
Dose-Dependent Effects of Lipopolysaccharide on the Endothelium—Sepsis versus Metabolic Endotoxemia-Induced Cellular Senescence


 ,
,  and
and _Haendeler.png)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Cardiovascular Diseases and Endothelial Dysfunction
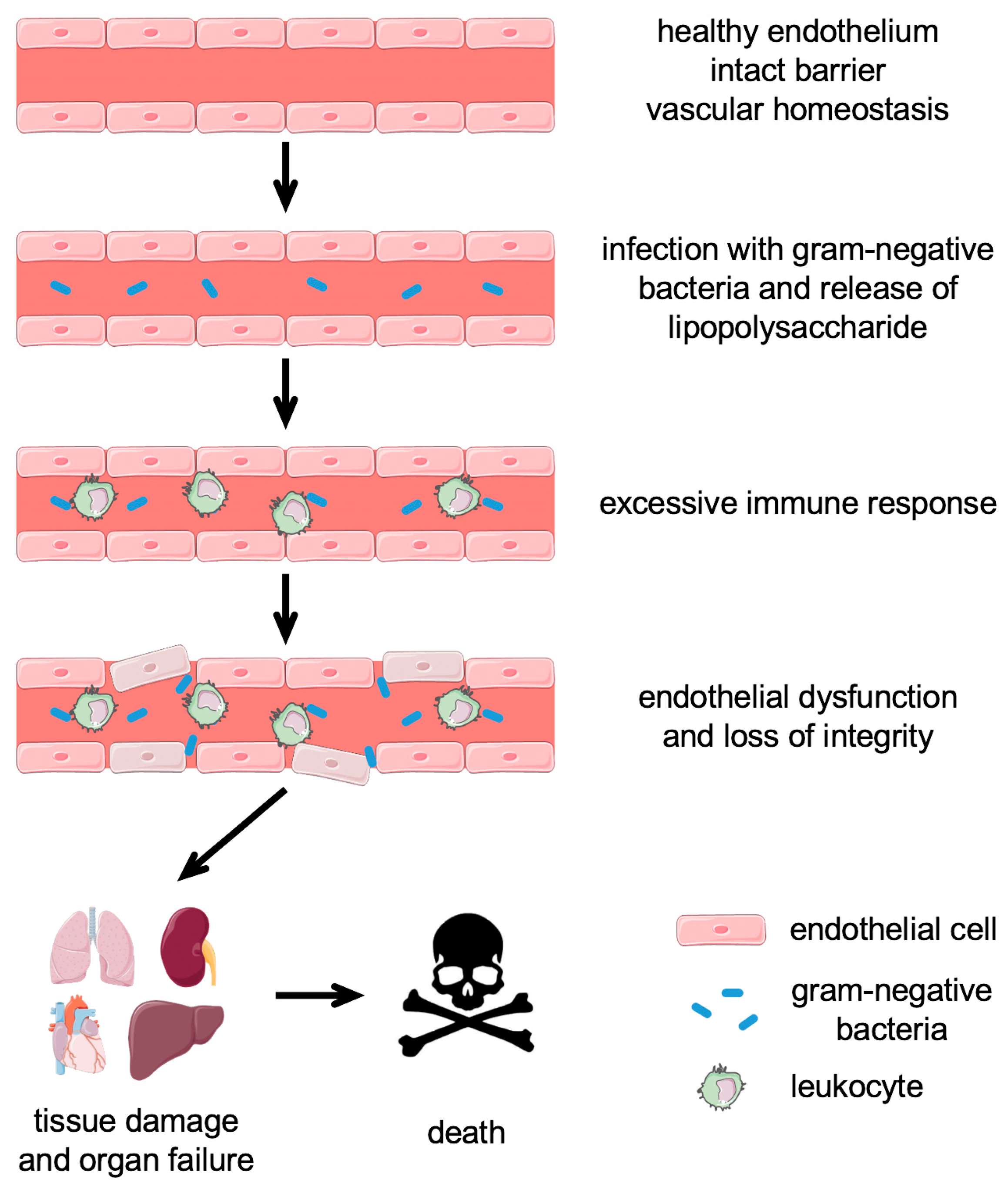
2. Sepsis
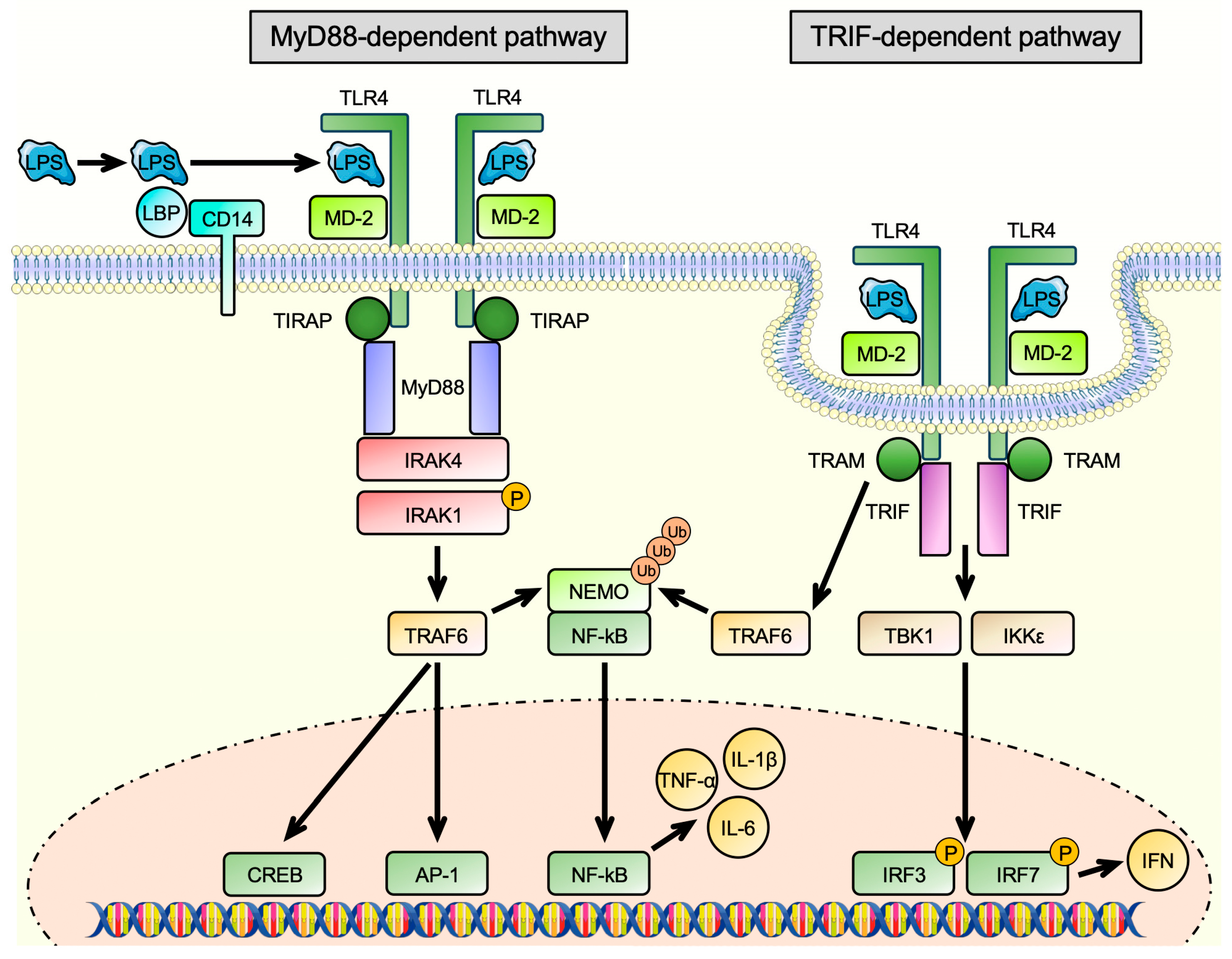
3. Toll-like Receptors
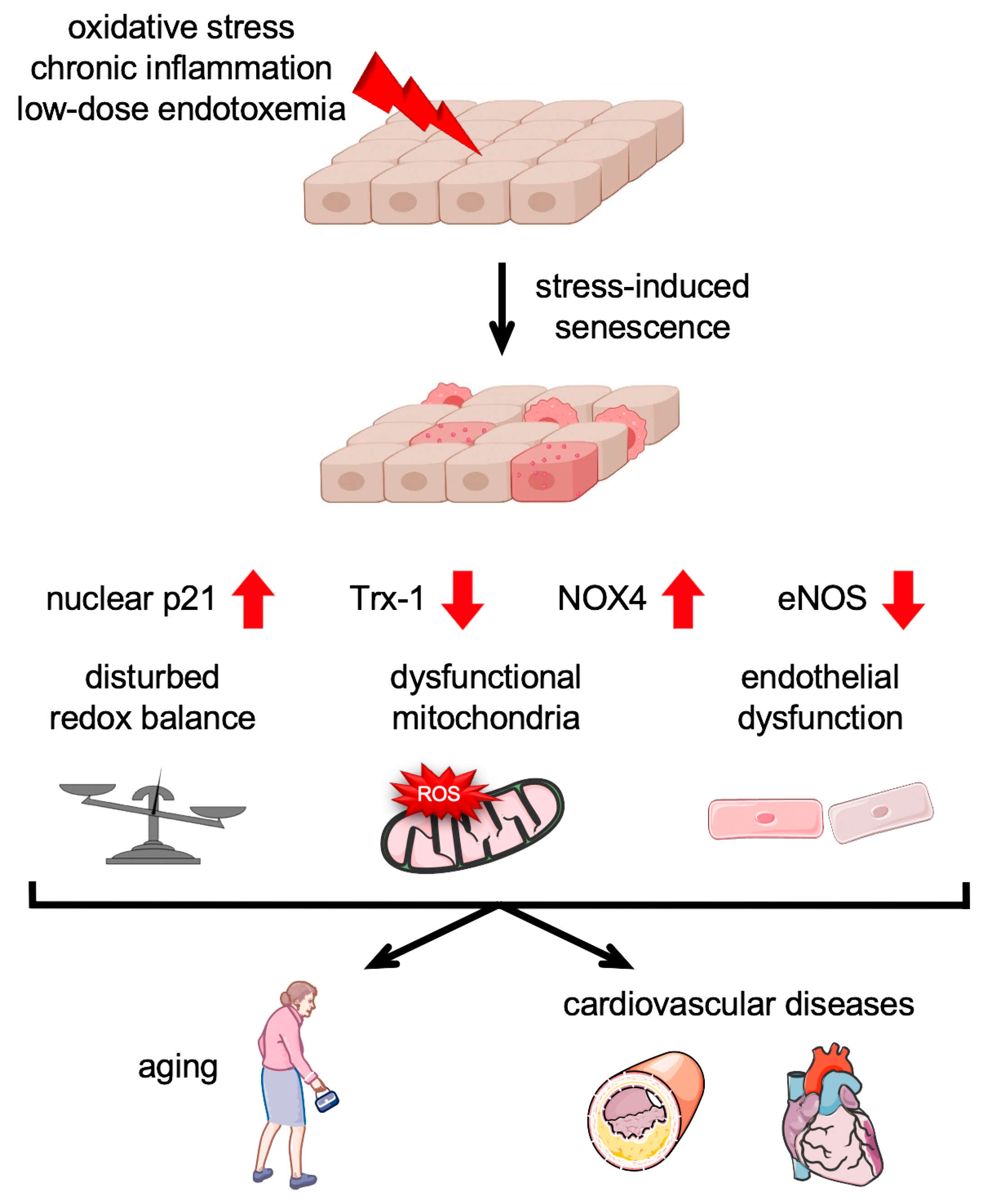
4. Stress-Induced Senescence in the Endothelium
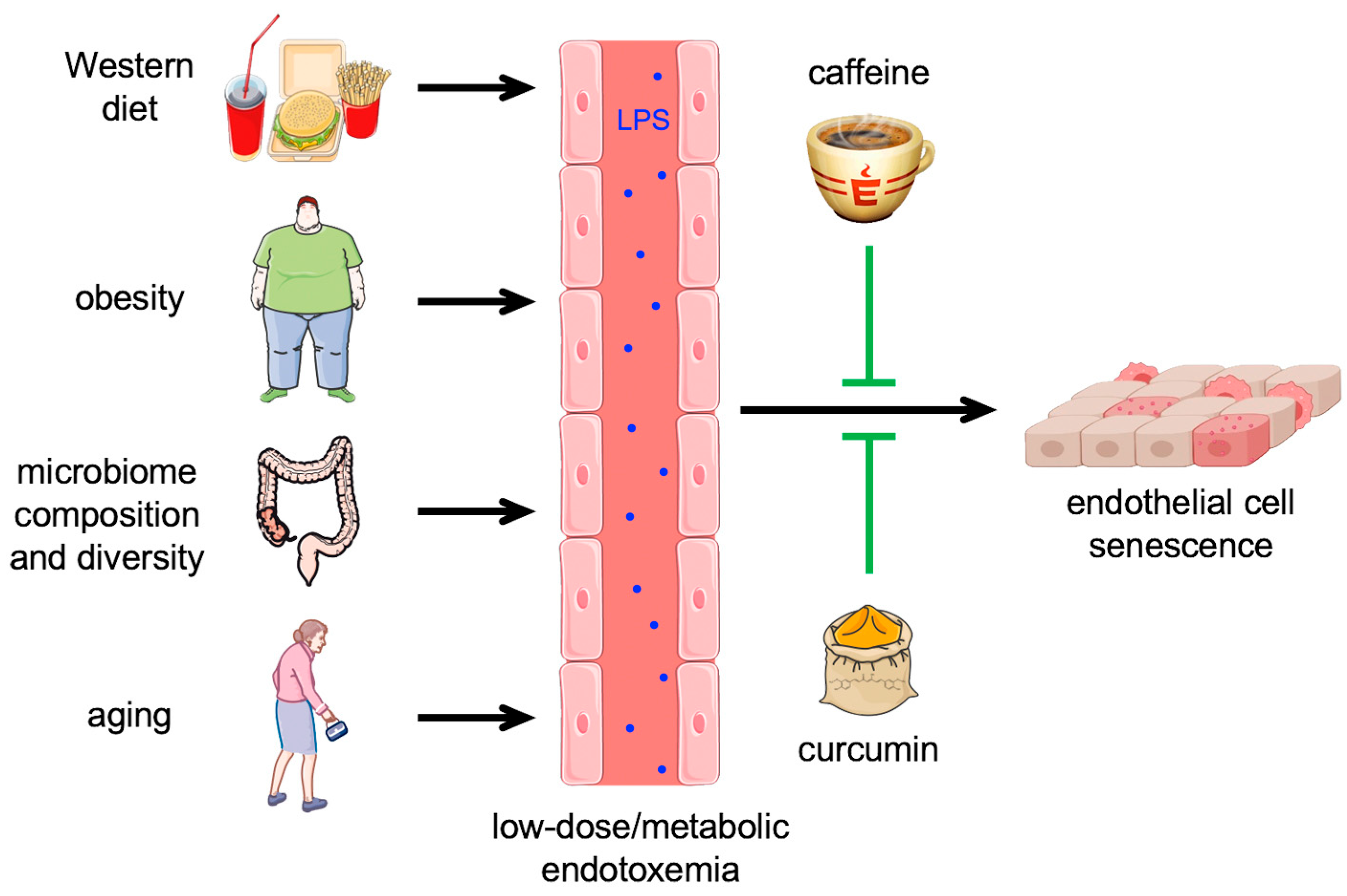
5. Low-Dose or Metabolic Endotoxemia—Role of the Gut Microbiome and the Endothelium
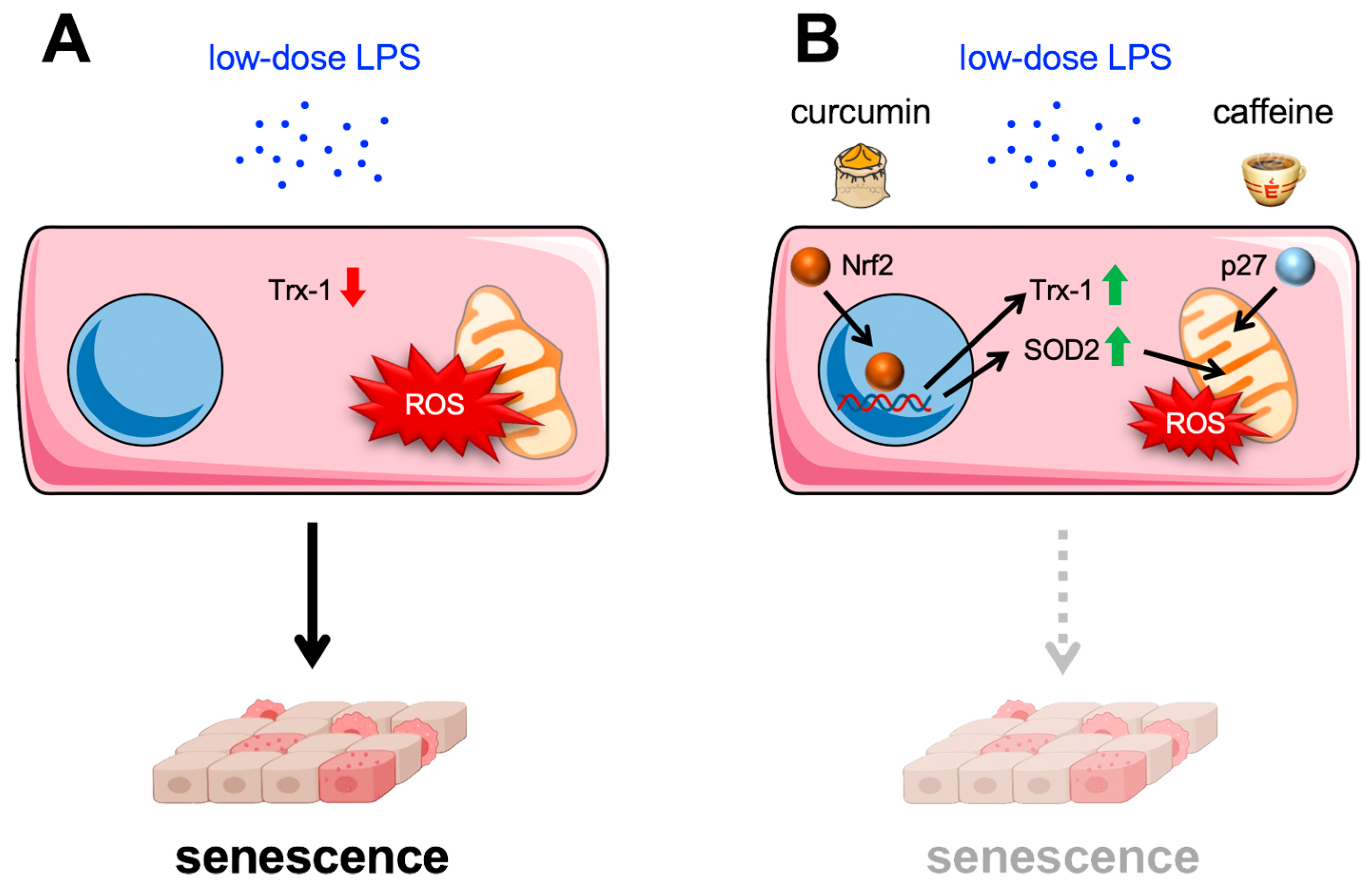
6. Impact of Curcumin and Caffeine on High-Dose and Metabolic Endotoxemia with Respect to the Endothelium
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, J.; Pan, X.; Li, G.; Chatterjee, E.; Xiao, J. Physical Exercise Protects Against Endothelial Dysfunction in Cardiovascular and Metabolic Diseases. J. Cardiovasc. Transl. Res. 2022, 15, 604–620. [Google Scholar] [CrossRef]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar]
- Pober, J.S.; Min, W.; Bradley, J.R. Mechanisms of endothelial dysfunction, injury, and death. Annu. Rev. Pathol. 2009, 4, 71–95. [Google Scholar] [CrossRef]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef]
- Boatright, K.M.; Salvesen, G.S. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 2003, 15, 725–731. [Google Scholar] [CrossRef]
- World Health Organization. Global Report on the Epidemiology and Burden of Sepsis: Current Evidence, Identifying Gaps and Future Directions; World Health Organization: Geneva, Switzerland, 2020; p. 55. [Google Scholar]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Virzi, G.M.; Mattiotti, M.; de Cal, M.; Ronco, C.; Zanella, M.; De Rosa, S. Endotoxin in Sepsis: Methods for LPS Detection and the Use of Omics Techniques. Diagnostics 2022, 13, 79. [Google Scholar] [CrossRef]
- Mayr, F.B.; Yende, S.; Angus, D.C. Epidemiology of severe sepsis. Virulence 2014, 5, 4–11. [Google Scholar] [CrossRef]
- Dauphinee, S.M.; Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Investig. 2006, 86, 9–22. [Google Scholar] [CrossRef]
- Delano, M.J.; Ward, P.A. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol. Rev. 2016, 274, 330–353. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C., Jr. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef]
- Page, M.J.; Kell, D.B.; Pretorius, E. The Role of Lipopolysaccharide-Induced Cell Signalling in Chronic Inflammation. Chronic Stress 2022, 6, 24705470221076390. [Google Scholar] [CrossRef]
- Molinaro, A.; Holst, O.; Di Lorenzo, F.; Callaghan, M.; Nurisso, A.; D’Errico, G.; Zamyatina, A.; Peri, F.; Berisio, R.; Jerala, R.; et al. Chemistry of lipid A: At the heart of innate immunity. Chemistry 2015, 21, 500–519. [Google Scholar] [CrossRef]
- Ho, J.; Chan, H.; Wong, S.H.; Wang, M.H.; Yu, J.; Xiao, Z.; Liu, X.; Choi, G.; Leung, C.C.; Wong, W.T.; et al. The involvement of regulatory non-coding RNAs in sepsis: A systematic review. Crit. Care 2016, 20, 383. [Google Scholar] [CrossRef]
- Merk, D.; Ptok, J.; Jakobs, P.; von Ameln, F.; Greulich, J.; Kluge, P.; Semperowitsch, K.; Eckermann, O.; Schaal, H.; Ale-Agha, N.; et al. Selenoprotein T Protects Endothelial Cells against Lipopolysaccharide-Induced Activation and Apoptosis. Antioxidants 2021, 10, 1427. [Google Scholar] [CrossRef]
- Opal, S.M.; Laterre, P.F.; Francois, B.; LaRosa, S.P.; Angus, D.C.; Mira, J.P.; Wittebole, X.; Dugernier, T.; Perrotin, D.; Tidswell, M.; et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The ACCESS randomized trial. JAMA 2013, 309, 1154–1162. [Google Scholar] [CrossRef]
- Marshall, J.C. Why have clinical trials in sepsis failed? Trends Mol. Med. 2014, 20, 195–203. [Google Scholar] [CrossRef]
- Guarino, M.; Perna, B.; Cesaro, A.E.; Maritati, M.; Spampinato, M.D.; Contini, C.; De Giorgio, R. 2023 Update on Sepsis and Septic Shock in Adult Patients: Management in the Emergency Department. J. Clin. Med. 2023, 12, 3188. [Google Scholar] [CrossRef]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.F. Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef]
- Vaure, C.; Liu, Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef]
- Gay, N.J.; Gangloff, M. Structure and function of Toll receptors and their ligands. Annu. Rev. Biochem. 2007, 76, 141–165. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Taylor, K.R.; Trowbridge, J.M.; Rudisill, J.A.; Termeer, C.C.; Simon, J.C.; Gallo, R.L. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J. Biol. Chem. 2004, 279, 17079–17084. [Google Scholar] [CrossRef]
- Sasu, S.; LaVerda, D.; Qureshi, N.; Golenbock, D.T.; Beasley, D. Chlamydia pneumoniae and chlamydial heat shock protein 60 stimulate proliferation of human vascular smooth muscle cells via toll-like receptor 4 and p44/p42 mitogen-activated protein kinase activation. Circ. Res. 2001, 89, 244–250. [Google Scholar] [CrossRef]
- Ghosh, S.; Lertwattanarak, R.; Garduno Jde, J.; Galeana, J.J.; Li, J.; Zamarripa, F.; Lancaster, J.L.; Mohan, S.; Hussey, S.; Musi, N. Elevated muscle TLR4 expression and metabolic endotoxemia in human aging. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 232–246. [Google Scholar] [CrossRef]
- Frantz, S.; Kobzik, L.; Kim, Y.D.; Fukazawa, R.; Medzhitov, R.; Lee, R.T.; Kelly, R.A. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J. Clin. Investig. 1999, 104, 271–280. [Google Scholar] [CrossRef]
- Kielian, T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J. Neurosci. Res. 2006, 83, 711–730. [Google Scholar] [CrossRef]
- Plociennikowska, A.; Hromada-Judycka, A.; Borzecka, K.; Kwiatkowska, K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci. 2015, 72, 557–581. [Google Scholar] [CrossRef]
- Ryu, J.K.; Kim, S.J.; Rah, S.H.; Kang, J.I.; Jung, H.E.; Lee, D.; Lee, H.K.; Lee, J.O.; Park, B.S.; Yoon, T.Y.; et al. Reconstruction of LPS Transfer Cascade Reveals Structural Determinants within LBP, CD14, and TLR4-MD2 for Efficient LPS Recognition and Transfer. Immunity 2017, 46, 38–50. [Google Scholar] [CrossRef]
- Meng, J.; Gong, M.; Bjorkbacka, H.; Golenbock, D.T. Genome-wide expression profiling and mutagenesis studies reveal that lipopolysaccharide responsiveness appears to be absolutely dependent on TLR4 and MD-2 expression and is dependent upon intermolecular ionic interactions. J. Immunol. 2011, 187, 3683–3693. [Google Scholar] [CrossRef]
- Funda, D.P.; Tuckova, L.; Farre, M.A.; Iwase, T.; Moro, I.; Tlaskalova-Hogenova, H. CD14 is expressed and released as soluble CD14 by human intestinal epithelial cells in vitro: Lipopolysaccharide activation of epithelial cells revisited. Infect. Immun. 2001, 69, 3772–3781. [Google Scholar] [CrossRef]
- Haziot, A.; Chen, S.; Ferrero, E.; Low, M.G.; Silber, R.; Goyert, S.M. The monocyte differentiation antigen, CD14, is anchored to the cell membrane by a phosphatidylinositol linkage. J. Immunol. 1988, 141, 547–552. [Google Scholar] [CrossRef]
- Frey, E.A.; Miller, D.S.; Jahr, T.G.; Sundan, A.; Bazil, V.; Espevik, T.; Finlay, B.B.; Wright, S.D. Soluble CD14 participates in the response of cells to lipopolysaccharide. J. Exp. Med. 1992, 176, 1665–1671. [Google Scholar] [CrossRef]
- Pugin, J.; Schurer-Maly, C.C.; Leturcq, D.; Moriarty, A.; Ulevitch, R.J.; Tobias, P.S. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc. Natl. Acad. Sci. USA 1993, 90, 2744–2748. [Google Scholar] [CrossRef]
- Golenbock, D.T.; Bach, R.R.; Lichenstein, H.; Juan, T.S.; Tadavarthy, A.; Moldow, C.F. Soluble CD14 promotes LPS activation of CD14-deficient PNH monocytes and endothelial cells. J. Lab. Clin. Med. 1995, 125, 662–671. [Google Scholar]
- Haziot, A.; Rong, G.W.; Silver, J.; Goyert, S.M. Recombinant soluble CD14 mediates the activation of endothelial cells by lipopolysaccharide. J. Immunol. 1993, 151, 1500–1507. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, X.; Bao, X.; Xiao, W.; Chen, G. Toll-like receptor 4 (TLR4) inhibitors: Current research and prospective. Eur. J. Med. Chem. 2022, 235, 114291. [Google Scholar] [CrossRef]
- Li, S.; Strelow, A.; Fontana, E.J.; Wesche, H. IRAK-4: A novel member of the IRAK family with the properties of an IRAK-kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 5567–5572. [Google Scholar] [CrossRef]
- Arch, R.H.; Gedrich, R.W.; Thompson, C.B. Tumor necrosis factor receptor-associated factors (TRAFs)—A family of adapter proteins that regulates life and death. Genes. Dev. 1998, 12, 2821–2830. [Google Scholar] [CrossRef]
- Shaywitz, A.J.; Greenberg, M.E. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 1999, 68, 821–861. [Google Scholar] [CrossRef]
- Zhao, D.; Kwon, S.H.; Chun, Y.S.; Gu, M.Y.; Yang, H.O. Anti-Neuroinflammatory Effects of Fucoxanthin via Inhibition of Akt/NF-kappaB and MAPKs/AP-1 Pathways and Activation of PKA/CREB Pathway in Lipopolysaccharide-Activated BV-2 Microglial Cells. Neurochem. Res. 2017, 42, 667–677. [Google Scholar] [CrossRef]
- Lu, Y.; Li, B.; Xu, A.; Liang, X.; Xu, T.; Jin, H.; Xie, Y.; Wang, R.; Liu, X.; Gao, X.; et al. NF-kappaB and AP-1 are required for the lipopolysaccharide-induced expression of MCP-1, CXCL1, and Cx43 in cultured rat dorsal spinal cord astrocytes. Front. Mol. Neurosci. 2022, 15, 859558. [Google Scholar] [CrossRef]
- Marongiu, L.; Gornati, L.; Artuso, I.; Zanoni, I.; Granucci, F. Below the surface: The inner lives of TLR4 and TLR9. J. Leukoc. Biol. 2019, 106, 147–160. [Google Scholar] [CrossRef]
- Tanimura, N.; Saitoh, S.; Matsumoto, F.; Akashi-Takamura, S.; Miyake, K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun. 2008, 368, 94–99. [Google Scholar] [CrossRef]
- Jiang, Z.; Georgel, P.; Du, X.; Shamel, L.; Sovath, S.; Mudd, S.; Huber, M.; Kalis, C.; Keck, S.; Galanos, C.; et al. CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 2005, 6, 565–570. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Verstak, B.; Stack, J.; Ve, T.; Mangan, M.; Hjerrild, K.; Jeon, J.; Stahl, R.; Latz, E.; Gay, N.; Kobe, B.; et al. The TLR signaling adaptor TRAM interacts with TRAF6 to mediate activation of the inflammatory response by TLR4. J. Leukoc. Biol. 2014, 96, 427–436. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Molloy, S. Host response: LPS goes non-canonical. Nat. Rev. Microbiol. 2013, 11, 599. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Russo, A.J.; Behl, B.; Banerjee, I.; Yankova, M.; Deshmukh, S.D.; Rathinam, V.A.K. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165, 1106–1119. [Google Scholar] [CrossRef]
- Hirschfeld, M.; Weis, J.J.; Toshchakov, V.; Salkowski, C.A.; Cody, M.J.; Ward, D.C.; Qureshi, N.; Michalek, S.M.; Vogel, S.N. Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect. Immun. 2001, 69, 1477–1482. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; O’Neill, L.A. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 2004, 113, 153–162. [Google Scholar] [CrossRef]
- Steimle, A.; Autenrieth, I.B.; Frick, J.S. Structure and function: Lipid A modifications in commensals and pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. [Google Scholar] [CrossRef]
- Mohr, A.E.; Crawford, M.; Jasbi, P.; Fessler, S.; Sweazea, K.L. Lipopolysaccharide and the gut microbiota: Considering structural variation. FEBS Lett. 2022, 596, 849–875. [Google Scholar] [CrossRef]
- Barochia, A.; Solomon, S.; Cui, X.; Natanson, C.; Eichacker, P.Q. Eritoran tetrasodium (E5564) treatment for sepsis: Review of preclinical and clinical studies. Expert. Opin. Drug Metab. Toxicol. 2011, 7, 479–494. [Google Scholar] [CrossRef]
- Abelli, J.; Mendez-Valdes, G.; Gomez-Hevia, F.; Bragato, M.C.; Chichiarelli, S.; Saso, L.; Rodrigo, R. Potential Antioxidant Multitherapy against Complications Occurring in Sepsis. Biomedicines 2022, 10, 3088. [Google Scholar] [CrossRef]
- Liang, Y.; Liang, N.; Yin, L.; Xiao, F. Cellular and molecular mechanisms of xenobiotics-induced premature senescence. Toxicol. Res. 2020, 9, 669–675. [Google Scholar] [CrossRef]
- Merk, D.; Greulich, J.; Vierkant, A.; Cox, F.; Eckermann, O.; von Ameln, F.; Dyballa-Rukes, N.; Altschmied, J.; Ale-Agha, N.; Jakobs, P.; et al. Caffeine Inhibits Oxidative Stress- and Low Dose Endotoxemia-Induced Senescence-Role of Thioredoxin-1. Antioxidants 2023, 12, 1244. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Andreo-López, M.C.; Contreras-Bolívar, V.; Muñoz-Torres, M.; García-Fontana, B.; García-Fontana, C. Influence of the Mediterranean Diet on Healthy Aging. Int. J. Mol. Sci. 2023, 24, 4491. [Google Scholar] [CrossRef]
- Suzuki, K.; Susaki, E.A.; Nagaoka, I. Lipopolysaccharides and Cellular Senescence: Involvement in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 11148. [Google Scholar] [CrossRef]
- Goy, C.; Czypiorski, P.; Altschmied, J.; Jakob, S.; Rabanter, L.L.; Brewer, A.C.; Ale-Agha, N.; Dyballa-Rukes, N.; Shah, A.M.; Haendeler, J. The imbalanced redox status in senescent endothelial cells is due to dysregulated Thioredoxin-1 and NADPH oxidase 4. Exp. Gerontol. 2014, 56, 45–52. [Google Scholar] [CrossRef]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arter. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef]
- Büchner, N.; Ale-Agha, N.; Jakob, S.; Sydlik, U.; Kunze, K.; Unfried, K.; Altschmied, J.; Haendeler, J. Unhealthy diet and ultrafine carbon black particles induce senescence and disease associated phenotypic changes. Exp. Gerontol. 2013, 48, 8–16. [Google Scholar] [CrossRef]
- Oh, S.T.; Park, H.; Yoon, H.J.; Yang, S.Y. Erratum to “Long-Term Treatment of Native LDL Induces Senescence of Cultured Human Endothelial Cells”. Oxid. Med. Cell Longev. 2017, 2017, 4576985. [Google Scholar] [CrossRef]
- Gonnissen, S.; Ptok, J.; Goy, C.; Jander, K.; Jakobs, P.; Eckermann, O.; Kaisers, W.; von Ameln, F.; Timm, J.; Ale-Agha, N.; et al. High Concentration of Low-Density Lipoprotein Results in Disturbances in Mitochondrial Transcription and Functionality in Endothelial Cells. Oxid. Med. Cell Longev. 2019, 2019, 7976382. [Google Scholar] [CrossRef]
- Han, Y.; Kim, S.Y. Endothelial senescence in vascular diseases: Current understanding and future opportunities in senotherapeutics. Exp. Mol. Med. 2023, 55, 1–12. [Google Scholar] [CrossRef]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of senescence and aging. Biochem. Med. 2019, 29, 030501. [Google Scholar] [CrossRef]
- Jendrach, M.; Pohl, S.; Vöth, M.; Kowald, A.; Hammerstein, P.; Bereiter-Hahn, J. Morpho-dynamic changes of mitochondria during ageing of human endothelial cells. Mech. Ageing Dev. 2005, 126, 813–821. [Google Scholar] [CrossRef]
- Berg, R.D. The indigenous gastrointestinal microflora. Trends Microbiol. 1996, 4, 430–435. [Google Scholar] [CrossRef]
- Carnevale, R.; Sciarretta, S.; Valenti, V.; di Nonno, F.; Calvieri, C.; Nocella, C.; Frati, G.; Forte, M.; d’Amati, G.; Pignataro, M.G.; et al. Low-grade endotoxaemia enhances artery thrombus growth via Toll-like receptor 4: Implication for myocardial infarction. Eur. Heart J. 2020, 41, 3156–3165. [Google Scholar] [CrossRef]
- Oliva, A.; Cammisotto, V.; Cangemi, R.; Ferro, D.; Miele, M.C.; De Angelis, M.; Cancelli, F.; Pignatelli, P.; Venditti, M.; Pugliese, F.; et al. Low-Grade Endotoxemia and Thrombosis in COVID-19. Clin. Transl. Gastroenterol. 2021, 12, e00348. [Google Scholar] [CrossRef]
- Wiedermann, C.J.; Kiechl, S.; Dunzendorfer, S.; Schratzberger, P.; Egger, G.; Oberhollenzer, F.; Willeit, J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: Prospective results from the Bruneck Study. J. Am. Coll. Cardiol. 1999, 34, 1975–1981. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in Plasma Endotoxin Concentrations and the Expression of Toll-Like Receptors and Suppressor of Cytokine Signaling-3 in Mononuclear Cells After a High-Fat, High-Carbohydrate Meal: Implications for insulin resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef]
- Boutagy, N.E.; McMillan, R.P.; Frisard, M.I.; Hulver, M.W. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie 2016, 124, 11–20. [Google Scholar] [CrossRef]
- Neves, A.L.; Coelho, J.; Couto, L.; Leite-Moreira, A.; Roncon-Albuquerque, R., Jr. Metabolic endotoxemia: A molecular link between obesity and cardiovascular risk. J. Mol. Endocrinol. 2013, 51, R51–R64. [Google Scholar] [CrossRef]
- Erlanson-Albertsson, C.; Stenkula, K.G. The Importance of Food for Endotoxemia and an Inflammatory Response. Int. J. Mol. Sci. 2021, 22, 9562. [Google Scholar] [CrossRef]
- Deopurkar, R.; Ghanim, H.; Friedman, J.; Abuaysheh, S.; Sia, C.L.; Mohanty, P.; Viswanathan, P.; Chaudhuri, A.; Dandona, P. Differential effects of cream, glucose, and orange juice on inflammation, endotoxin, and the expression of Toll-like receptor-4 and suppressor of cytokine signaling-3. Diabetes Care 2010, 33, 991–997. [Google Scholar] [CrossRef]
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A high-fat meal induces low-grade endotoxemia: Evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292. [Google Scholar] [CrossRef]
- Laugerette, F.; Vors, C.; Geloen, A.; Chauvin, M.A.; Soulage, C.; Lambert-Porcheron, S.; Peretti, N.; Alligier, M.; Burcelin, R.; Laville, M.; et al. Emulsified lipids increase endotoxemia: Possible role in early postprandial low-grade inflammation. J. Nutr. Biochem. 2011, 22, 53–59. [Google Scholar] [CrossRef]
- Ghoshal, S.; Witta, J.; Zhong, J.; de Villiers, W.; Eckhardt, E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J. Lipid Res. 2009, 50, 90–97. [Google Scholar] [CrossRef]
- Tomassen, M.M.M.; Govers, C.; Vos, A.P.; de Wit, N.J.W. Dietary fat induced chylomicron-mediated LPS translocation in a bicameral Caco-2cell model. Lipids Health Dis. 2023, 22, 4. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Do, M.H.; Lee, E.; Oh, M.-J.; Kim, Y.; Park, H.-Y. High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients 2018, 10, 761. [Google Scholar] [CrossRef]
- Guerville, M.; Leroy, A.; Sinquin, A.; Laugerette, F.; Michalski, M.C.; Boudry, G. Western-diet consumption induces alteration of barrier function mechanisms in the ileum that correlates with metabolic endotoxemia in rats. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E107–E120. [Google Scholar] [CrossRef]
- Zhou, X.; Han, D.; Xu, R.; Li, S.; Wu, H.; Qu, C.; Wang, F.; Wang, X.; Zhao, Y. A model of metabolic syndrome and related diseases with intestinal endotoxemia in rats fed a high fat and high sucrose diet. PLoS ONE 2014, 9, e115148. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Cani, P.D.; Neyrinck, A.M.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.M.; Gibson, G.R.; Delzenne, N.M. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007, 50, 2374–2383. [Google Scholar] [CrossRef]
- Anhe, F.F.; Barra, N.G.; Cavallari, J.F.; Henriksbo, B.D.; Schertzer, J.D. Metabolic endotoxemia is dictated by the type of lipopolysaccharide. Cell Rep. 2021, 36, 109691. [Google Scholar] [CrossRef]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferrieres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar] [CrossRef]
- Ahola, A.J.; Lassenius, M.I.; Forsblom, C.; Harjutsalo, V.; Lehto, M.; Groop, P.H. Dietary patterns reflecting healthy food choices are associated with lower serum LPS activity. Sci. Rep. 2017, 7, 6511. [Google Scholar] [CrossRef]
- Lyte, J.M.; Gabler, N.K.; Hollis, J.H. Postprandial serum endotoxin in healthy humans is modulated by dietary fat in a randomized, controlled, cross-over study. Lipids Health Dis. 2016, 15, 186. [Google Scholar] [CrossRef]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated With Endotoxemia That Originates From the Gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef]
- Mehta, N.N.; McGillicuddy, F.C.; Anderson, P.D.; Hinkle, C.C.; Shah, R.; Pruscino, L.; Tabita-Martinez, J.; Sellers, K.F.; Rickels, M.R.; Reilly, M.P. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes 2010, 59, 172–181. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Jeffery, I.B.; Lynch, D.B.; O’Toole, P.W. Composition and temporal stability of the gut microbiota in older persons. ISME J. 2016, 10, 170–182. [Google Scholar] [CrossRef]
- Langille, M.G.; Meehan, C.J.; Koenig, J.E.; Dhanani, A.S.; Rose, R.A.; Howlett, S.E.; Beiko, R.G. Microbial shifts in the aging mouse gut. Microbiome 2014, 2, 50. [Google Scholar] [CrossRef]
- Xu, C.; Zhu, H.; Qiu, P. Aging progression of human gut microbiota. BMC Microbiol. 2019, 19, 236. [Google Scholar] [CrossRef]
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J. Endocr. Soc. 2020, 4, bvz039. [Google Scholar] [CrossRef]
- Violi, F.; Cammisotto, V.; Bartimoccia, S.; Pignatelli, P.; Carnevale, R.; Nocella, C. Gut-derived low-grade endotoxaemia, atherothrombosis and cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 24–37. [Google Scholar] [CrossRef]
- Mayr, F.B.; Spiel, A.O.; Leitner, J.M.; Firbas, C.; Sieghart, W.; Jilma, B. Effects of low dose endotoxemia on endothelial progenitor cells in humans. Atherosclerosis 2007, 195, e202–e206. [Google Scholar] [CrossRef]
- Freedman, N.D.; Park, Y.; Abnet, C.C.; Hollenbeck, A.R.; Sinha, R. Association of coffee drinking with total and cause-specific mortality. N. Engl. J. Med. 2012, 366, 1891–1904. [Google Scholar] [CrossRef]
- Gunter, M.J.; Murphy, N.; Cross, A.J.; Dossus, L.; Dartois, L.; Fagherazzi, G.; Kaaks, R.; Kuhn, T.; Boeing, H.; Aleksandrova, K.; et al. Coffee Drinking and Mortality in 10 European Countries: A Multinational Cohort Study. Ann. Intern. Med. 2017, 167, 236–247. [Google Scholar] [CrossRef]
- Spyridopoulos, I.; Fichtlscherer, S.; Popp, R.; Toennes, S.W.; Fisslthaler, B.; Trepels, T.; Zernecke, A.; Liehn, E.A.; Weber, C.; Zeiher, A.M.; et al. Caffeine enhances endothelial repair by an AMPK-dependent mechanism. Arter. Thromb. Vasc. Biol. 2008, 28, 1967–1974. [Google Scholar] [CrossRef]
- Ale-Agha, N.; Goy, C.; Jakobs, P.; Spyridopoulos, I.; Gonnissen, S.; Dyballa-Rukes, N.; Aufenvenne, K.; von Ameln, F.; Zurek, M.; Spannbrucker, T.; et al. CDKN1B/p27 is localized in mitochondria and improves respiration-dependent processes in the cardiovascular system-New mode of action for caffeine. PLoS Biol. 2018, 16, e2004408. [Google Scholar] [CrossRef]
- Cox, F.F.; Misiou, A.; Vierkant, A.; Ale-Agha, N.; Grandoch, M.; Haendeler, J.; Altschmied, J. Protective Effects of Curcumin in Cardiovascular Diseases-Impact on Oxidative Stress and Mitochondria. Cells 2022, 11, 342. [Google Scholar] [CrossRef]
- Scazzocchio, B.; Minghetti, L.; D’Archivio, M. Interaction between Gut Microbiota and Curcumin: A New Key of Understanding for the Health Effects of Curcumin. Nutrients 2020, 12, 2499. [Google Scholar] [CrossRef]
- Chen, L.; Wang, X.J.; Chen, J.X.; Yang, J.C.; Ling, L.; Cai, X.B.; Chen, Y.S. Caffeine ameliorates the metabolic syndrome in diet-induced obese mice through regulating the gut microbiota and serum metabolism. Diabetol. Metab. Syndr. 2023, 15, 37. [Google Scholar] [CrossRef]
- Olszanecki, R.; Gebska, A.; Korbut, R. The role of haem oxygenase-1 in the decrease of endothelial intercellular adhesion molecule-1 expression by curcumin. Basic. Clin. Pharmacol. Toxicol. 2007, 101, 411–415. [Google Scholar] [CrossRef]
- Nugent, W.H.; Carr, D.A.; Friedman, J.; Song, B.K. Novel transdermal curcumin therapeutic preserves endothelial barrier function in a high-dose LPS rat model. Artif. Cells Nanomed. Biotechnol. 2023, 51, 33–40. [Google Scholar] [CrossRef]
- Karimi, A.; Naeini, F.; Niazkar, H.R.; Tutunchi, H.; Musazadeh, V.; Mahmoodpoor, A.; Asghariazar, V.; Mobasseri, M.; Tarighat-Esfanjani, A. Nano-curcumin supplementation in critically ill patients with sepsis: A randomized clinical trial investigating the inflammatory biomarkers, oxidative stress indices, endothelial function, clinical outcomes and nutritional status. Food Funct. 2022, 13, 6596–6612. [Google Scholar] [CrossRef]
- Wang, J.; Ghosh, S.S.; Ghosh, S. Curcumin improves intestinal barrier function: Modulation of intracellular signaling, and organization of tight junctions. Am. J. Physiol. Cell Physiol. 2017, 312, C438–C445. [Google Scholar] [CrossRef]
- Panahi, Y.; Hosseini, M.S.; Khalili, N.; Naimi, E.; Simental-Mendia, L.E.; Majeed, M.; Sahebkar, A. Effects of curcumin on serum cytokine concentrations in subjects with metabolic syndrome: A post-hoc analysis of a randomized controlled trial. Biomed. Pharmacother. 2016, 82, 578–582. [Google Scholar] [CrossRef]
- Dei Cas, M.; Ghidoni, R. Dietary Curcumin: Correlation between Bioavailability and Health Potential. Nutrients 2019, 11, 2147. [Google Scholar] [CrossRef]
- Lukosz, M.; Jakob, S.; Buchner, N.; Zschauer, T.C.; Altschmied, J.; Haendeler, J. Nuclear redox signaling. Antioxid. Redox Signal 2010, 12, 713–742. [Google Scholar] [CrossRef]
- Brinkmann, V.; Romeo, M.; Larigot, L.; Hemmers, A.; Tschage, L.; Kleinjohann, J.; Schiavi, A.; Steinwachs, S.; Esser, C.; Menzel, R.; et al. Aryl Hydrocarbon Receptor-Dependent and -Independent Pathways Mediate Curcumin Anti-Aging Effects. Antioxidants 2022, 11, 613. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merk, D.; Cox, F.F.; Jakobs, P.; Prömel, S.; Altschmied, J.; Haendeler, J. Dose-Dependent Effects of Lipopolysaccharide on the Endothelium—Sepsis versus Metabolic Endotoxemia-Induced Cellular Senescence. Antioxidants 2024, 13, 443. https://doi.org/10.3390/antiox13040443
Merk D, Cox FF, Jakobs P, Prömel S, Altschmied J, Haendeler J. Dose-Dependent Effects of Lipopolysaccharide on the Endothelium—Sepsis versus Metabolic Endotoxemia-Induced Cellular Senescence. Antioxidants. 2024; 13(4):443. https://doi.org/10.3390/antiox13040443
Chicago/Turabian StyleMerk, Dennis, Fiona Frederike Cox, Philipp Jakobs, Simone Prömel, Joachim Altschmied, and Judith Haendeler. 2024. "Dose-Dependent Effects of Lipopolysaccharide on the Endothelium—Sepsis versus Metabolic Endotoxemia-Induced Cellular Senescence" Antioxidants 13, no. 4: 443. https://doi.org/10.3390/antiox13040443
APA StyleMerk, D., Cox, F. F., Jakobs, P., Prömel, S., Altschmied, J., & Haendeler, J. (2024). Dose-Dependent Effects of Lipopolysaccharide on the Endothelium—Sepsis versus Metabolic Endotoxemia-Induced Cellular Senescence. Antioxidants, 13(4), 443. https://doi.org/10.3390/antiox13040443