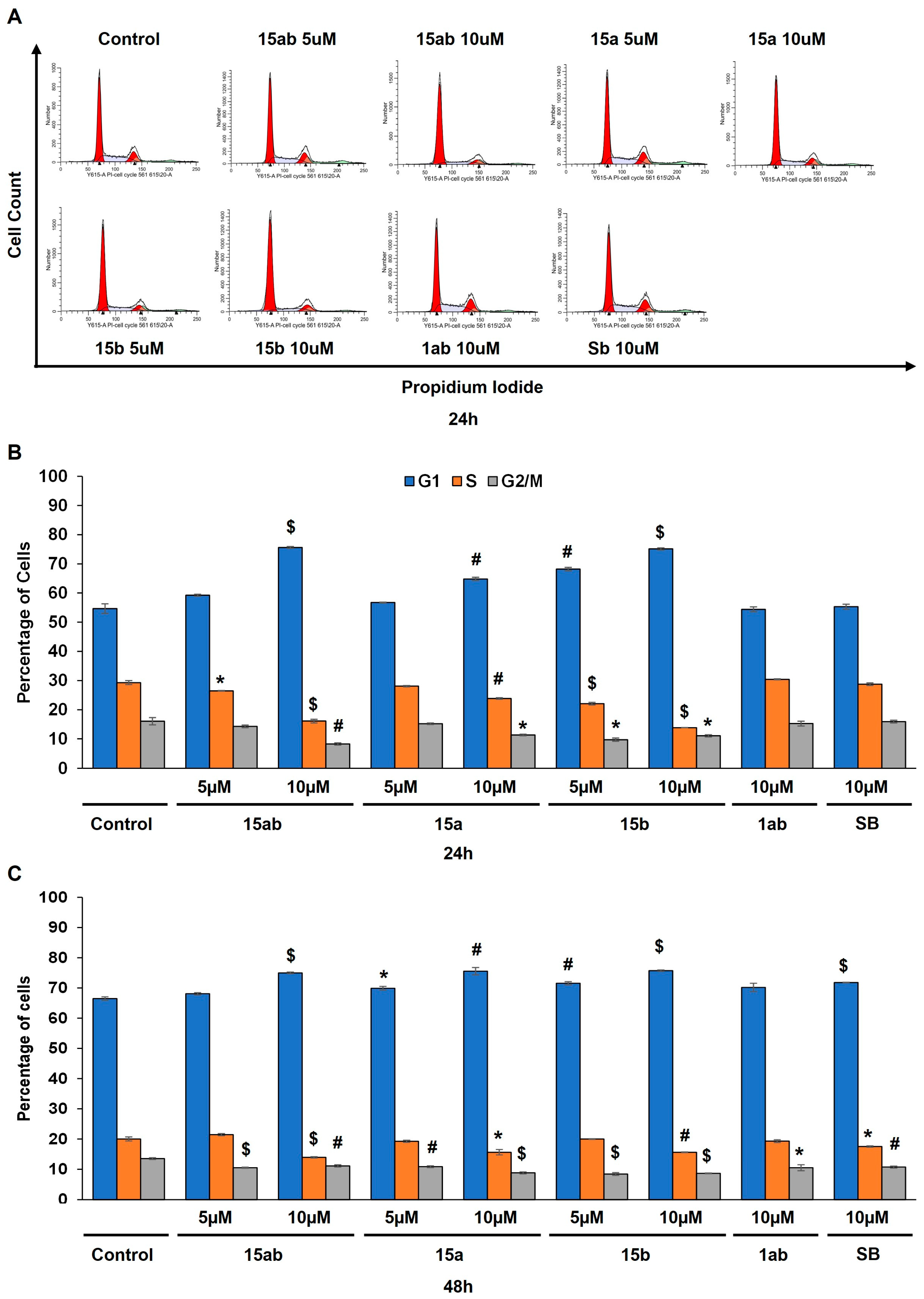
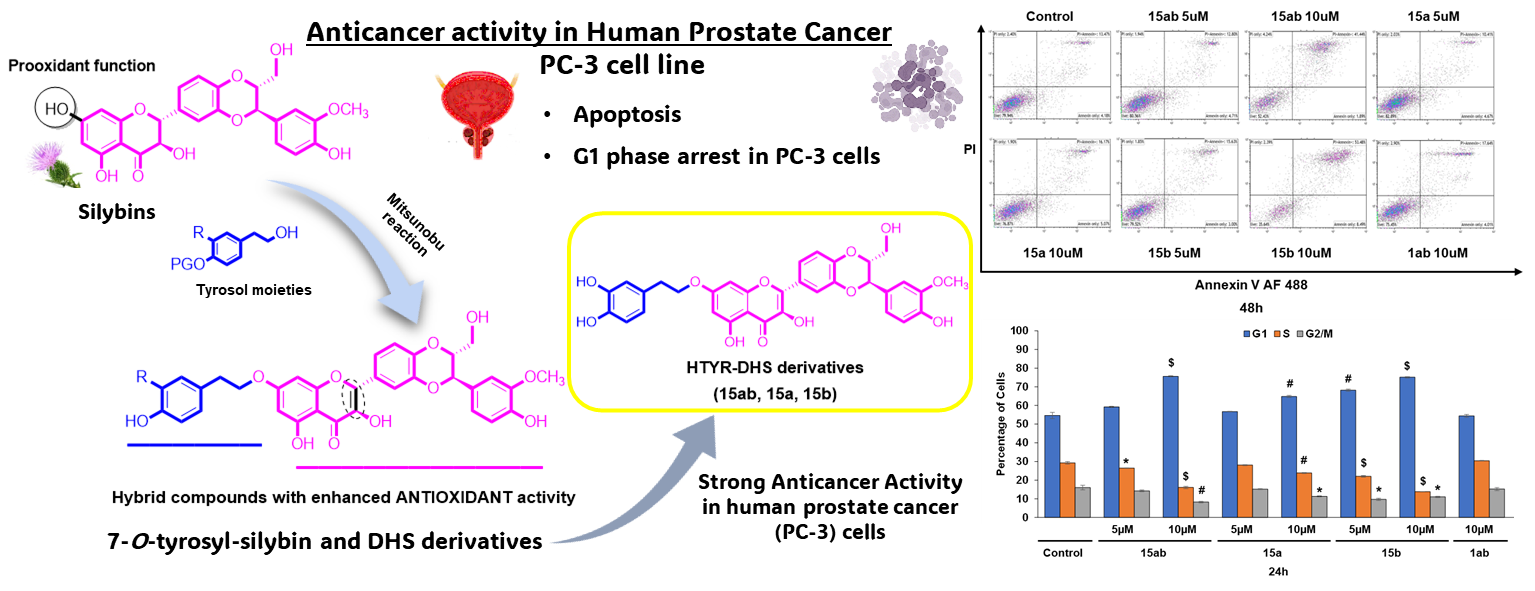
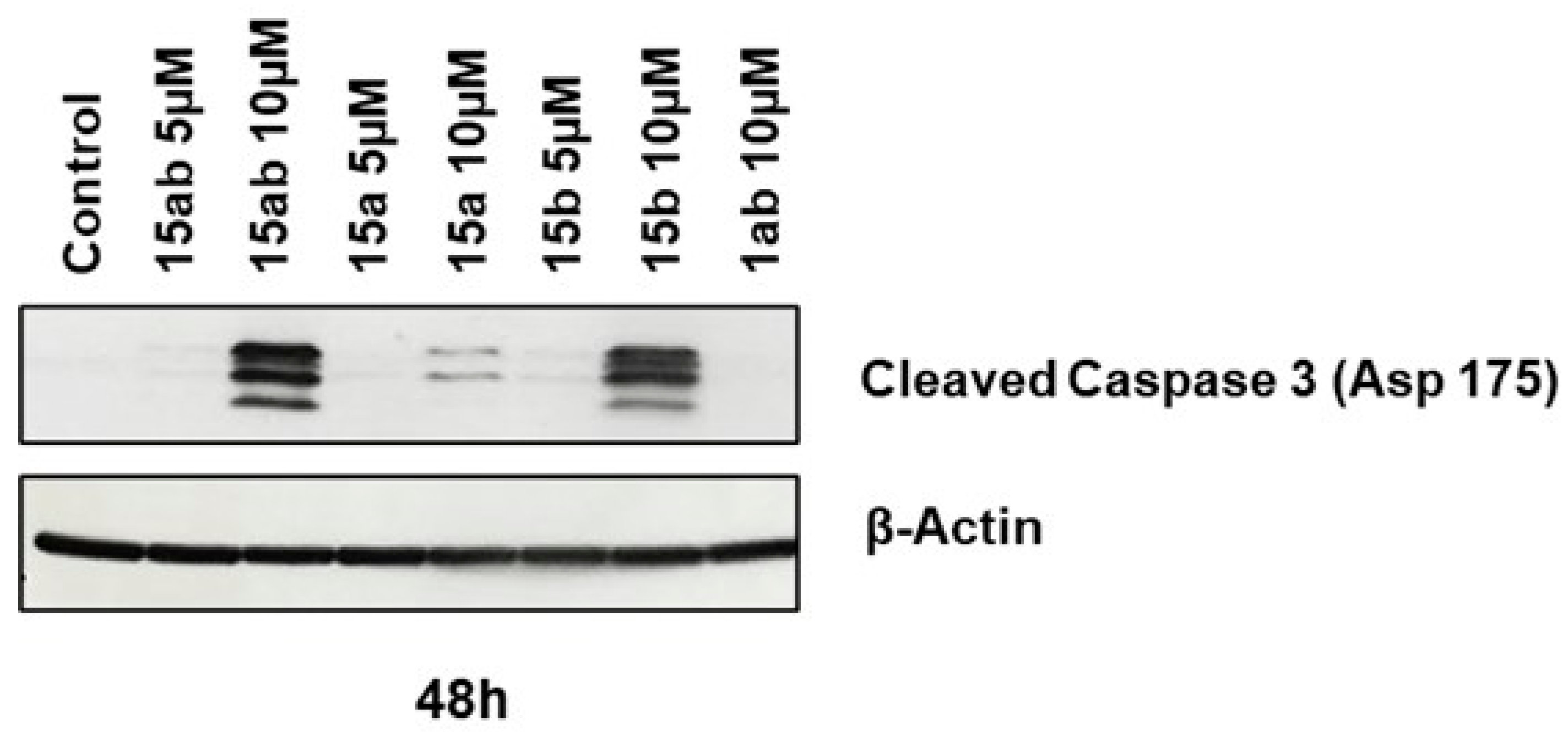
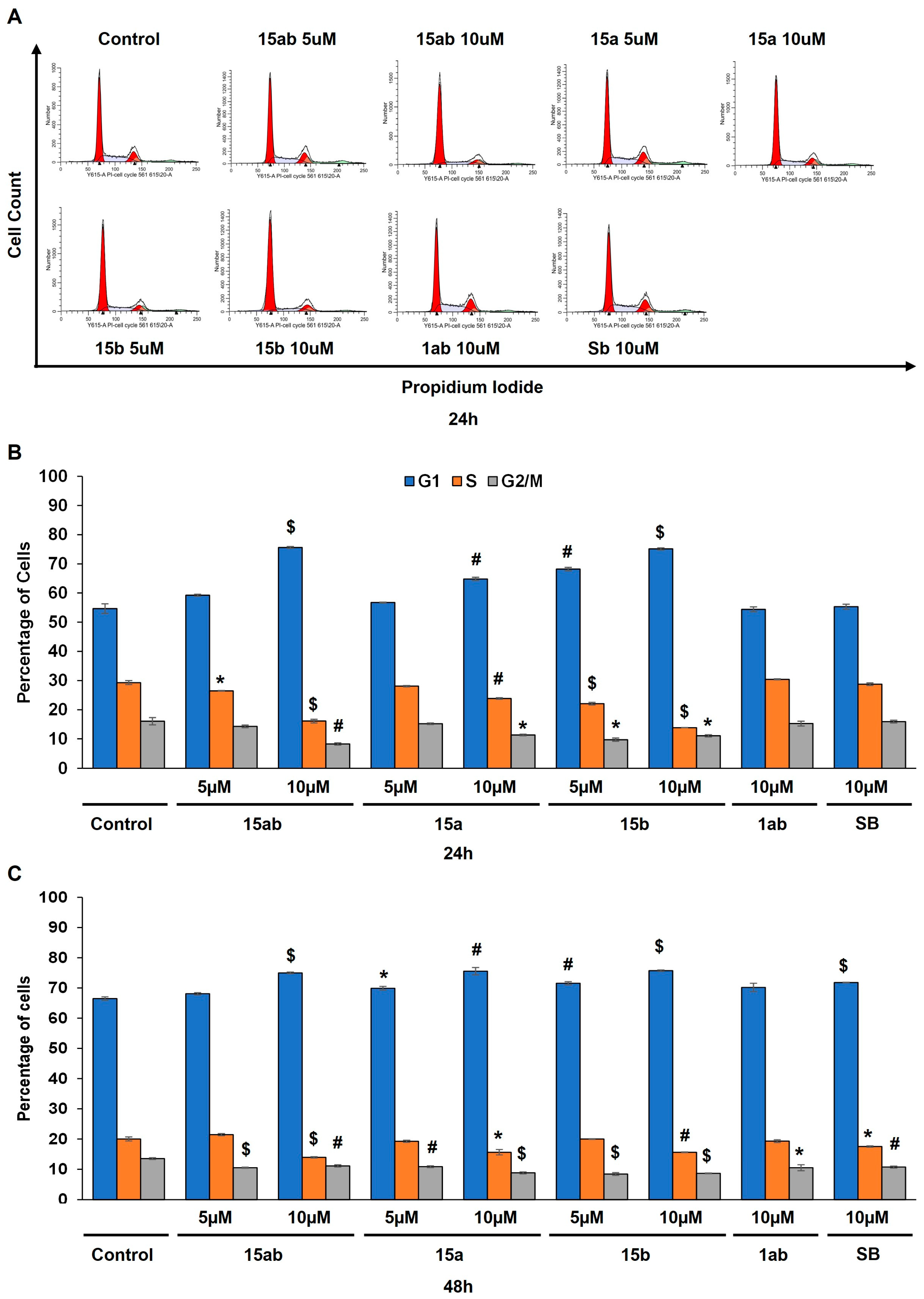
3.4. Effects of 7-O-tyrosyl 2,3-dehydro-silybin Derivatives on PC-3 Apoptosis and Cell Cycle Progression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, D.J. Natural Products as Leads to Potential Drugs: An Old Process or the New Hope for Drug Discovery? J. Med. Chem. 2008, 51, 2589–2599. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The Re-Emergence of Natural Products for Drug Discovery in the Genomics Era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chem. Int. Ed. 2011, 50, 586–621. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, E.; Clausen, M.H.; Laraia, L. Small-Molecule Inhibitors of Reactive Oxygen Species Production. J. Med. Chem. 2021, 64, 5252–5275. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Milelli, A.; Minarini, A.; Melchiorre, C. Oxidative Stress in Alzheimer’s Disease: Are We Connecting the Dots? J. Med. Chem. 2014, 57, 2821–2831. [Google Scholar] [CrossRef] [PubMed]
- Kirtonia, A.; Sethi, G.; Garg, M. The Multifaceted Role of Reactive Oxygen Species in Tumorigenesis. Cell. Mol. Life Sci. 2020, 77, 4459–4483. [Google Scholar] [CrossRef]
- Luo, M.; Zhou, L.; Huang, Z.; Li, B.; Nice, E.C.; Xu, J.; Huang, C. Antioxidant Therapy in Cancer: Rationale and Progress. Antioxidants 2022, 11, 1128. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on Natural Products for Drug Design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef]
- Gazak, R.; Walterova, D.; Kren, V. Silybin and Silymarin-New and Emerging Applications in Medicine. Curr. Med. Chem. 2007, 14, 315–338. [Google Scholar] [CrossRef]
- Kim, N.-C.; Graf, T.N.; Sparacino, C.M.; Wani, M.C.; Wall, M.E. Complete Isolation and Characterization of Silybins and Isosilybins from Milk Thistle (Silybum marianum). Org. Biomol. Chem. 2003, 1, 1684. [Google Scholar] [CrossRef]
- Pyszková, M.; Biler, M.; Biedermann, D.; Valentová, K.; Kuzma, M.; Vrba, J.; Ulrichová, J.; Sokolová, R.; Mojović, M.; Popović-Bijelić, A.; et al. Flavonolignan 2,3-Dehydroderivatives: Preparation, Antiradical and Cytoprotective Activity. Free Radic. Biol. Med. 2016, 90, 114–125. [Google Scholar] [CrossRef]
- Valentová, K.; Havlík, J.; Kosina, P.; Papoušková, B.; Jaimes, J.D.; Káňová, K.; Petrásková, L.; Ulrichová, J.; Křen, V. Biotransformation of Silymarin Flavonolignans by Human Fecal Microbiota. Metabolites 2020, 10, 29. [Google Scholar] [CrossRef]
- Platzer, M.; Kiese, S.; Tybussek, T.; Herfellner, T.; Schneider, F.; Schweiggert-Weisz, U.; Eisner, P. Radical Scavenging Mechanisms of Phenolic Compounds: A Quantitative Structure-Property Relationship (QSPR) Study. Front. Nutr. 2022, 9, 882458. [Google Scholar] [CrossRef]
- Gažák, R.; Svobodová, A.; Psotová, J.; Sedmera, P.; Přikrylová, V.; Walterová, D.; Křen, V. Oxidised Derivatives of Silybin and Their Antiradical and Antioxidant Activity. Bioorg. Med. Chem. 2004, 12, 5677–5687. [Google Scholar] [CrossRef]
- Polachi, N.; Bai, G.; Li, T.; Chu, Y.; Wang, X.; Li, S.; Gu, N.; Wu, J.; Li, W.; Zhang, Y.; et al. Modulatory Effects of Silibinin in Various Cell Signaling Pathways against Liver Disorders and Cancer—A Comprehensive Review. Eur. J. Med. Chem. 2016, 123, 577–595. [Google Scholar] [CrossRef]
- Flaig, T.W.; Glodé, M.; Gustafson, D.; van Bokhoven, A.; Tao, Y.; Wilson, S.; Su, L.-J.; Li, Y.; Harrison, G.; Agarwal, R.; et al. A Study of High-Dose Oral Silybin-Phytosome Followed by Prostatectomy in Patients with Localized Prostate Cancer. Prostate 2010, 70, 848–855. [Google Scholar] [CrossRef]
- Han, Y.H.; Lou, H.X.; Ren, D.M.; Sun, L.R.; Ma, B.; Ji, M. Stereoselective Metabolism of Silybin Diastereoisomers in the Glucuronidation Process. J. Pharm. Biomed. Anal. 2004, 34, 1071–1078. [Google Scholar] [CrossRef]
- Morazzoni, P.; Montalbetti, A.; Malandrino, S.; Pifferi, G. Comparative Pharmacokinetics of Silipide and Silymarin in Rats. Eur. J. Drug Metab. Pharmacokinet. 1993, 18, 289–297. [Google Scholar] [CrossRef]
- Wu, J.-W.; Lin, L.-C.; Hung, S.-C.; Chi, C.-W.; Tsai, T.-H. Analysis of Silibinin in Rat Plasma and Bile for Hepatobiliary Excretion and Oral Bioavailability Application. J. Pharm. Biomed. Anal. 2007, 45, 635–641. [Google Scholar] [CrossRef]
- Vue, B.; Zhang, S.; Zhang, X.; Parisis, K.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q.H. Silibinin Derivatives as Anti-Prostate Cancer Agents: Synthesis and Cell-Based Evaluations. Eur. J. Med. Chem. 2016, 109, 36–46. [Google Scholar] [CrossRef]
- Karas, D.; Gažák, R.; Valentová, K.; Chambers, C.S.; Pivodová, V.; Biedermann, D.; Křenková, A.; Oborná, I.; Kuzma, M.; Cvačka, J.; et al. Effects of 2,3-Dehydrosilybin and Its Galloyl Ester and Methyl Ether Derivatives on Human Umbilical Vein Endothelial Cells. J. Nat. Prod. 2016, 79, 812–820. [Google Scholar] [CrossRef]
- Huber, A.; Thongphasuk, P.; Erben, G.; Lehmann, W.-D.; Tuma, S.; Stremmel, W.; Chamulitrat, W. Significantly Greater Antioxidant Anticancer Activities of 2,3-Dehydrosilybin than Silybin. Biochim. Biophys. Acta 2008, 1780, 837–847. [Google Scholar] [CrossRef]
- Vue, B.; Zhang, X.; Lee, T.; Nair, N.; Zhang, S.; Chen, G.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q.-H. 5- or/and 20- O -Alkyl-2,3-Dehydrosilybins: Synthesis and Biological Profiles on Prostate Cancer Cell Models. Bioorg. Med. Chem. 2017, 25, 4845–4854. [Google Scholar] [CrossRef]
- Zhang, S.; Vue, B.; Huang, M.; Zhang, X.; Lee, T.; Chen, G.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q.-H. 3-O-Alkyl-2,3-Dehydrosilibinins: Two Synthetic Approaches and in Vitro Effects toward Prostate Cancer Cells. Bioorg. Med. Chem. Lett. 2016, 26, 3226–3231. [Google Scholar] [CrossRef]
- Křen, V.; Valentová, K. Silybin and Its Congeners: From Traditional Medicine to Molecular Effects. Nat. Prod. Rep. 2022, 39, 1264–1281. [Google Scholar] [CrossRef]
- Křen, V. Chirality Matters: Biological Activity of Optically Pure Silybin and Its Congeners. Int. J. Mol. Sci. 2021, 22, 7885. [Google Scholar] [CrossRef]
- Gažák, R.; Sedmera, P.; Vrbacký, M.; Vostálová, J.; Drahota, Z.; Marhol, P.; Walterová, D.; Křen, V. Molecular Mechanisms of Silybin and 2,3-Dehydrosilybin Antiradical Activity—Role of Individual Hydroxyl Groups. Free Radic. Biol. Med. 2009, 46, 745–758. [Google Scholar] [CrossRef]
- Sciacca, M.F.; Romanucci, V.; Zarrelli, A.; Monaco, I.; Lolicato, F.; Spinella, N.; Galati, C.; Grasso, G.; D’Urso, L.; Romeo, M.; et al. Inhibition of Aβ Amyloid Growth and Toxicity by Silybins: The Crucial Role of Stereochemistry. ACS Chem. Neurosci. 2017, 8, 1767–1778. [Google Scholar] [CrossRef]
- García-Viñuales, S.; Ilie, I.M.; Santoro, A.M.; Romanucci, V.; Zarrelli, A.; Di Fabio, G.; Caflisch, A.; Milardi, D. Silybins Inhibit Human IAPP Amyloid Growth and Toxicity through Stereospecific Interactions. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2022, 1870, 140772. [Google Scholar] [CrossRef]
- Persico, M.; García-Viñuales, S.; Santoro, A.M.; Lanza, V.; Tundo, G.R.; Sbardella, D.; Coletta, M.; Romanucci, V.; Zarrelli, A.; Di Fabio, G.; et al. Silybins Are Stereospecific Regulators of the 20S Proteasome. Bioorg. Med. Chem. 2022, 66, 116813. [Google Scholar] [CrossRef]
- Vilchez, D.; Saez, I.; Dillin, A. The Role of Protein Clearance Mechanisms in Organismal Ageing and Age-Related Diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Santoro, A.M.; Coletta, A.; Oddone, F.; Grasso, G.; Milardi, D.; Lacal, P.M.; Marini, S.; Purrello, R.; et al. The Proteasome as a Druggable Target with Multiple Therapeutic Potentialities: Cutting and Non-Cutting Edges. Pharmacol. Ther. 2020, 213, 107579. [Google Scholar] [CrossRef]
- García-Viñuales, S.; Ahmed, R.; Sciacca, M.F.M.; Lanza, V.; Giuffrida, M.L.; Zimbone, S.; Romanucci, V.; Zarrelli, A.; Bongiorno, C.; Spinella, N.; et al. Trehalose Conjugates of Silybin as Prodrugs for Targeting Toxic Aβ Aggregates. ACS Chem. Neurosci. 2020, 11, 2566–2576. [Google Scholar] [CrossRef]
- Romanucci, V.; Giordano, M.; Pagano, R.; Zimbone, S.; Giuffrida, M.L.; Milardi, D.; Zarrelli, A.; Di Fabio, G. Investigation on the Solid-Phase Synthesis of Silybin Prodrugs and Their Timed-Release. Bioorg. Med. Chem. 2021, 50, 116478. [Google Scholar] [CrossRef]
- Romanucci, V.; Giordano, M.; De Tommaso, G.; Iuliano, M.; Bernini, R.; Clemente, M.; Garcia-Viñuales, S.; Milardi, D.; Zarrelli, A.; Di Fabio, G. Synthesis of New Tyrosol-Based Phosphodiester Derivatives: Effect on Amyloid β Aggregation and Metal Chelation Ability. ChemMedChem 2021, 16, 1172–1183. [Google Scholar] [CrossRef]
- Kandhari, K.; Mishra, J.P.N.; Agarwal, R.; Singh, R.P. Acacetin Induces Sustained ERK1/2 Activation and RIP1-Dependent Necroptotic Death in Breast Cancer Cells. Toxicol. Appl. Pharmacol. 2023, 462, 116409. [Google Scholar] [CrossRef]
- Biedermann, D.; Vavříková, E.; Cvak, L.; Křen, V. Chemistry of Silybin. Nat. Prod. Rep. 2014, 31, 1138. [Google Scholar] [CrossRef]
- Džubák, P.; Hajdúch, M.; Gažák, R.; Svobodová, A.; Psotová, J.; Walterová, D.; Sedmera, P.; Křen, V. New Derivatives of Silybin and 2,3-Dehydrosilybin and Their Cytotoxic and P-Glycoprotein Modulatory Activity. Bioorg. Med. Chem. 2006, 14, 3793–3810. [Google Scholar] [CrossRef]
- Althagafy, H.S.; Graf, T.N.; Sy-Cordero, A.A.; Gufford, B.T.; Paine, M.F.; Wagoner, J.; Polyak, S.J.; Croatt, M.P.; Oberlies, N.H. Semisynthesis, Cytotoxicity, Antiviral Activity, and Drug Interaction Liability of 7-O-Methylated Analogues of Flavonolignans from Milk Thistle. Bioorg. Med. Chem. 2013, 21, 3919–3926. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.X.; Huang, K.X.; Li, H.B.; Gong, J.X.; Wang, F.; Feng, Y.B.; Tao, Q.F.; Wu, Y.H.; Li, X.K.; Wu, X.M.; et al. Design, Synthesis, and Examination of Neuron Protective Properties of Alkenylated and Amidated Dehydro-Silybin Derivatives. J. Med. Chem. 2009, 52, 7732–7752. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S. The Mitsunobu Reaction in the 21 St Century. Org. Chem. Front. 2015, 2, 739–752. [Google Scholar] [CrossRef]
- Huang, G.; Schramm, S.; Heilmann, J.; Biedermann, D.; Křen, V.; Decker, M. Unconventional Application of the Mitsunobu Reaction: Selective Flavonolignan Dehydration Yielding Hydnocarpins. Beilstein J. Org. Chem. 2016, 12, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Trouillas, P.; Marsal, P.; Svobodová, A.; Vostálová, J.; Gažák, R.; Hrbáč, J.; Sedmera, P.; Křen, V.; Lazzaroni, R.; Duroux, J.; et al. Mechanism of the Antioxidant Action of Silybin and 2,3-Dehydrosilybin Flavonolignans: A Joint Experimental and Theoretical Study. J. Phys. Chem. A 2008, 112, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Maruca, A.; Catalano, R.; Bagetta, D.; Mesiti, F.; Ambrosio, F.A.; Romeo, I.; Moraca, F.; Rocca, R.; Ortuso, F.; Artese, A.; et al. The Mediterranean Diet as Source of Bioactive Compounds with Multi-Targeting Anti-Cancer Profile. Eur. J. Med. Chem. 2019, 181, 111579. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; He, X.-W.; Jiang, J.-G.; Xu, X.-L. Hydroxytyrosol and Its Potential Therapeutic Effects. J. Agric. Food Chem. 2014, 62, 1449–1455. [Google Scholar] [CrossRef]
- Deep, G.; Singh, R.P.; Agarwal, C.; Kroll, D.J.; Agarwal, R. Silymarin and Silibinin Cause G1 and G2-M Cell Cycle Arrest via Distinct Circuitries in Human Prostate Cancer PC3 Cells: A Comparison of Flavanone Silibinin with Flavanolignan Mixture Silymarin. Oncogene 2006, 25, 1053–1069. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Yield (%) a | ORAC (TE) b | DPPH (EC50, µM) |
|---|---|---|---|
| Sil (1ab) | – | 4.76 ± 0.20 | 620 ± 0.2 |
| SilA (1a) | – | 4.21 ± 0.16 | 360 ± 0.2 |
| SilB (1b) | – | 4.36 ± 0.15 | 580 ± 0.5 |
| DHS (2ab) | – | 3.57 ± 0.23 | 29.6 ± 0.18 |
| DHSA (2a) | – | 3.88 ± 0.20 | 15.5 ± 0.3 |
| DHSB (2b) | – | 3.86 ± 0.13 | 11.6 ± 0.4 |
| TYR (3) | – | 2.18 ± 0.12 | >1000 |
| MTYR (4) | – | 2.90 ± 0.21 | 31.0 ± 2.6 |
| HTYR (5) | – | 7.40 ± 0.17 | 12.3 ± 1.0 |
| 10a | 35 | 4.81 ± 0.45 | 290 ± 0.2 |
| 11a | 28 | 2.50 ± 0.29 | 13.5 ± 0.6 |
| 12a | 26 | 5.57 ± 0.17 | 6.53 ± 0.6 |
| 13a | 95 | 1.16 ± 0.05 | 17.1 ± 0.1 |
| 14a | 78 | 1.25 ± 0.02 | 9.01 ± 0.5 |
| 15a | 95 | 1.28 ± 0.02 | 4.98 ± 0.4 |
| 10b | 83 | 4.89 ± 0.22 | 220.0 ± 0.1 |
| 11b | 21 | 3.22 ± 0.52 | 17.8 ± 0.6 |
| 12b | 65 | 5.38 ± 0.17 | 5.65 ± 0.5 |
| 13b | 72 | 1.15 ± 0.02 | 19.3 ± 0.1 |
| 14b | 82 | 1.32 ± 0.04 | 9.10 ± 0.6 |
| 15b | 81 | 1.25 ± 0.04 | 4.76 ± 0.4 |
| 10ab | 51 | 4.88 ± 0.09 | 200 ± 0.2 |
| 11ab | 33 | 4.91 ± 0.17 | 9.4 ± 0.5 |
| 12ab | 40 | 5.45 ± 0.54 | 4.9 ± 0.6 |
| 13ab | 90 | 1.47 ± 0.05 | 26.9 ± 0.2 |
| 14ab | 76 | 1.01 ± 0.12 | 9.27 ± 0.6 |
| 15ab | 74 | 1.44 ± 0.14 | 6.19 ± 0.7 |
| Position | 10a (DMSO-d6) | 11a (CD3OD) | 12a (CD3OD) | |||
|---|---|---|---|---|---|---|
| δH, (J in Hz) | δC Type | δH, (J, Hz) | δC Type | δH, (J, Hz) | δC Type | |
| 2 | 5.13, d (11.3) | 83.1 | 4.99, d (11.3) | 83.4 | 4.97, d (11.5) | 83.1 |
| 3 | 4.65, dd (11.3) | 72.0 | 4.52, d (11.3) | 72.3 | 4.50, d (11.5) | 72.0 |
| 3-OH | 4.9, s | – | – | |||
| 4 | – | 198.7 | – | 197.3 | – | 198.7 |
| 4a | – | 101.8 | – | 101.1 | – | 101.7 |
| 5 | – | 163.4 | – | 163.5 | – | 163.4 |
| 6 | 6.10, d (2.3) | 95.7 | 6.06, d (2.1) | 95.2 | 6.05, d (2.0) | 95.7 |
| 7 | – | 167.2 | – | 167.6 | – | 167.2 |
| 8 | 6.08, d (2.3) | 94.6 | 6.01, d (2.1) | 94.1 | 6.00, d (2.0) | 94.6 |
| 8a | – | 162.8 | 162.8 | – | 162.8 | |
| 1′ | – | 130.4 | 130.0 | – | 130.4 | |
| 2′ | 7.09, s | 117.1 | 7.11, d (1.5) | 116.1 | 7.10, d (1.5) | 117.1 |
| 3′ | – | 143.7 | – | 143.7 | – | 143.7 |
| 4′ | – | 144.1 | – | 144.1 | – | 144.1 |
| 5′ | 6.98, d (8.3) | 116.7 | 7.02–6.96, m | 116.4 | 6.98, d (8.3) | 116.7 |
| 6′ | 7.04–7.00, m | 121.6 | 7.03, dd (8.2, 1.8) | 120.8 | 7.03, dd (8.3, 1.8) | 121.6 |
| 1″ | – | 127.9 | – | 127.9 | – | 127.9 |
| 2″ | 7.04–7.00, m | 112.0 | 7.02–6.96, m | 110.6 | 7.01, d (1.7) | 112.0 |
| 3″ | – | 148.0 | – | 147.5 | – | 148.0 |
| 4″ | – | 147.4 | – | 146.9 | – | 147.4 |
| 5″ | 6.80, d (8.1) | 115.9 | 6.87–6.83, m | 114.8 | 6.84, d (8.2) | 115.9 |
| 6″ | 6.87, dd (8.1, 1.7) | 120.9 | 6.91, dd (8.0, 1.7) | 120.2 | 6.90, dd (8.2, 1.7) | 120.9 |
| 7″ | 4.92, d (7.9) | 76.3 | 4.95–4.89, m | 76.2 | 4.93–4.83, m | 76.3 |
| 8″ | 4.23–4.12, m | 78.5 | 4.09–4.03, m | 78.6 | 4.08–4.02, m | 78.5 |
| 9″ | H-9″a 3.54, d (12.1) H-9″b 3.39–3.30, m | 60.6 | H-9″a 3.71, dd (12.4, 2.4) H-9″b 3.49, dd (12.3, 4.3) | 60.6 | H-9″a 3.70, dd (12.4, 2.2) H-9″b 3.49 dd (12.3, 4.4) | 60.6 |
| 3″-OMe | 3.78, s | 56.1 | 3.82, s | 54.9 | 3.86, s | 56.1 |
| 1‴ | 128.2 | 129.3 | 129.0 | |||
| 2‴ | 7.08, d (8.6) | 130.2 | 6.87–6.83, m | 112.2 | 6.71, d (1.7) | 116.7 |
| 3‴ | 6.68, d (8.6) | 115.5 | – | 147.8 | – | 145.5 |
| 4‴ | – | 156.3 | – | 144.7 | – | 144.2 |
| 5‴ | 6.68, d (8.6) | 115.5 | 6.72, d (8.0) | 114.7 | 6.69, d (8.0) | 115.7 |
| 6‴ | 7.08, d (8.6) | 130.2 | 6.69, dd (8.0, 1.6) | 121.1 | 6.57, dd (8.0, 1.7) | 120.0 |
| 7‴ | 2.88, t (6.9) | 34.1 | 2.95, t (6.8) | 34.5 | 2.88, t (6.8) | 34.5 |
| 8‴ | 4.23–4.12, m | 69.6 | 4.15, t, (6.8) | 69.3 | 4.10, t (6.8) | 69.6 |
| 3‴-OMe | – | - | 3.87, s | 55.0 | – | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanucci, V.; Pagano, R.; Kandhari, K.; Zarrelli, A.; Petrone, M.; Agarwal, C.; Agarwal, R.; Di Fabio, G. 7-O-tyrosyl Silybin Derivatives as a Novel Set of Anti-Prostate Cancer Compounds. Antioxidants 2024, 13, 418. https://doi.org/10.3390/antiox13040418
Romanucci V, Pagano R, Kandhari K, Zarrelli A, Petrone M, Agarwal C, Agarwal R, Di Fabio G. 7-O-tyrosyl Silybin Derivatives as a Novel Set of Anti-Prostate Cancer Compounds. Antioxidants. 2024; 13(4):418. https://doi.org/10.3390/antiox13040418
Chicago/Turabian StyleRomanucci, Valeria, Rita Pagano, Kushal Kandhari, Armando Zarrelli, Maria Petrone, Chapla Agarwal, Rajesh Agarwal, and Giovanni Di Fabio. 2024. "7-O-tyrosyl Silybin Derivatives as a Novel Set of Anti-Prostate Cancer Compounds" Antioxidants 13, no. 4: 418. https://doi.org/10.3390/antiox13040418
APA StyleRomanucci, V., Pagano, R., Kandhari, K., Zarrelli, A., Petrone, M., Agarwal, C., Agarwal, R., & Di Fabio, G. (2024). 7-O-tyrosyl Silybin Derivatives as a Novel Set of Anti-Prostate Cancer Compounds. Antioxidants, 13(4), 418. https://doi.org/10.3390/antiox13040418