1. Introduction
The separation between the maternal and fetal placentas after fetal delivery is a prerequisite for the smooth expulsion of the placenta [
1,
2]. During the expulsion of the placenta, the ability to induce the proliferation, invasion, and migration of trophoblast and endometrial epithelial cells is weakened, increasing cell death and causing the fetal cotyledon to detach from the maternal uterine caruncle, thereby promoting placental expulsion [
3]. Research has found that, compared to pregnancy, a large number of apoptotic factors immediately appear in the placenta after fetal expulsion. During normal placental expulsion, placental cell death increases, whereas in cows with a retained placenta (RP), an increase in the placental cell count is observed during the prenatal period [
4]. This indicates that the death of placental cells plays a crucial role in the normal separation and expulsion of membranes. It is known that ferroptosis is a programmed cell death (PCD) pattern that facilitates the development of organisms [
5,
6] and is characterized by the accumulation of a large, oxidative, toxic, labile iron pool, which induces the generation of intracellular lipid reactive oxygen species (ROS); excessive oxidative stress leads to lipid peroxidation and subsequent cell death [
7], which can inhibit the invasive ability of trophoblast cells [
8]. Erastin is an efficient inducer of ferroptosis, mainly through the inhibition of cysteine/glutamate antiporter activity, ultimately leading to cell death [
7]. However, information on the involvement of ferroptosis in regulating the expulsion of fetal membranes is lacking.
Circular RNAs (circRNAs) and micro RNAs (miRNAs) are involved in regulating the process of cell ferroptosis at different developmental stages, as well as regulating cell invasion and migration abilities [
9,
10,
11]. For example, regulating miR-30b-5p/SLC7A11 plays a crucial role in ferroptosis in pre-eclampsia [
9]. Circle_0008433 knockdown suppresses the angiogenesis, migration, and proliferation of endometrial stromal cells and promotes apoptosis [
10]. Circle_0000848 is an miR-6768-5p sponge that regulates the apoptosis, invasion, and migration of trophoblast cells and, thus, helps control the development and function of the placenta [
11]. Therefore, elucidating the mechanism by which circRNA regulates placental expulsion through ferroptosis would be a novel strategy for treating RP.
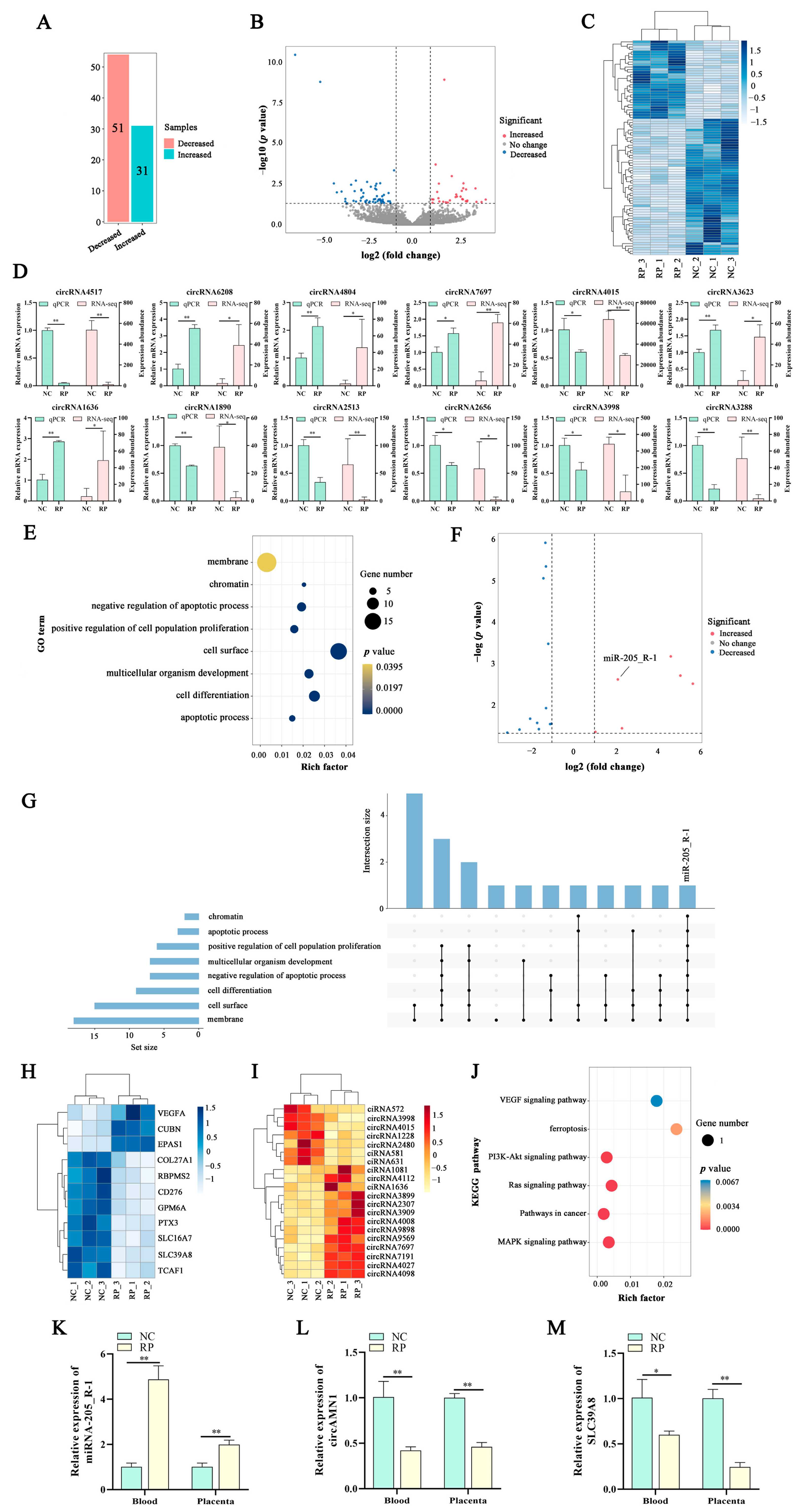
In this study, we aimed to identify candidate differentially expressed (DE) genes related to PCD in cows with normal fetal membrane discharge (NC) or RP based on previous research [
1], RNA sequencing (RNA-Seq), GSE214588 dataset, and bioinformatic analysis. Furthermore, we aimed to elucidate the mechanisms associated with ferroptosis in trophoblast cells in vitro. Our results provide insights into the promotion of placenta expulsion by ferroptosis, which can help develop novel methods for treating RP.
2. Materials and Methods
2.1. Animals
Holstein cows (age 3–4 years; 500 ± 10 kg; 2–3 parities) without any diseases were chosen as experimental animals (Zhangye, China) and allocated to two groups: normal fetal membrane discharge (NC; n = 3) and no expulsion of the fetal membrane within 12 h after delivery (RP; n = 3). Caudal vein blood and placental tissue samples were labeled NC-1–3 and RP-1–3, respectively.
The animal experiments were approved by the Animal Protection Committee of Gansu Agricultural University (Lanzhou, China; approval No.: GSAU-Eth-LST-2021-003). All institutional and national guidelines for the care and use of laboratory animals were followed.
2.2. RNA Sequencing
Total RNA was extracted and purified from NC and RP blood samples using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. The circRNA libraries were generated using the Ribo-Zero rRNA Removal Kit (Illumina, San Diego, CA, USA), according to the manufacturer’s protocol. Screened circRNAs with
p < 0.05 and |log2 fold-change| > 1 were considered DE genes [
12]. The Targetscan and miRanda databases were used to predict the targeting relationship between genes [
13]. The functions of these genes were assessed using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) using DAVID 6.7 (
https://david.ncifcrf.gov/summary.jsp, accessed on 30 October 2021).
2.3. Quantitative PCR Analysis (qPCR)
The cDNA was synthesized for qPCR using the Evo M-MLV RT Kit with gDNA Clean (Accurate Biotechnology Co., Ltd., Changsha, China). A miRNA first-strand cDNA synthesis kit (Accurate Biotechnology Co., Ltd.) was subsequently used for miRNA reverse transcription. The qPCR was conducted using the 2× SYBR green qPCR Master Mix (Bimake, Houston, TX, USA) and a LightCycler 96 Real-Time System (Roche, Basel, Switzerland);
GAPDH and
U6 levels were used for normalization. Each experiment of qPCR was repeated three times, and relative expression was calculated using the 2
−ΔΔCt method [
14]. The primer sequences of all genes are shown in
Tables S1 and S2.
2.4. Cell Lines
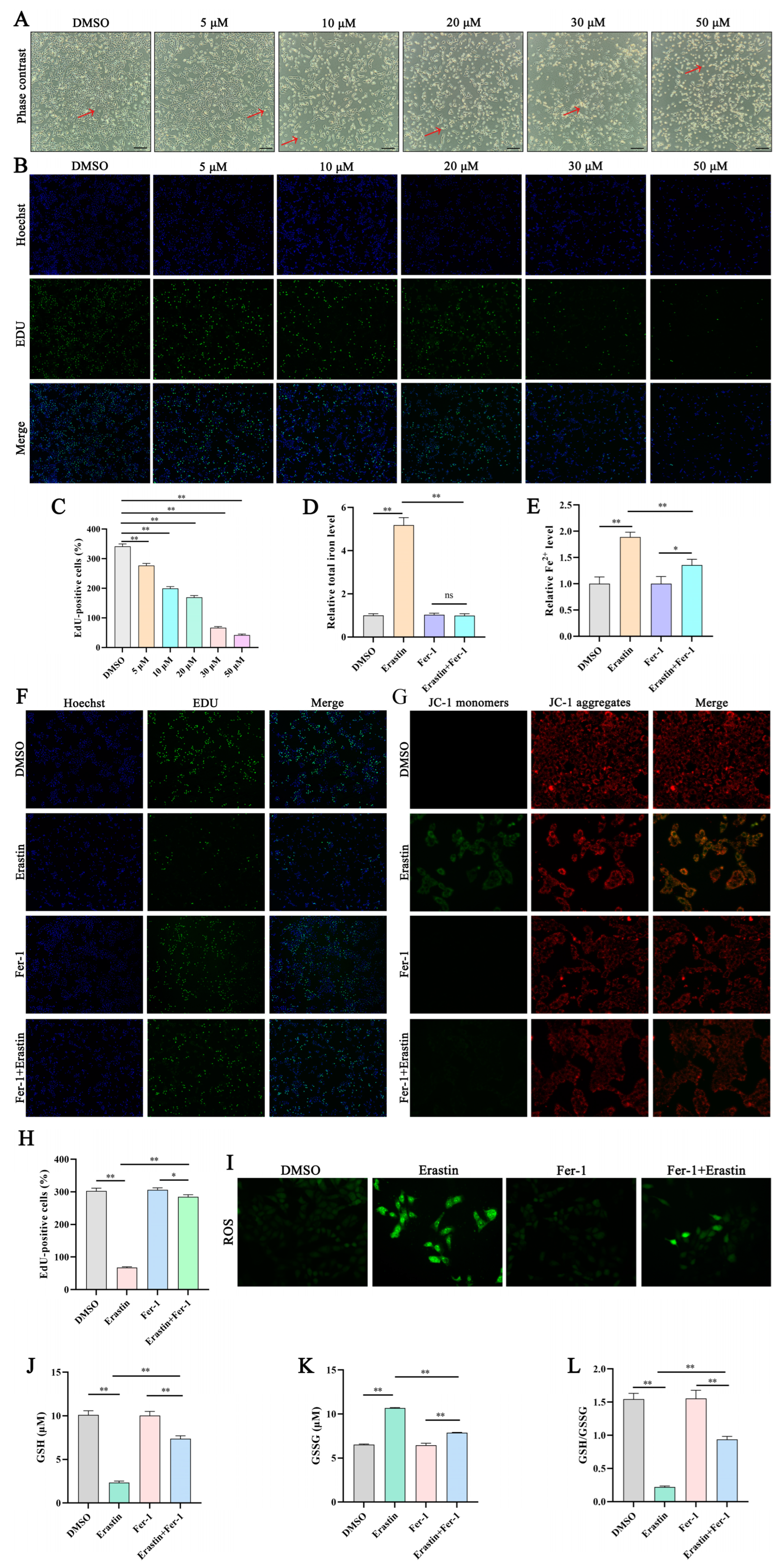
Trophoblast cells were donated by Prof. Longfei Xiao from the Beijing University of Agriculture and screened for Mycoplasma contamination using a Mycoplasma staining assay kit (C0296; Beyotime, Shanghai, China). Cells were cultivated in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) in a humidified incubator at 37 °C and 5% CO2. These cells were treated with erastin (5, 10, 15, 20, 30, and 50 µM) or 10 µM Fer-1 (Selleck, Houston, TX, USA), with 0.2% dimethyl sulfoxide (DMSO) as the negative control.
The circAMN1-overexpressing plasmids (pcDNA3.1 circAMN1), silencing RNA (si-circAMN1), and their negative controls (NC; pcDNA3.1 NC and si-NC) were constructed by GenePharma (Shanghai, China). The miR-205_R-1 mimic/inhibitor and NC were constructed by Sangon Biotech (Shanghai, China). These plasmids and RNA were transfected into trophoblast cells at different concentrations using Lipofectamine 2000 (Thermo Fisher Scientific), according to the manufacturer’s protocol. The RNA sequences are presented in
Table S3.
2.5. EdU Assay
EdU was diluted with cell culture medium at a ratio of 1:500 to achieve a final concentration of 20 μM. Cells were removed from the culture medium and incubated with 1 mL of diluted EdU at 37 °C for 2 h. Cell proliferation was analyzed using the BeyoClick EdU-488 Cell Proliferation Detection kit (C0071S; Beyotime), according to the manufacturer’s protocol. The corresponding cell fluorescence was detected using a fluorescence microscope (ECHO, Chicago, IL, USA).
2.6. JC-1 and ROS Staining
After cell transfection, the fluorescent probe DCFH-DA (S0033S; Beyotime) was diluted with serum-free culture medium at a ratio of 1:1000, and the fluorescent probe JC-1 (C2003S; Beyotime) was diluted with JC-1 staining buffer at a ratio of 1:200, both resulting in a final concentration of 10 μM. Cells were removed from the culture medium and incubated with 1 mL of diluted DCFH-DA or JC-1 at 37 °C for 20 min to detect intracellular ROS or mitochondrial membrane potential, respectively. The corresponding cell fluorescence was detected using a fluorescence microscope (ECHO).
2.7. Measurement of Total Iron, Fe2+, GSH, and Glutathione Disulfide (GSSG) Levels
Total iron and Fe2+ contents in trophoblast cells were measured using a Cell Total Iron Colorimetric Assay Kit (E-BC-K880-M; Elabscience, Wuhan, China) and Cell Ferrous Iron Colorimetric Assay Kit (E-BC-K881-M; Elabscience), respectively, and a ReadMax 1900 microplate reader (Shanghai Flash, Shanghai, China) was used to measure absorbance at 593 nm. GSH and GSSG concentrations were detected using a GSH and GSSG Assay Kit (S0053; Beyotime) and by measuring the cell absorbance at 412 nm.
2.8. Wound-Healing Assay
Transfected cells were cultured in a 6-well plate to 90% confluence. Subsequently, scratch wounds were created using a 200 μL plastic pipette tip. After 24 h of cultivation, the cell culture was washed with fresh culture medium to remove non-adherent cells. Images of the cell culture at 0 h and 24 h were obtained under a fluorescence microscope (ECHO). The migration distance of cells was determined by migration rate = (0 h scratch width − 24 h scratch width)/0 h scratch width.
2.9. Immunofluorescence
Cells were treated with 4% paraformaldehyde for 30 min, permeabilized with 0.5% Triton X-100 for 10 min, and blocked with 5% bovine serum albumin for 30 min. The cells were incubated overnight at 4 °C with rabbit anti-ZIP8 (No. 20459-1-AP, 1:400; Proteintech, Rosemont, IL, USA), and then with the secondary antibody fluorescein-conjugated affinipure goat anti-rabbit IgG (H + L) (No. SA00003-2, 1:500; Proteintech) for 1.5 h at 37 °C. To detect the nuclei, all samples were treated with 4′,6-diamidino-2-phenylindole (Beyotime). The results were then visualized using an ECHO fluorescence microscope.
2.10. Cell Viability Assay
Cell suspensions were inoculated in a 96-well plate, and 10 μL of Cell Counting Kit-8 (CCK-8) solution (BS350A; Biosharp, Hefei, China) was added before incubation for 2 h. A ReadMax 1900 microplate reader (Shanghai Flash) was used to measure absorbance at 450 nm.
2.11. Subcellular Localization
The nuclei and cytoplasm of isolated cells were extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific), according to the manufacturer’s protocol. Nuclei and cytoplasmic RNA were extracted using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions.
circAMN1 expression levels were analyzed by qPCR, according to the methods described in
Section 2.3.
2.12. RNase R Treatment
Total RNA (1 μg) was incubated with 4 U/μg RNase R (R7092S; Beyotime) at 37 °C for 30 min. The reaction was terminated by incubation at 70 °C for 10 min. AMN1 and circAMN1 mRNA expression levels were detected using 15% polyacrylamide gel electrophoresis.
2.13. Dual-Luciferase Reporter Assay
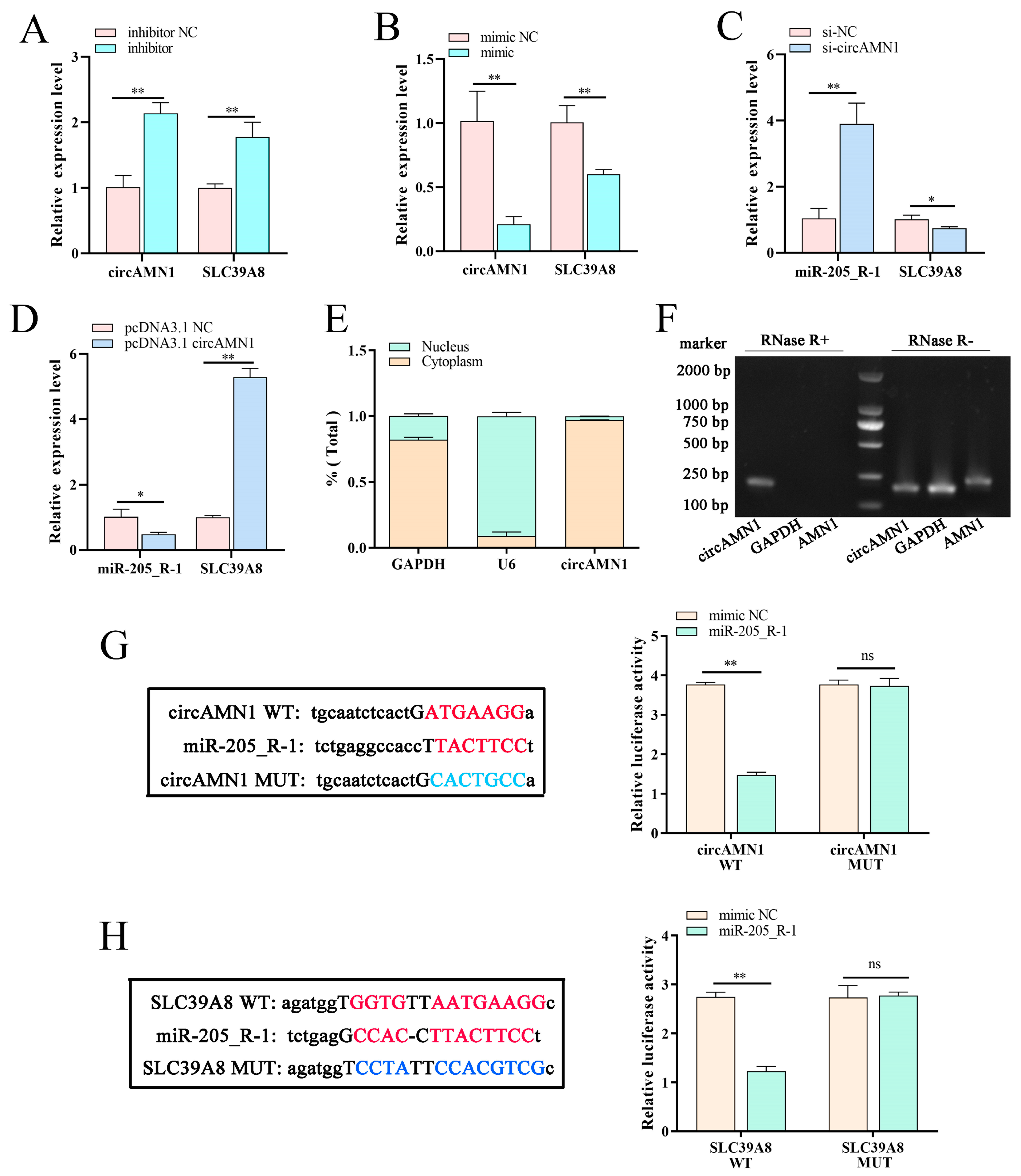
Dual-luciferase reporter vectors for wild- and mut-type circAMN1 and SLC39A8 were constructed by AZENTA Life Sciences (Nanjing, China). These vectors were co-transfected into trophoblast cells with miR-205_R-1 mimic or mimic NC and incubated for 12 h. Luciferase activity was detected using the Dual-Luciferase Reporter Assay Kit (Promega, Madison, WI, USA), according to the manufacturer’s protocol.
2.14. Transmission Electron Microscopy
Trophoblast cells were presoaked in a 2.5% glutaraldehyde solution and then fixed in 1% osmium tetroxide at 4 °C for 2 h. The samples were dehydrated by gradient alcohol and gradient acetone, and double-stained with uranyl acetate-lead citrate. Randomly selected fields of view were photographed under a transmission electron microscope (Hitachi HT7700, Tokyo, Japan).
2.15. Transwell Assay
Following transfection, the cells were seeded into Transwell chambers with an 8 μm pore size (Corning, New York, NY, USA) for migration assays, with inserts that were pre-coated with Matrigel (BD Pharmingen, San Jose, CA, USA) for invasion assays. After incubation at 37 °C for 12 h, the cells were treated with 4% paraformaldehyde for 15 min before staining with 0.1% crystal violet. Images of the migrated or invaded areas were recorded in three randomly selected areas using a fluorescence microscope (ECHO).
2.16. Quantification and Statistical Analysis
Data are presented as mean and SD. Statistical comparisons were performed using Student’s t-test with GraphPad Prism version 9.0 (GraphPad Software, Boston, MA, USA). A p-value of p < 0.05 was considered statistically significant; p < 0.01, highly significant; and p > 0.05, not significant.
4. Discussion
The mammalian uterus and placenta undergo cell remodeling during each cycle; hence, systematic cell renewal is a fundamental event of uterine and placental cell change, including PCD and cell renewal caused by cell proliferation [
15]. Maturation and shedding of the placenta are inseparable from PCD [
3]. To elucidate the molecular mechanism by which PCD regulates the shedding of the placenta in Holstein cows, we constructed a circAMN1/miR-205_R-1/SLC39A8 signaling network axis related to PCD using the RNA-Seq and GSE214588 databases. The miR-205 is associated with apoptosis and proliferation, and miR-205_R-1 was named based on the missing base from the right of miR-205 [
16,
17,
18]. Chen et al. [
16] found that miR-205-5p/PTK7 regulates the proliferation, invasion, and migration ability of colorectal cancer cells. Inhibition of miR-205 expression promotes Rho family GTPase 3 activity in cardiac ischemia/reperfusion injury, regulating the function of the heart and mitochondria and, thus, reducing oxidative stress and apoptosis [
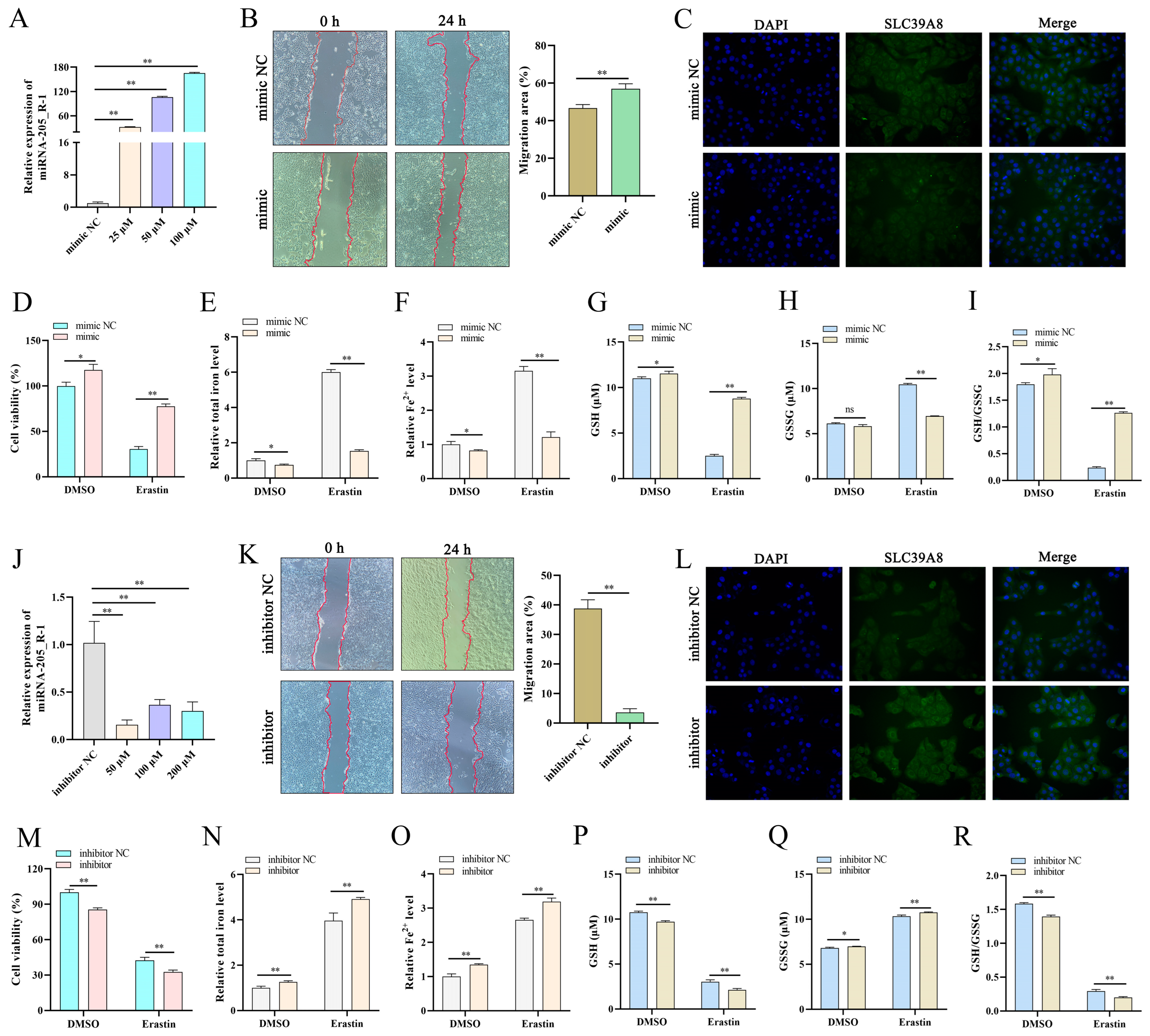
18]. These results indicate that miR-205_R-1 is closely related to PCD; therefore, it was selected as a candidate gene that promotes the expulsion of the placenta.
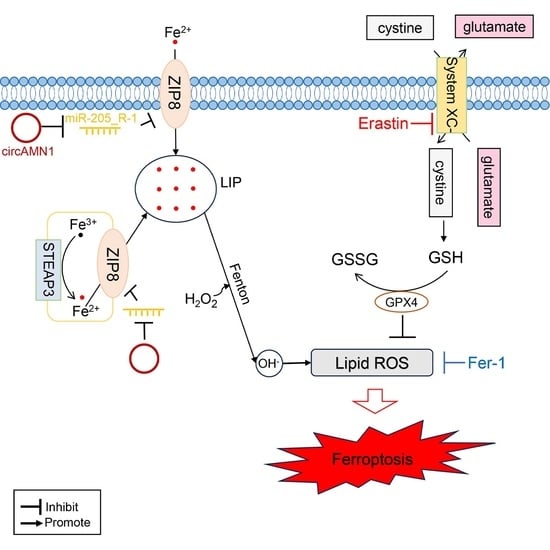
Expulsion of the placenta results from the PCD of endometrial epithelial and trophoblast cells after placental maturation, reducing cell migration and invasion, weakening the ability of the fetal cotyledonary to grasp the uterus, and promoting the detachment of the fetal lobe from the uterine caruncle. Ferroptosis, a type of PCD, plays a vital role in biological development, especially in the expulsion of fetal membranes [
3]. In this study, KEGG pathway analysis revealed that the miR-205_R-1 target gene,
SLC39A8, was ultimately involved in the ferroptosis pathway. SLC39A8, also known as ZRT/IRT-like protein 8, is a transmembrane, divalent, metal transporter protein originally recognized as a zinc importer [
19]. However, it was later found to transport iron with high affinity, thereby influencing the iron ion level and metabolism in vivo [
20,
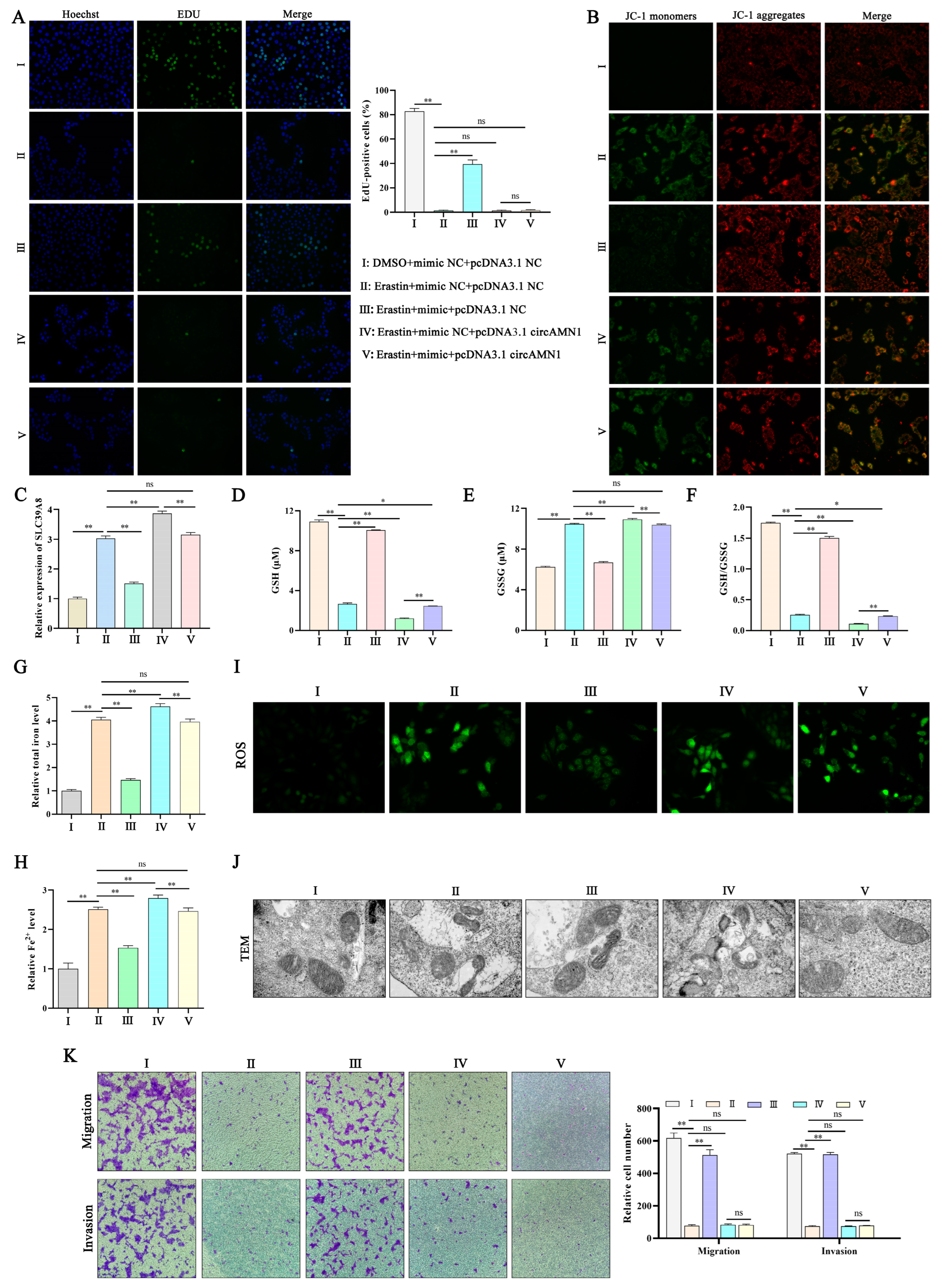
21]. In this study, SLC39A8 expression was significantly reduced in the RP group, which may have weakened the ability of the body to transport iron ions. We speculate that, after fetal delivery, the mature placenta and uterus have less iron ion accumulation and trophoblast cells and endometrial epithelial cells cannot undergo ferroptosis, thereby preventing the expulsion of the placenta and causing RP.
Erastin can induce intracellular GSH depletion and ROS accumulation, leading to ferroptosis by inhibiting system xc
− activity [
22]. Therefore, in this study, we used erastin to construct a ferroptosis model and detected ferroptosis-related indicators. We observed a dose-dependent effect of erastin on trophoblast cell death, with increases in intracellular levels of total iron, Fe
2+, and ROS. Excessive accumulation of ROS can induce cell damage, which is closely related to GSH [
23]. GSH, as a substrate for glutathione peroxidase and glutathione transferase, can eliminate harmful free radicals (mainly oxygen radicals) and lipid peroxides in the body by converting them into fatty acids and water as it is oxidized to GSSG [
24]. In this study, erastin induced increases in intracellular GSSG levels and decreases in GSH levels and GSH/GSSG. The depletion of GSH and increase in GSSG levels indicate a decrease in the intracellular ability to clear lipid peroxides, leading to ferroptosis. Oxidative stress causes cell ferroptosis, which can lead to mitochondrial damage, resulting in the reduction or disappearance of mitochondrial cristae and a decrease in mitochondrial membrane potential [
25]. In the current study, a decrease in mitochondrial membrane potential induced by erastin was detected. Fer-1 is a lipophilic iron death inhibitor that clears ROS through a reduction reaction, inhibits the Fenton reaction by reducing unstable iron in cells, and regulates ferroptosis [
26]. The current study showed that Fer-1 reversed the changes induced by erastin. These results confirmed that a trophoblast ferroptosis model was constructed using erastin.
The non-coding RNAs (ncRNAs) play vital roles in various forms of PCD [
27,
28,
29]. For example, lncRNA Mir9-3hg suppresses cardiomyocyte ferroptosis in ischemia–reperfusion mice via the Pum2/PRDX6 axis [
27]. Furthermore, MiR-134-3p targets HMOX1 to inhibit ferroptosis in granulosa cells of sheep follicles [
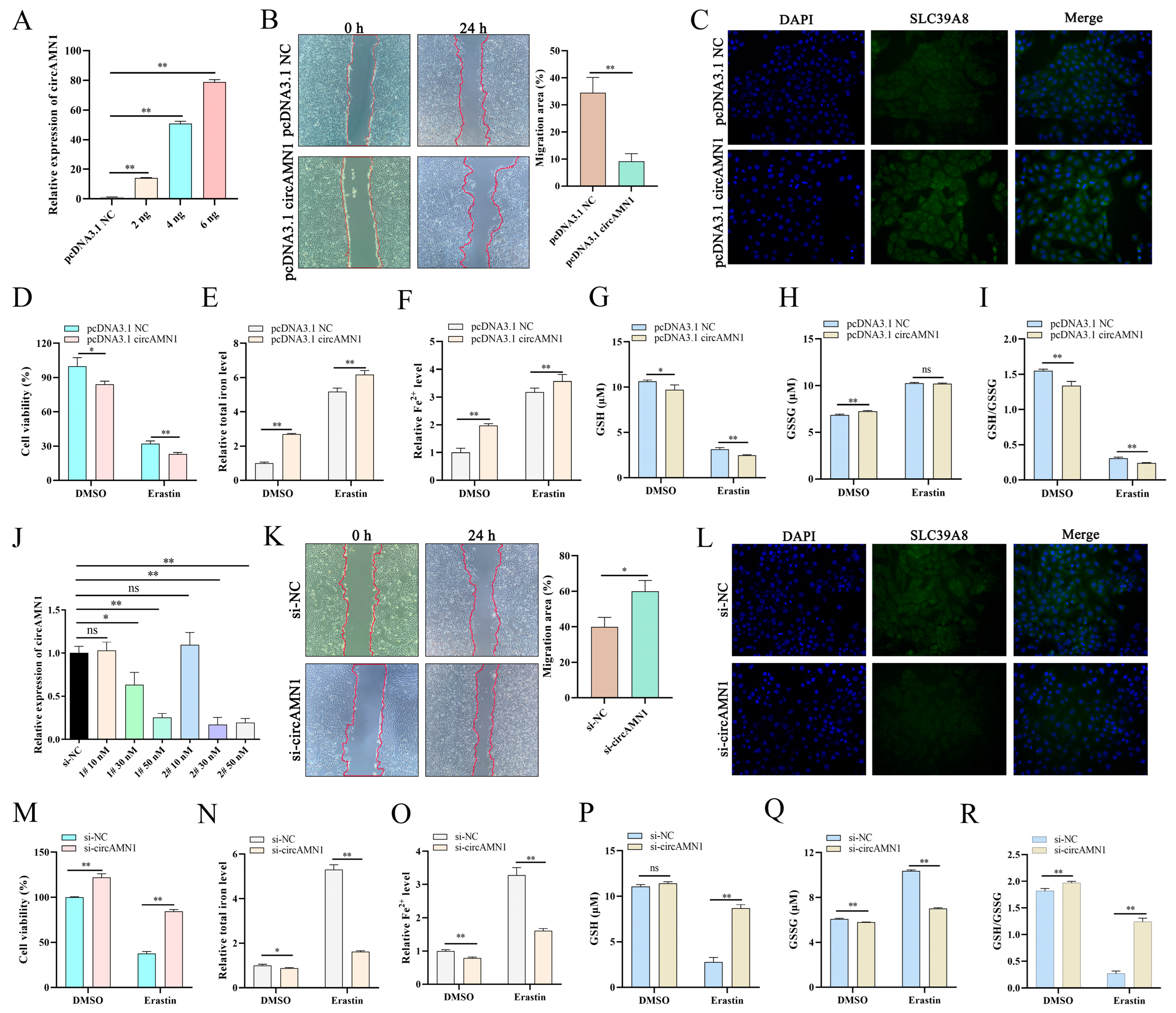
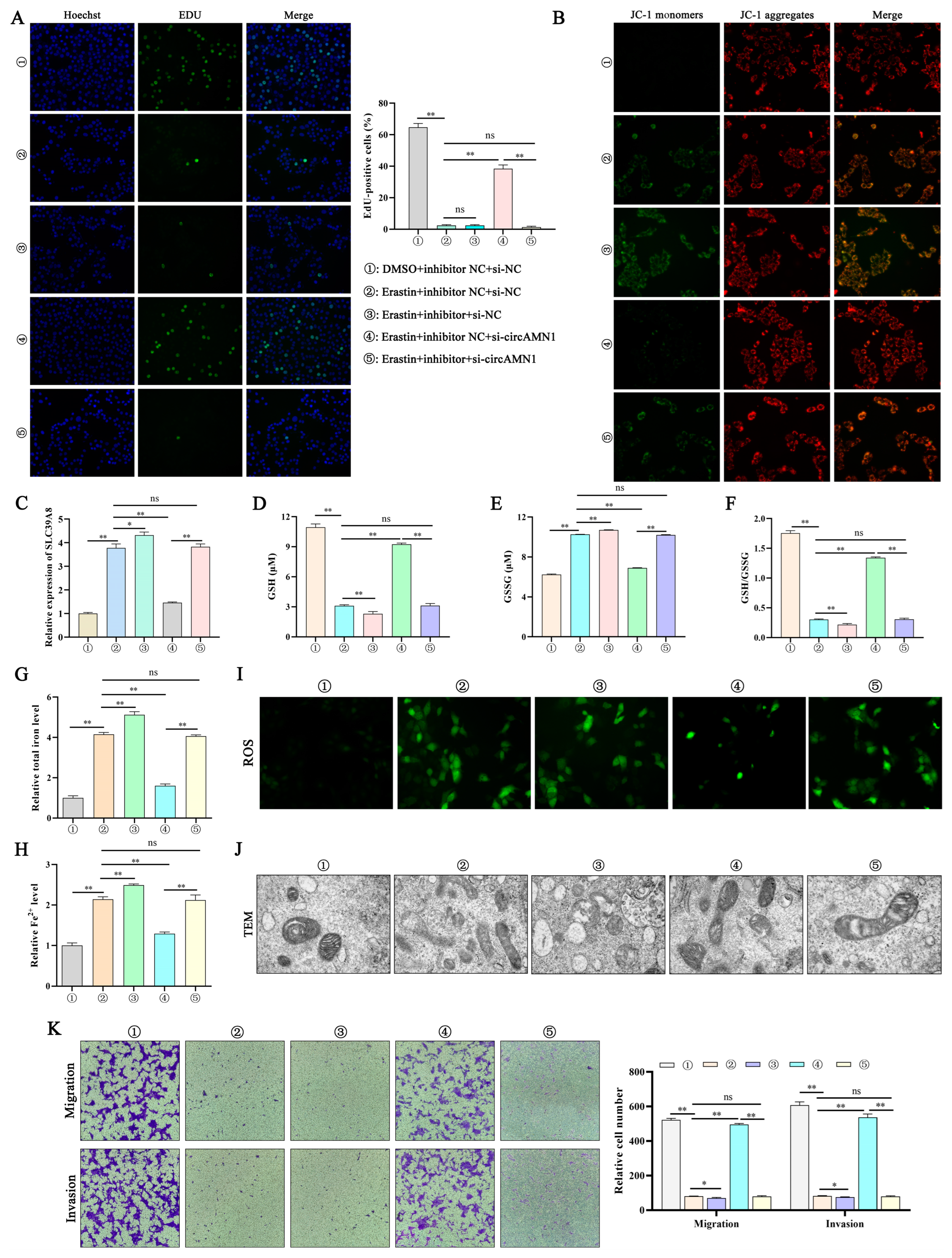
29]. In this study, regulating the expression of circAMN1 or miR-205_R-1 changed the migration ability of trophoblast cells. The si-circAMN1 or the miR-205_R-1 mimic inhibited erastin-induced ferroptosis. In contrast, pcDNA3.1 circAMN1 or the miR-205_R-1 inhibitor promoted erastin-induced ferroptosis. This indicates that circRNAs and miRNAs have regulatory effects on cells and are closely involved in the ferroptosis of trophoblast cells.
To demonstrate the regulatory relationship between circAMN1, miR-205_R-1, and SLC39A8, the current study confirmed the binding sites between circAMN1 and miR-205_R-1 and between miR-205_R-1 and SLC39A8 using dual-luciferase reporter assays. The circAMN1 was primarily expressed in the cytoplasm of trophoblast cells, rather than in the nucleus. It was also found that circAMN1 had a circular characteristic and was not easily degraded by the exonuclease, RNase R. These results indicate that circAMN1 regulates the expression of SLC39A8 through a ceRNA mechanism. In the subsequent results, in erastin-treated cells, si-circAMN1 was transfected into the trophoblast cells, reducing cell ferroptosis and eliminating the effect of the miR-205_R-1 inhibitor. pcDNA3.1 circAMN1 was transfected into trophoblast cells, exacerbating cell ferroptosis and thus eliminating the effect of the miR-205_R-1 mimic. In short, circAMN1 acted as an miR-205_R-1 sponge, regulating the expression of SLC39A8 and erastin-induced in vitro ferroptosis. SLC39A8 mainly transports iron ions, which are essential metal elements involved in several biological processes, from oxygen transportation to protein synthesis and enzymatic reactions [
30]. Abnormalities in iron metabolism, including increased iron absorption and decreased iron output, can lead to ferroptosis by generating iron-mediated ROS via the Fenton reaction [
31]. Extracellular trivalent Fe
3+ forms a complex with transferrin, which binds to transferrin receptor 1 on the cell membrane and enters the cell through endocytosis to form an endosome. Fe
3+ is subsequently reduced to Fe
2+ by six-transmembrane epithelial antigen of prostate 3 and transported to the cytoplasm through SLC39A8, where it is introduced into a labile iron pool [
19]. SLC39A8 can also transport extracellular free Fe
2+ to intracellular, labile iron pools [
19]. Through the Fenton reaction, free iron interacts with H
2O
2 to form highly reactive hydroxyl radicals [
8]. The H
2O
2 and superoxide radicals are decomposed through chain reactions, producing a large amount of molecular oxygen, hydroxyl radicals, and hydroxyl anions, promoting lipid peroxidation in the cell membrane and resulting in ferroptosis progression [
31], thereby leading to the reduced invasion and migration ability of endometrial epithelial and trophoblast cells and causing fetal membrane shedding.
To the best of our knowledge, this is the first study to emphasize the regulatory effect of circAMN1 against erastin-induced ferroptosis and to demonstrate its potential as an miR-205_R-1 sponge to regulate SLC39A8 expression, thereby mediating iron transport and regulating ferroptosis induced by erastin in trophoblast cells. Furthermore, the observed synergistic effects of circAMN1/miR-205_R-1 in the context of erastin-induced ferroptosis provide insights into potential biomarkers and present a novel treatment strategy for RP.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






