
Potential New Therapies “ROS-Based” in CLL: An Innovative Paradigm in the Induction of Tumor Cell Apoptosis


 and
and
Abstract
1. Introduction
1.1. General Considerations on Chronic Lymphocytic Leukemia
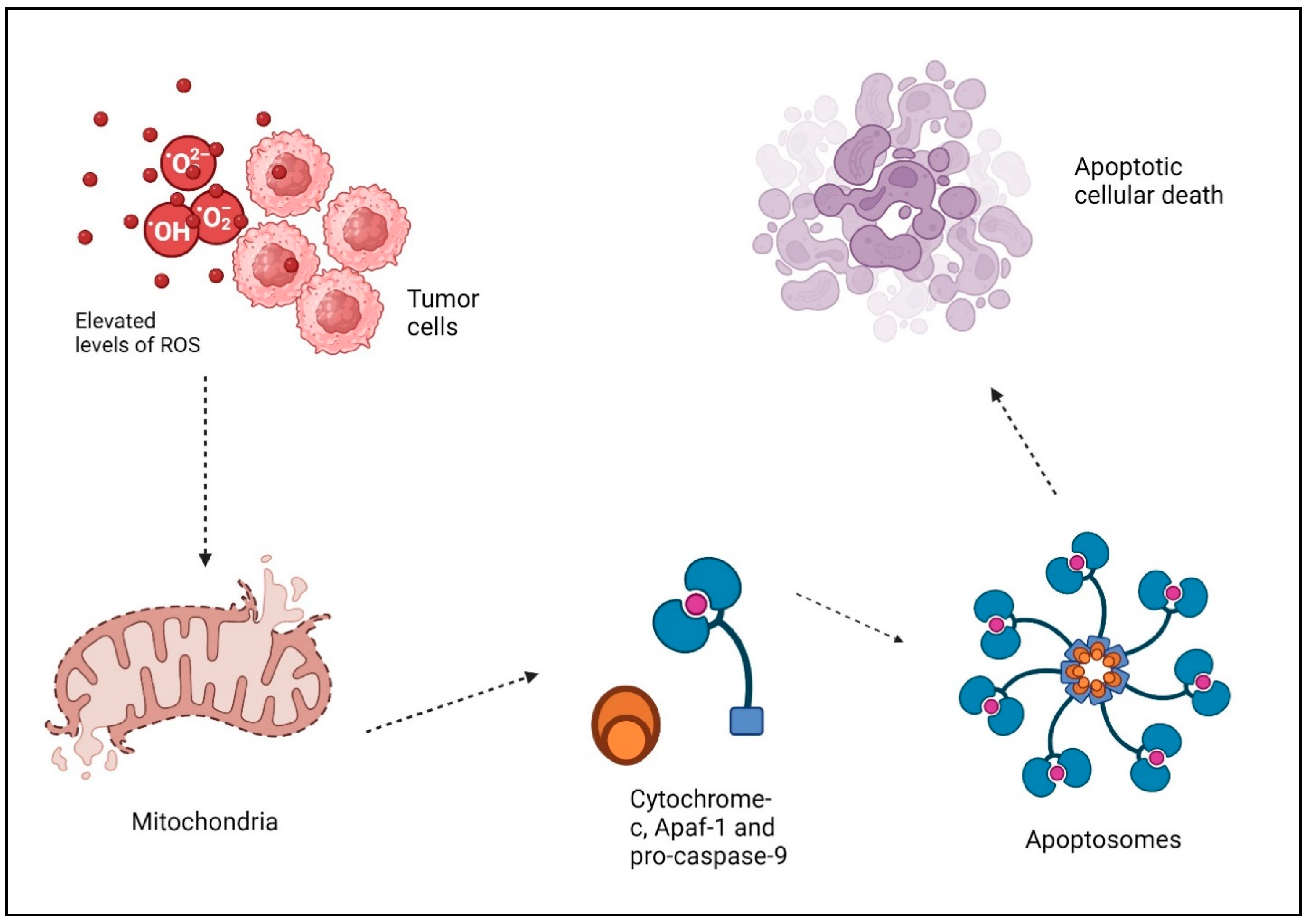
1.2. ROS, Cancer, and Apoptosis
1.3. Cancer Therapy and ROS
1.3.1. Role of ROS and Other Mechanisms of Cell Death
Necroptosis
Ferroptosis
2. CLL and Oxidative Stress
2.1. CLL and ROS—General Considerations
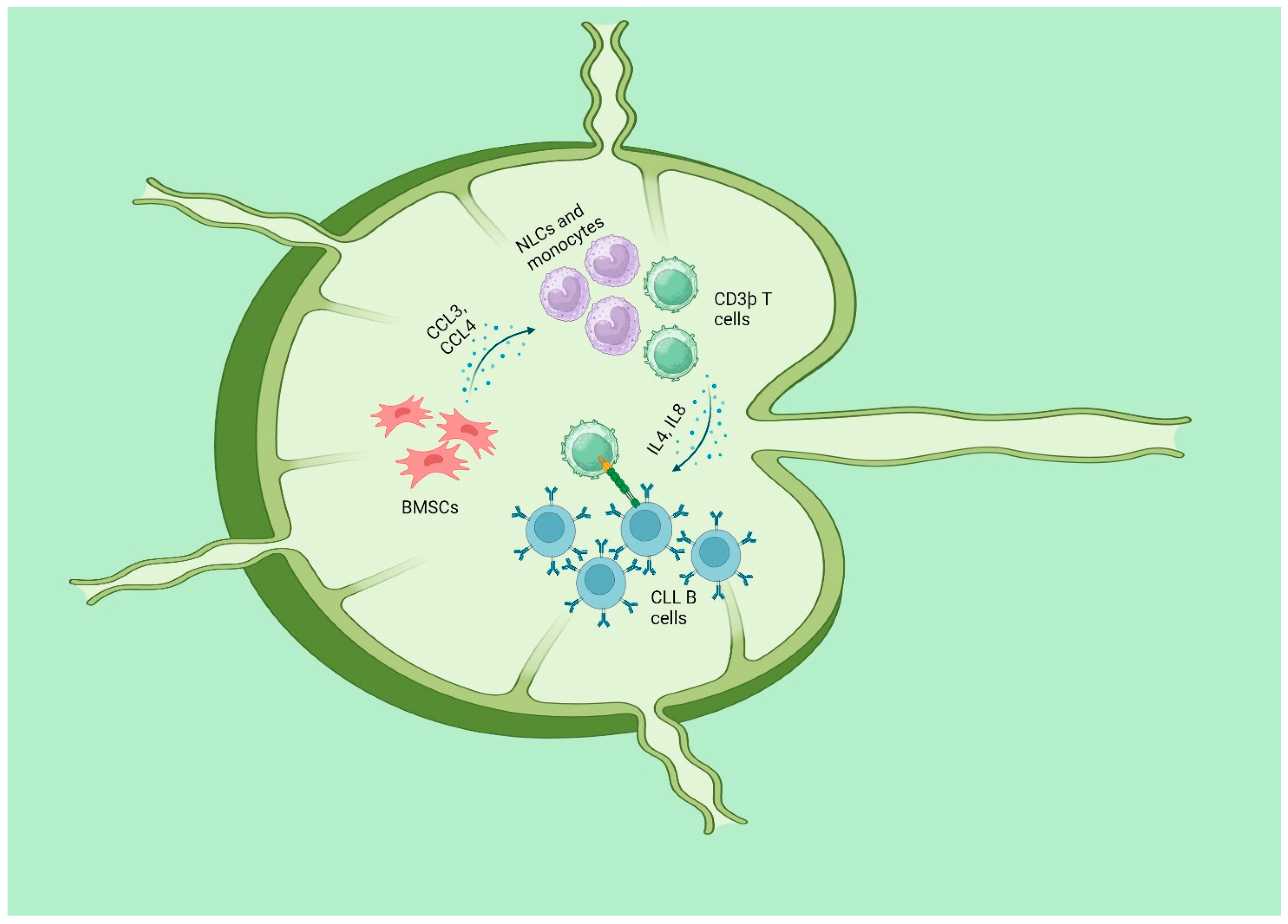
2.1.1. Microenvironment and Oxidative Stress in CLL
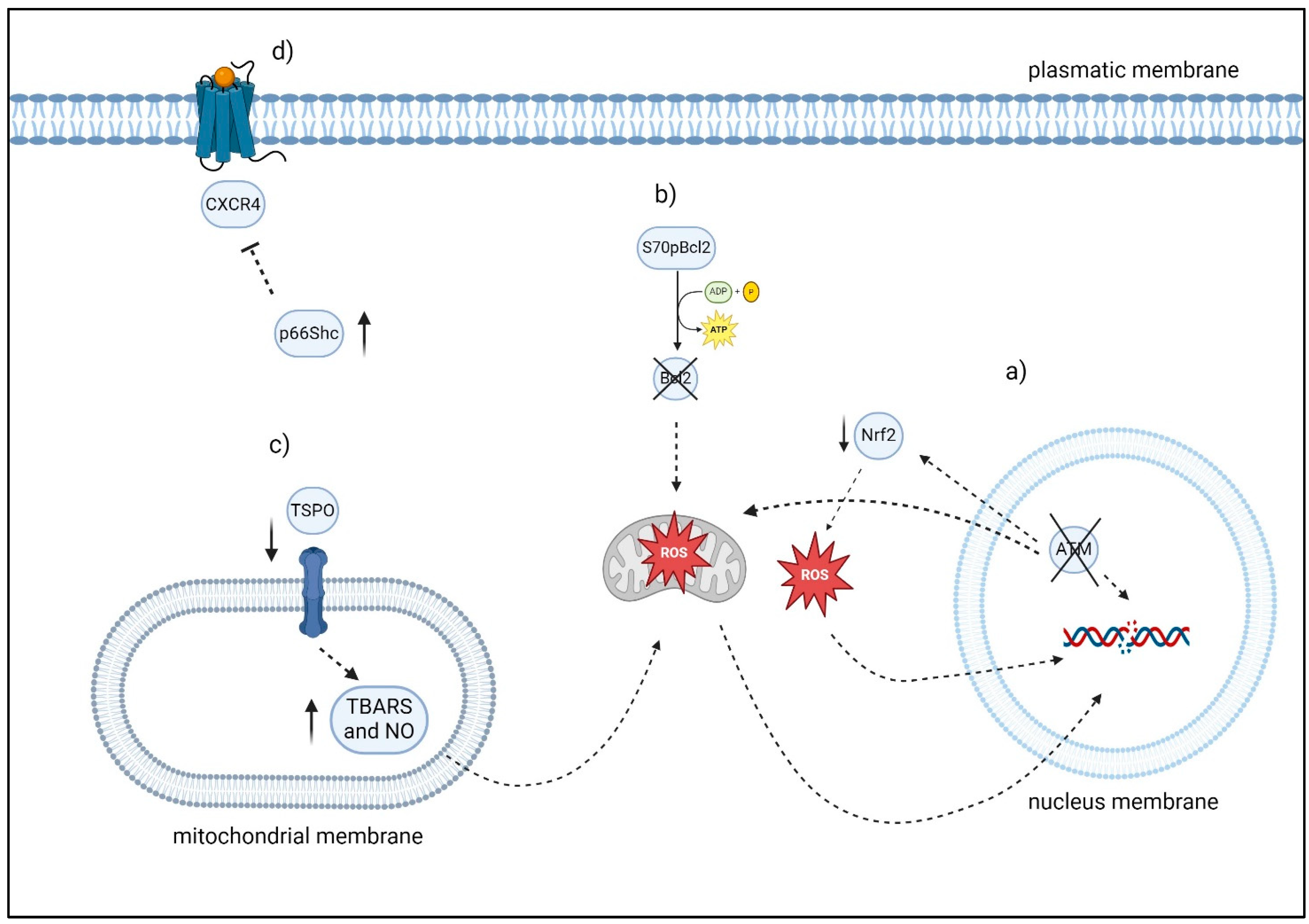
2.1.2. ATM Gene and Oxidative Stress
2.1.3. S70pBcl2
2.1.4. BACH2, PRDM1
2.1.5. p66Shc
2.1.6. TSPO
2.1.7. Prognosis, Oxidative Stress and CLL
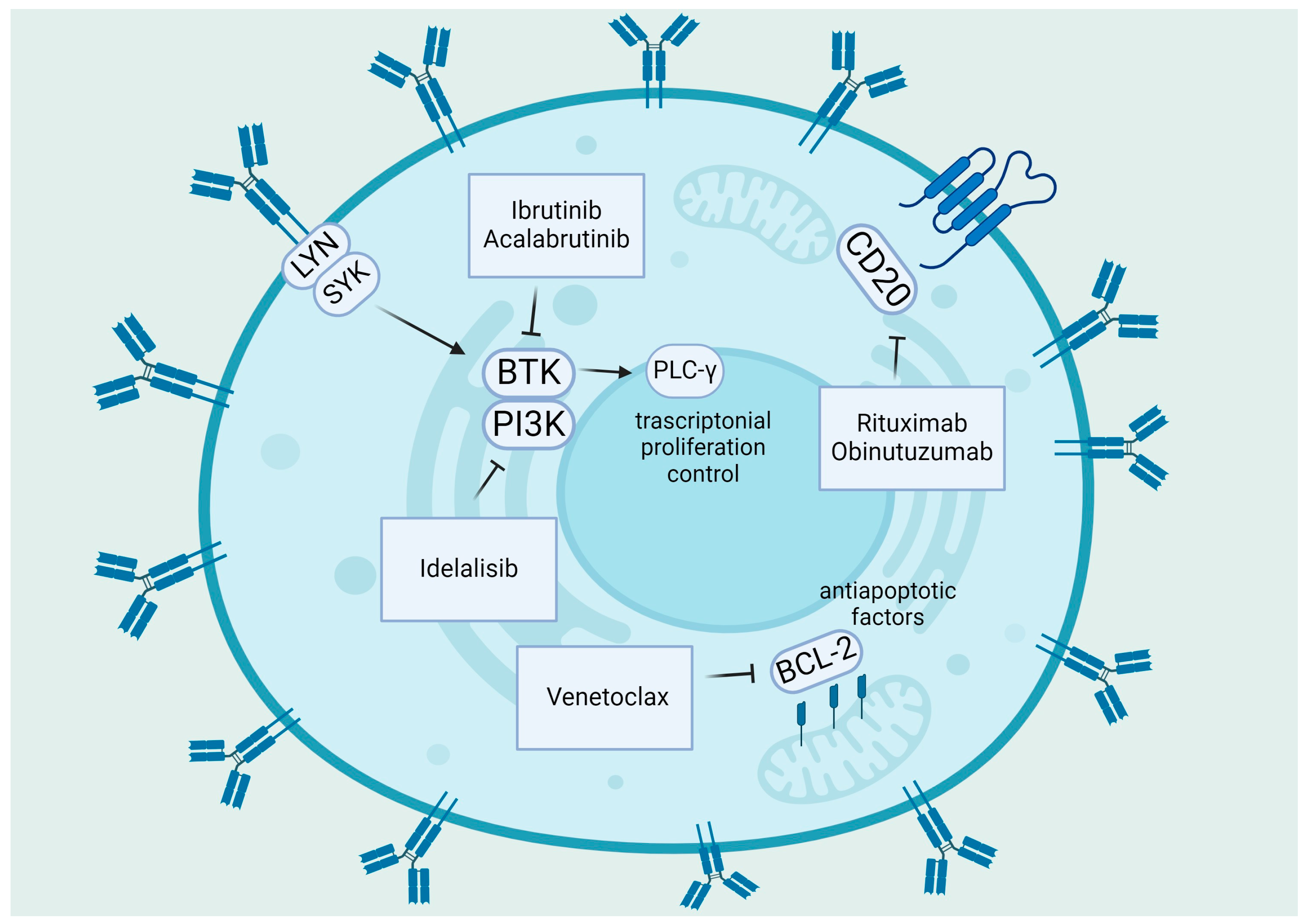
3. Old and Potential New Therapies “ROS-Based” in CLL
3.1. The Role of Adaphostin
3.2. Role of ROS in Bendamustine Treatment
3.3. Auranofin
3.4. Acacetin
3.5. Conus Textile
3.6. Motexafin Gadolinium
3.7. Rigosertib
3.8. Arsenic Trioxide

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs/Agents | Biological Mechanisms/Target |
|---|---|
| Adaphostin | Disruptions in cell signaling processes, such as the secretion of cytochrome c and apoptosis inhibiting factor (AIF) in a dose-dependent manner, deactivation of protective pathways (Raf1, MEK, extracellular ERK1/2 and Akt), activation of stress-induced pathways (JNK and MAPK), and removal of phosphate groups from RB [159] |
| Bendamustine | Cytotoxicity p53-dependent and p53-independent mechanism; increased expression of the pro-apoptotic proteins PUMA and NOXA [177] |
| Auranofin | Inhibit TrxRs [181] |
| Acacetin | Phosphorylation of JNK and c-Jun [190] |
| Conux Texile | Release of a cardiotoxin III-like peptide [195] |
| Motexafin gadolinium | Futile redox cycling [197] |
| Rigosertib | Inhibit PI3K/AKT and induce ROS, leading to oxidative stress response by activating AP-1, c-Jun NH2-terminal kinase, and ATF3, ultimately resulting in the overexpression of NOXA [87] |
| Arsenic trioxide | Degrade the PML-RARα fusion protein [201] Increase HMOX1 [213] |
4. New Perspectives: Nanotechnology
New Prospectives: PROTACs
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Lee Harris, N.; Stein, H.; Siebert, R.; Advani, T.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, B.; Robak, T.; Montserrat, E.; Ghia, P.; Niemann, C.U.; Kater, A.P.; Gregor, M.; Cymbalista, F.; Buske, C.; Hillmen, P.; et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Tambaro, F.P.; Wierda, W.G. Tumour Lysis Syndrome in Patients with Chronic Lymphocytic Leukaemia Treated with BCL-2 Inhibitors: Risk Factors, Prophylaxis, and Treatment Recommendations. Lancet Haematol. 2020, 7, e168–e176. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Kreuzer, K.A.; Soosapilla, A.; Spacek, M.; Stehlikova, O.; Gambell, P.; McIver-Brown, N.; Villamor, N.; Psarra, K.; Arroz, M.; et al. Reproducible diagnosis of chronic lymphocytic leukemia by flow cytometry: An European Research Initiative on CLL (ERIC) & European Society for Clinical Cell Analysis (ESCCA) Harmonisation project. Cytom. B Clin. Cytom. 2018, 94, 121–128. [Google Scholar] [CrossRef]
- Wang, E.; Mi, X.; Thompson, M.C.; Montoya, S.; Notti, R.Q.; Afaghani, J.; Durham, B.H.; Penson, A.; Witkowski, M.T.; Lu, S.X.; et al. Mechanisms of Resistance to Noncovalent Bruton’s Tyrosine Kinase Inhibitors. N. Engl. J. Med. 2022, 386, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Ishikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yoshimoto, G.; Mori, Y.; Iino, T.; Yamauchi, T.; Eto, T.; et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011, 20, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survivalin chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Fischer, K.; Fingerle-Rowson, G.; Fink, A.M.; Busch, R.; Mayer, J.; Hensel, M.; Hopfinger, G.; Hess, G.; von Grünhagen, U.; et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: A randomised, open-label, phase 3 trial. Lancet 2010, 376, 1164–1174. [Google Scholar] [CrossRef]
- Knisbacher, B.A.; Lin, Z.; Hahn, C.K.; Nadeu, F.; Duran-Ferrer, M.; Stevenson, K.E.; Tausch, E.; Delgado, J.; Barbera-Mourelle, A.; Taylor-Weiner, A.; et al. Molecular map of chronic lymphocytic leukemia and its impact on outcome. Nat. Genet. 2022, 54, 1664–1674. [Google Scholar] [CrossRef]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Zenz, T.; Vollmer, D.; Trbusek, M.; Smardova, J.; Benner, A.; Soussi, T.; Helfrich, H.; Heuberger, M.; Hoth, P.; Fuge, M.; et al. TP53 mutation profile in chronic lymphocytic leukemia: Evidence for a disease specific profile from a comprehensive analysis of 268 mutations. Leukemia 2010, 24, 2072–2079. [Google Scholar] [CrossRef]
- Seiffert, M.; Dietrich, S.; Jethwa, A.; Glimm, H.; Lichter, P.; Zenz, T. Exploiting biological diversity and genomic aberrations in chronic lymphocytic leukemia. Leuk. Lymphoma 2012, 53, 1023–1031. [Google Scholar] [CrossRef]
- Hamilton, E.; Pearce, L.; Morgan, L.; Robinson, S.; Ware, V.; Brennan, P.; Thomas, N.S.; Yallop, D.; Devereux, S.; Fegan, C.; et al. Mimicking the tumour microenvironment: Three different co-culture systems induce a similar phenotype but distinct proliferative signals in primary chronic lymphocytic leukaemia cells. Br. J. Haematol. 2012, 158, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, F.K.; Krysov, S.; Davies, A.J.; Steele, A.J.; Packham, G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood 2011, 118, 4313–4320. [Google Scholar] [CrossRef]
- Decker, T.; Schneller, F.; Kronschnabl, M.; Dechow, T.; Lipford, G.B.; Wagner, H.; Peschel, C. Immunostimulatory CpG-oligonucleotides induce functional high affinity IL-2 receptors on B-CLL cells: Costimulation with IL-2 results in a highly immunogenic phenotype. Exp. Hematol. 2000, 28, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.J.; Vogler, M.; Samuel, J.; Jayne, S.; Wagner, S.; Pritchard, C.; Macip, S. Precision medicines for B-cell leukaemias and lymphomas; progress and potential pitfalls. Br. J. Haematol. 2013, 160, 725–733. [Google Scholar] [CrossRef]
- Vogler, M.; Butterworth, M.; Majid, A.; Walewska, R.J.; Sun, X.M.; Dyer, M.J.; Cohen, G.M. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 2009, 113, 4403–4413. [Google Scholar] [CrossRef]
- Roberts, A.W.; Stilgenbauer, S.; Seymour, J.F.; Huang, D.C. Venetoclax in Patients with Previously Treated Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 4527–4533. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.C.; Lowe, S.W. Bcl-2 mediates chemoresistance in matched pairs of primary E(mu)-myc lymphomas in vivo. Blood Cells Mol. Dis. 2001, 27, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Itchaki, G.; Brown, J.R. The potential of venetoclax (ABT-199) in chronic lymphocytic leukemia. Ther. Adv. Hematol. 2016, 7, 270–287. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; El Rouby, S.; Reed, J.C.; Krajewski, S.; Silber, R.; Potmesil, M.; Newcomb, E.W. Drug-induced apoptosis in B-cell chronic lymphocytic leukemia: Relationship between p53 mutation and bcl-2/bax proteins in drug resistance. Oncogene 1996, 12, 1055–1062. [Google Scholar] [PubMed]
- Kitada, S.; Andersen, J.; Akar, S.; Zapata, J.M.; Takayama, S.; Krajewski, S.; Wang, H.G.; Zhang, X.; Bullrich, F.; Croce, C.M. Expression of apoptotis-regulating proteins in chronic lymphocytic leukemia: Correlations with in vitro and in vivo chemoresponses. Blood 1998, 91, 3379–3389. [Google Scholar] [CrossRef] [PubMed]
- Beekman, R.; Chapaprieta, V.; Russinol, N.; Vilarrasa-Blasi, R.; Verdaguer-Dot, N.; Martens, J.H.A.; Duran-Ferrer, M.; Kulis, M.; Serra, F.; Javierre, B.M. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Clement, K.; Ziller, M.J.; Boyle, P.; Fan, J.; Gu, H.; Stevenson, K.; Sougnez, C.; Wang, L.; Li, S.; et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell 2014, 26, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Baliakas, P.; Hadzidimitriou, A.; Sutton, L.-A.; Rossi, D.; Minga, E.; Villamor, N.; Larrayoz, M.; Kminkova, J.; Agathangelidis, A.; Davis, Z.; et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia 2015, 29, 329–336. [Google Scholar] [CrossRef]
- Rossi, D.; Rasi, S.; Spina, V.; Bruscaggin, A.; Monti, S.; Ciardullo, C.; Deambrogi, C.; Khiabanian, H.; Serra, R.; Bertoni, F.; et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013, 121, 1403–1412. [Google Scholar] [CrossRef]
- Rai, K.R.; Sawitsky, A.; Cronkite, E.P.; Chanana, A.D.; Levy, R.N.; Pasternack, B.S. Clinical staging of chronic lymphocytic leukemia. Blood 1975, 46, 219–234. [Google Scholar] [CrossRef]
- Binet, J.L.; Auquier, A.; Dighiero, G.; Chastang, C.; Piguet, H.; Goasguen, J.; Vaugier, G.; Potron, G.; Colona, P.; Oberling, F.; et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981, 48, 198–206. [Google Scholar] [CrossRef]
- Rai, K.R. A critical analysis of staging in CLL. In Chronic Lymphocytic Leukemia: Recent Progress and Future Directions; Gale, R.P., Rai, K.R., Eds.; Alan R. Liss: New York, NY, USA, 1987; pp. 253–264. [Google Scholar]
- Shustik, C.; Mick, R.; Silver, R.; Sawitsky, A.; Rai, K.; Shapiro, L. Treatment of early chronic lymphocytic leukemia: Intermittent chlorambucil versus observation. Hematol. Oncol. 1988, 6, 7–12. [Google Scholar] [CrossRef]
- CLL trialists collaborative group. Chemotherapeutic options in chronic lymphocytic leukemia: A meta-analysis of the randomized trials. J. Natl. Cancer Inst. 1999, 91, 861–868. [Google Scholar] [CrossRef]
- Fischer, K.; Al-Sawaf, O.; Bahlo, J.; Fink, A.M.; Tandon, M.; Dixon, M.; Robrecht, S.; Warburton, S.; Humphrey, K.; Samoylova, O.; et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N. Engl. J. Med. 2019, 380, 2225–2236. [Google Scholar] [CrossRef]
- Ferrer, G.; Montserrat, E. Critical molecular pathways in CLL therapy. Mol. Med. 2018, 24, 9. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cel. Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Huang, R.; Chen, H.; Liang, J.; Li, Y.; Yang, J.; Luo, C.; Tang, Y.; Ding, Y.; Liu, X.; Yuan, Q.; et al. Dual role of reactive oxygen species and their application in cancer therapy. J. Cancer 2021, 12, 5543–5561. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Mohan, G.M.; Shailender, G.; Malla, R.R. Reactive oxygen species: A key constituent in cancer survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Saikolappan, S.; Kumar, B.; Shishodia, G.; Koul, S.; Koul, H.K. Reactive oxygen species and cancer: A complex interaction. Cancer Lett. 2019, 452, 132–143. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Article, R.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 2, 3945–3952. [Google Scholar] [CrossRef]
- Ramalingam, V.; Rajaram, R. A paradoxical role of reactive oxygen species in cancer signaling pathway: Physiology and pathology. Process Biochem. 2021, 100, 69–81. [Google Scholar] [CrossRef]
- Morris, G.; Gevezova, M.; Sarafian, V.; Maes, M. Redox regulation of the immune response. Cell. Mol. Immunol. 2022, 19, 1079–1101. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Shen, P.; Song, Y.; Huang, Y.; Tu, S. Reactive Oxygen Species in Autoimmune Cells: Function, Differentiation, and Metabolism. Front. Immunol. 2021, 12, 635021. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic radioresistance: Can ROS be the key to overcome it? Cancers 2019, 11, 112. [Google Scholar] [CrossRef]
- Howard, D.; Sebastian, S.; Le, Q.V.C.; Thierry, B.; Kempson, I. Chemical mechanisms of nanoparticle radiosensitization and radioprotection: A review of structure-function relationships influencing reactive oxygen species. Int. J. Mol. Sci. 2020, 21, 579. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive oxygen species in cancer stem cells. Antioxid. Redox Signal. 2012, 16, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Cie, A.; Yue, J.; Lee, H.C.; Skonieczna, M.; Wei, Y.H. ROS and oxidative stress in stem cells. Oxid. Med. Cell. Longev. 2017, 2017, 5047168. [Google Scholar] [CrossRef]
- Allegra, A.; Pioggia, G.; Tonacci, A.; Musolino, C.; Gangemi, S. Oxidative Stress and Photodynamic Therapy of Skin Cancers: Mechanisms, Challenges and Promising Developments. Antioxidants 2020, 9, 448. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Chen, H. Cardiotoxicity of Anticancer Therapeutics. Front. Cardiovasc. Med. 2018, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Cardinale, D.; Dent, S.; Criscitiello, C.; Aseyev, O.; Lenihan, D.; Cipolla, C.M. Cardiotoxicity of anticancer treatments: Epidemiology, detection, and management. CA Cancer J. Clin. 2016, 66, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.S.; Steinauer, K.K.; Hornung, B.; Irish, J.M.; Lecane, P.; Birrell, G.W.; Peehl, D.M.; Knox, S.J. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002, 9, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, and apoptosis. Am. J. Med. Genet. 2001, 106, 62–70. [Google Scholar] [CrossRef]
- Hileman, E.O.; Liu, J.; Albitar, M.; Keating, M.J.; Huang, P. Intrinsic oxidative stress in cancer cells: A biochemical basis for therapeutic selectivity. Cancer Chemother. Pharmacol. 2004, 53, 209–219. [Google Scholar] [CrossRef]
- Gartenhaus, R.B.; Prachand, S.N.; Paniaqua, M.; Li, Y.; Gordon, L.I. Arsenic trioxide cytotoxicity in steroid and chemotherapy-resistant myeloma cell lines: Enhancement of apoptosis by manipulation of cellular redox state. Clin. Cancer Res. 2002, 8, 566–572. [Google Scholar]
- Grad, J.M.; Bahlis, N.J.; Reis, I.; Oshiro, M.M.; Dalton, W.S.; Boise, L.H. Ascorbic acid enhances arsenic trioxide-induced cytotoxicity in multiple myeloma cells. Blood 2001, 98, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Bahlis, N.J.; McCafferty-Grad, J.; Jordan-McMurry, I.; Neil, J.; Reis, I.; Kharfan-Dabaja, M.; Eckman, J.; Goodman, M.; Fernandez, H.F.; Boise, L.H.; et al. Feasibility and correlates of arsenic trioxide combined with ascorbic acid-mediated depletion of intracellular glutathione for the treatment of relapsed/refractory multiple myeloma. Clin. Cancer Res. 2002, 8, 3658–3668. [Google Scholar] [PubMed]
- Bellosillo, B.; Villamor, N.; López-Guillermo, A.; Marcé, S.; Esteve, J.; Campo, E.; Colomer, D.; Montserrat, E. Complement-mediated cell death induced by rituximab in B-cell lymphoproliferative disorders is mediated in vitro by a caspase-independent mechanism involving the generation of reactive oxygen species. Blood 2001, 98, 2771–2777. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Kirtonia, A.; Sethi, G.; Garg, M.; Artesunate, A.R.T. The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol. Life Sci. 2020, 77, 4459–4483. [Google Scholar] [CrossRef] [PubMed]
- Najafov, A.; Chen, H.; Yuan, J. Necroptosis and Cancer. Trends Cancer 2017, 3, 294–301. [Google Scholar] [CrossRef]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef]
- Schmidt, S.V.; Seibert, S.; Walch-Rückheim, B.; Vicinus, B.; Kamionka, E.M.; Pahne-Zeppenfeld, J.; Solomayer, E.F.; Kim, Y.J.; Bohle, R.M.; Smola, S. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1α release, and efficient paracrine dendritic cell activation. Oncotarget 2015, 6, 8635–8647, Erratum in Oncotarget 2019, 10, 4503–4504. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wang, F.; Guo, Q.; Li, M.; Wang, L.; Zhang, Z.; Jiang, S.; Jin, H.; Chen, A.; Tan, S.; et al. Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in hepatic stellate cells. Redox Biol. 2018, 19, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [PubMed]
- Raza, M.H.; Siraj, S.; Arshad, A.; Waheed, U.; Aldakheel, F.; Alduraywish, S.; Arshad, M. ROS-modulated therapeutic approaches in cancer treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 1789–1890. [Google Scholar]
- Hsu, S.K.; Chang, W.T.; Lin, I.L.; Chen, Y.F.; Padalwar, N.B.; Cheng, K.C.; Teng, Y.N.; Wang, C.H.; Chiu, C.C. The Role of Necroptosis in ROS-Mediated Cancer Therapies and Its Promising Applications. Cancers 2020, 12, 2185. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.V.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Friedmann Angeli, J.P.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.M.; Sun, X.; Roschewski, M.; Aue, G.; Farooqui, M.; Stennett, L.; Gibellini, F.; Arthur, D.; Pérez-Galán, P.; Wiestner, A. ON 01910.Na is selectively cytotoxic for chronic lymphocytic leukemia cells through a dual mechanism of action involving PI3K/AKT inhibition and induction of oxidative stress. Clin. Cancer Res. 2012, 18, 1979–1991. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells byROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Zhang, H.; Zhang, W.; Feng, L.; Du, M.; Zhou, Y.; Chen, Z.; Pelicano, H.; Plunkett, W.; Wierda, W.G.; et al. Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood 2008, 112, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef] [PubMed]
- Avery, S. Molecular targets of oxidative stress. Biochem. J. 2011, 434, 201–210. [Google Scholar] [CrossRef]
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.; Vourc’h, C.; Matthias, P.; et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007, 21, 2172–2181. [Google Scholar] [CrossRef]
- Matthias, P.; Yoshida, M.; Khochbin, S. HDAC6 a new cellular stress surveillance factor. Cell Cycle 2008, 7, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Nalluri, S.; Kolhe, R.; Yang, Y.; Fiskus, W.; Chen, J.; Ha, K.; Buckley, K.M.; Balusu, R.; Coothankandaswamy, V.; et al. Treatment with panobinostat induces glucose-regulated protein 78 acetylation and endoplasmic reticulum stress in breast cancer cells. Mol. Cancer Ther. 2010, 9, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rao, R.; Shen, J.; Tang, Y.; Fiskus, W.; Nechtman, J.; Atadja, P.; Bhalla, K. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 2008, 68, 4833–4842. [Google Scholar] [CrossRef] [PubMed]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–539. [Google Scholar] [CrossRef]
- Moulick, K.; Ahn, J.H.; Zong, H.; Rodina, A.; Cerchietti, L.; Gomes DaGama, E.M.; Caldas-Lopes, E.; Beebe, K.; Perna, F.; Hatzi, K.; et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 2011, 7, 818–826. [Google Scholar] [CrossRef]
- Rao, R.; Fiskus, W.; Yang, Y.; Herger, B.; Joshi, R.; Fernandez, P.; Mandawat, A.; Atadja, P.; Bradner, J.E.; Bhalla, K. HDAC6 inhibition enhances 17-AAG-mediated abrogation of HSP90 Chaper-one function in human leukemia cells. Blood 2008, 112, 1886–1893. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.E.; Prada, C.E.; Loria, O.; Kamal, A.; Chen, L.; Burrows, F.J.; Kipps, T.J. ZAP-70 is a novel conditional heat shock protein 90 (Hsp90) client: Inhibition of Hsp90 leads to ZAP-70 degradation, apoptosis, and impaired signaling in chronic lymphocytic leukemia. Blood 2005, 106, 2506–2512. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Nawrocki, S.T.; Xu, R.H.; Dunner, K.; McConkey, D.J.; Wierda, W.G.; Keating, M.J.; Huang, P. Increased mitochondrial biogenesis in primary leukemia cells: The role of endogenous nitric oxide and impact on sensitivity to fludarabine. Leukemia 2004, 18, 1934–1940. [Google Scholar] [CrossRef]
- Jitschin, R.; Hofmann, A.D.; Bruns, H.; Giessl, A.; Bricks, J.; Berger, J.; Saul, D.; Eckart, M.J.; Mackensen, A.; Mougiakakos, D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014, 123, 2663–2672. [Google Scholar] [CrossRef]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Wiestner, A. Targeting B cell receptor signalling in cancer: Preclinical and clinical advances. Nat. Rev. Cancer 2018, 18, 148–167. [Google Scholar] [CrossRef] [PubMed]
- Herishanu, Y.; Pérez-Galán, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Herndon, T.M.; Chen, S.S.; Saba, N.S.; Valdez, J.; Emson, C.; Gatmaitan, M.; Tian, X.; Hughes, T.E.; Sun, C.; Arthur, D.C.; et al. Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia 2017, 31, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. Treatment of chronic lymphocytic leukemia. N. Engl. J. Med. 2020, 383, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Caligaris-Cappio, F.; Ghia, P. Novel insights in chronic lymphocytic leukemia: Are we getting closer to understanding the pathogenesis of the disease? J. Clin. Oncol. 2008, 26, 4497–4503. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.J.; Verschuer, L.A.; Harmon, B.V.; Prentice, R.L.; Pope, J.H.; Kerr, J.F. Spontaneous programmed death (apoptosis) of B-chronic lymphocytic leukaemia cells following their culture in vitro. Br. J. Haematol. 1989, 71, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Panayiotidis, P.; Jones, D.; Ganeshaguru, K.; Foroni, L.; Hoffbrand, A.V. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br. J. Haematol. 1996, 92, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [CrossRef]
- Burger, J.A.; Quiroga, M.P.; Hartmann, E.; Bürkle, A.; Wierda, W.G.; Keating, M.J.; Rosenwald, A. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood 2009, 113, 3050–3058. [Google Scholar] [CrossRef]
- Ghia, P.; Strola, G.; Granziero, L.; Geuna, M.; Guida, G.; Sallusto, F.; Ruffing, N.; Montagna, L.; Piccoli, P.; Chilosi, M.; et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4þ, CD40Lþ T cells by producing CCL22. Eur. J. Immunol. 2002, 32, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Pascutti, M.F.; Jak, M.; Tromp, J.M.; Derks, I.A.; Remmerswaal, E.B.; Thijssen, R.; van Attekum, M.H.A.; van Bochove, G.G.; Luijks, D.M.; Pals, S.T.; et al. IL-21 and CD40L signals from autologous T cells can induce antigen independent proliferation of CLL cells. Blood 2013, 122, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Ahearne, M.J.; Willimott, S.; Piñon, L.; Kennedy, D.B.; Miall, F.; Dyer, M.J.; Wagner, S.D. Enhancement of CD154/IL4 proliferation by the T follicular helper (Tfh) cytokine, IL21 and increased numbers of circulating cells resembling Tfh cells in chronic lymphocytic leukaemia. Br. J. Haematol. 2013, 162, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Willimott, S.; Baou, M.; Naresh, K.; Wagner, S.D. CD154 induces a switch in pro-survival Bcl-2 family members in chronic lymphocytic leukaemia. Br. J. Haematol. 2007, 138, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Kitada, S.; Zapata, J.M.; Andreeff, M.; Reed, J.C. Bryostatin and CD40-ligand enhance apoptosis resistance and induce expression of cell survival genes in B-cell chronic lymphocytic leukaemia. Br. J. Haematol. 1999, 106, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Buske, C.; Gogowski, G.; Schreiber, K.; Rave-Frank, M.; Hiddemann, W.; Wormann, B. Stimulation of B-chronic lymphocytic leukemia cells by murine fibroblasts, IL-4, anti-CD40 antibodies, and the soluble CD40 ligand. Exp. Hematol. 1997, 25, 329–337. [Google Scholar]
- Yosifov, D.Y.; Idler, I.; Bhattacharya, N.; Reichenzeller, M.; Close, V.; Ezerina, D.; Scheffold, A.; Jebaraj, B.M.C.; Kugler, S.; Bloehdorn, J.; et al. Oxidative stress as candidate therapeutic target to overcome microenvironmental protection of CLL. Leukemia 2020, 34, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Braun, M.; Qorraj, M.; Saul, D.; Le Blanc, K.; Zenz, T.; Mougiakakos, D. Stromal cell-mediated glycolytic switch in CLL cells involves Notch-c-Myc signaling. Blood 2015, 125, 3432–3436. [Google Scholar] [CrossRef] [PubMed]
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxy-gen species in p53-mediated cell fate decisions. Mol. Cell. Biol. 2003, 23, 8576–8585. [Google Scholar] [CrossRef]
- Carrera, S.; de Verdier, P.J.; Khan, Z.; Zhao, B.; Mahale, A.; Bowman, K.J.; Zainol, M.; Jones, G.D.D.; Lee, S.W.; Aaronson, S.A.; et al. Protection of cells in physiological oxygen tensions against DNA damage-induced apoptosis. J. Biol. Chem. 2010, 285, 13658–13665. [Google Scholar] [CrossRef]
- Milczarek, G.J.; Martinez, J.; Bowden, G.T. p53 Phosphorylation: Biochemical and functional consequences. Life Sci. 1997, 60, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Samuel, J.; Jayne, S.; Chen, Y.; Majid, A.; Wignall, A.; Wormull, T.; Najeeb, H.; Luo, J.L.; Jones, G.D.; Macip, S.; et al. Posttranscriptional Upregulation of p53 by Reactive Oxygen Species in Chronic Lymphocytic Leukemia. Cancer Res. 2016, 76, 6311–6319. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Watters, D.; Kedar, P.; Spring, K.; Bjorkman, J.; Chen, P.; Gatei, M.; Birrell, G.; Garrone, B.; Srinivasa, P.; Crane, P.I.; et al. Localization of a portion of extranuclear ATM to peroxisomes. J. Biol. Chem. 1999, 274, 34277–34282. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jonnalagadda, J.C.; Douglas, P.; Young, D.; Ye, R.; Moorhead, G.B.G.; Lees-Miller, S.P.; Khanna, K.K. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004, 23, 4451–4461. [Google Scholar] [CrossRef] [PubMed]
- Austen, B.; Skowronska, A.; Baker, C.; Powell, J.E.; Gardiner, A.; Oscier, D.; Majid, A.; Dyer, M.; Siebert, R.; Taylor, A.M.; et al. Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J. Clin. Oncol. 2007, 25, 5448–5457. [Google Scholar] [CrossRef] [PubMed]
- Valentin-Vega, Y.A.; Maclean, K.H.; Tait-Mulder, J.; Milasta, S.; Steeves, M.; Dorsey, F.C.; Cleveland, J.L.; Green, D.R.; Kastan, M.B. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012, 119, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef]
- Li, B.; Wang, X.; Rasheed, N.; Hu, Y.; Boast, S.; Ishii, T.; Nakayama, K.; Nakayama, K.I.; Goff, S.P. Distinct roles of c-Abl and Atm in oxidative stress response are mediated by protein kinase C delta. Genes Dev. 2004, 18, 1824–1837. [Google Scholar] [CrossRef] [PubMed]
- Agathanggelou, A.; Weston, V.J.; Perry, T.; Davies, N.J.; Skowronska, A.; Payne, D.T.; Fossey, J.S.; Oldreive, C.E.; Wei, W.; Pratt, G.; et al. Targeting the Ataxia Telangiectasia Mutated-null phenotype in chronic lymphocytic leukemia with pro-oxidants. Haematologica 2015, 100, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.J.F.; Iskandar, K.; Lai, J.X.H.; Qu, J.; Raman, D.; Valentin, R.; Herbaux, C.; Collins, M.; Low, I.C.C.; Loh, T.; et al. Serine-70 phosphorylated Bcl-2 prevents oxidative stress-induced DNA damage by modulating the mitochondrial redox metabolism. Nucleic Acids Res. 2020, 48, 12727–12745. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Huang, C.; Geng, H.; Chen, Z.; Harvey, R.; Kang, H.; Ng, C.; Titz, B.; Hurtz, C.; Sadiyah, M.F.; et al. BACH2 mediates negative selection and p53-dependent tumor suppression at the pre-B cell receptor checkpoint. Nat. Med. 2013, 19, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Chi, V.L.D.; Garaud, S.; De Silva, P.; Thibaud, V.; Stamatopoulos, B.; Berehad, M.; Gu-Trantien, C.; Krayem, M.; Duvillier, H.; Lodewyckx, J.N.; et al. Age-related changes in the BACH2 and PRDM1 genes in lymphocytes from healthy donors and chronic lymphocytic leukemia patients. BMC Cancer 2019, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Finetti, F.; Petronilli, V.; Ulivieri, C.; Giusti, F.; Lupetti, P.; Giorgio, M.; Pelicci, P.G.; Bernardi, P.; Baldari, C.T. p66SHC promotes T cell apoptosis by inducing mitochondrial dysfunction and impaired Ca2+ homeostasis. Cell Death Differ. 2007. [Google Scholar] [CrossRef] [PubMed]
- Pacini, S.; Pellegrini, M.; Migliaccio, E.; Patrussi, L.; Ulivieri, C.; Ventura, A.; Carraro, F.; Naldini, A.; Lanfrancone, L.; Pelicci, P.; et al. p66SHC promotes apoptosis and antagonizes mitogenic signaling in T cells. Mol. Cell. Biol. 2004, 24, 1747–1757. [Google Scholar] [CrossRef]
- Patrussi, L.; Giommoni, N.; Pellegrini, M.; Gamberucci, A.; Baldari, C.T. p66Shc-dependent apoptosis requires Lck and CamKII activity. Apoptosis 2012, 17, 174–186. [Google Scholar] [CrossRef]
- Capitani, N.; Lucherini, O.M.; Sozzi, E.; Ferro, M.; Giommoni, N.; Finetti, F.; De Falco, G.; Cencini, E.; Raspadori, D.; Pelicci, P.G.; et al. Impaired expression of p66Shc, a novel regulator of B-cell survival, in chronic lymphocytic leukemia. Blood 2010, 115, 3726–3736. [Google Scholar] [CrossRef][Green Version]
- Patrussi, L.; Capitani, N.; Cattaneo, F.; Manganaro, N.; Gamberucci, A.; Frezzato, F.; Martini, V.; Visentin, A.; Pelicci, P.G.; D’Elios, M.M.; et al. p66Shc deficiency enhances CXCR4 and CCR7 recycling in CLL B cells by facilitating their dephosphorylation-dependent release from β-arrestin at early endosomes. Oncogene 2018, 37, 1534–1550. [Google Scholar] [CrossRef] [PubMed]
- Capitani, N.; Patrussi, L.; Trentin, L.; Lucherini, O.M.; Cannizzaro, E.; Migliaccio, E.; Frezzato, F.; Gattazzo, C.; Forconi, F.; Pelicci, P.G.; et al. S1P1 expression is controlled by the pro-oxidant activity of p66Shc and is impaired in B-CLL patients with unfavorable prognosis. Blood 2012, 120, 4391–4399. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patrussi, L.; Capitani, N.; Cannizzaro, E.; Finetti, F.; Lucherini, O.M.; Pelicci, P.G.; Baldari, C.T. Negative regulation of chemokine receptor signaling and B-cell chemotaxis by p66Shc. Cell Death Dis. 2014, 5, e1068. [Google Scholar] [CrossRef] [PubMed]
- Patrussi, L.; Capitani, N.; Martini, V.; Pizzi, M.; Trimarco, V.; Frezzato, F.; Marino, F.; Semenzato, G.; Trentin, L.; Baldari, C.T. Enhanced Chemokine Receptor Recycling and Impaired S1P1 Expression Promote Leukemic Cell Infiltration of Lymph Nodes in Chronic Lymphocytic Leukemia. Cancer Res. 2015, 75, 4153–4163. [Google Scholar] [CrossRef] [PubMed]
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.A.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960. [Google Scholar] [CrossRef] [PubMed]
- Patrussi, L.; Capitani, N.; Ulivieri, C.; Manganaro, N.; Granai, M.; Cattaneo, F.; Kabanova, A.; Mundo, L.; Gobessi, S.; Frezzato, F.; et al. p66Shc deficiency in the Eμ-TCL1 mouse model of chronic lymphocytic leukemia enhances leukemogenesis by altering the chemokine receptor landscape. Haematologica 2019, 104, 2040–2052. [Google Scholar] [CrossRef] [PubMed]
- Pavlasova, G.; Borsky, M.; Seda, V.; Cerna, K.; Osickova, J.; Doubek, M.; Mayer, J.; Calogero, R.; Trbusek, M.; Pospisilova, S.; et al. Ibrutinib inhibits CD20 upregulation on CLL B cells mediated by the CXCR4/SDF-1 axis. Blood 2016, 128, 1609–1613. [Google Scholar] [CrossRef] [PubMed]
- DERosa, A.; Zappavigna, S.; Villa, M.R.; Improta, S.; Cesario, E.; Mastrullo, L.; Caraglia, M.; Stiuso, P. Prognostic role of translocator protein and oxidative stress markers in chronic lymphocytic leukemia patients treated with bendamustine plus rituximab. Oncol. Lett. 2015, 9, 1327–1332. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Alonci, A.; Saija, A.; Russo, S.; Cannavò, A.; Cristani, M.; Centorrino, R.; Saitta, S.; Alibrandi, A.; et al. Carbonyl group serum levels are associated with CD38 expression in patients with B chronic lymphocytic leukemia. Clin. Biochem. 2011, 44, 1487–1490. [Google Scholar] [CrossRef]
- Penna, G.; Allegra, A.; Alonci, A.; Aguennouz, M.; Garufi, A.; Cannavò, A.; Gerace, D.; Alibrandi, A.; Musolino, C. MDR-1 polymorphisms (G2677T and C3435T) in B-chronic lymphocytic leukemia: An impact on susceptibility and prognosis. Med. Oncol. 2011, 28, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Sabry, S.A.; El-Senduny, F.F.; Abousamra, N.K.; Salah El-Din, M.; Youssef, M.M. Oxidative stress in CLL patients leads to activation of Th9 cells: An experimental and comprehensive survey. Immunol. Med. 2020, 43, 36–46. [Google Scholar] [CrossRef] [PubMed][Green Version]
- D’Arena, G.; Vitale, C.; Perbellini, O.; Coscia, M.; La Rocca, F.; Ruggieri, V.; Visco, C.; Di Minno, N.M.D.; Innocenti, I.; Pizza, V.; et al. Prognostic relevance of oxidative stress measurement in chronic lymphocytic leukaemia. Eur. J. Haematol. 2017, 99, 306–314. [Google Scholar] [CrossRef] [PubMed]
- D’Arena, G.; Seneca, E.; Migliaccio, I.; De Feo, V.; Giudice, A.; La Rocca, F.; Capunzo, M.; Calapai, G.; Festa, A.; Caraglia, M.; et al. Oxidative stress in chronic lymphocytic leukemia: Still a matter of debate. Leuk. Lymphoma 2019, 60, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Svingen, P.A.; Tefferi, A.; Kottke, T.; Kaur, G.; Narayanan, V.L.; Sausville, E.A.; Kaufmann, S.H. Effects of the bcr/abl kinase inhibitors AG957 and NSC 680410 on chronic myelogenous leukemia cells in vitro. Clin. Cancer Res. 2000, 6, 237–249. [Google Scholar] [PubMed]
- Levitzki, A. Tyrphostins: Tyrosine kinase blockers as novel antiproliferative agents and dissectors of signal transduction. FASEB J. 1992, 6, 3275–3282. [Google Scholar] [CrossRef]
- Yu, C.; Rahmani, M.; Almenara, J.; Sausville, E.A.; Dent, P.; Grant, S. Induction of apoptosis in human leukemia cells by the tyrosine kinase inhibitor adaphostin proceeds through a RAF-1/MEK/ERK- and AKT-dependent process. Oncogene 2004, 23, 1364–1376. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; Hackbarth, J.; Le, S.; Loegering, D.; Bone, N.; Bruzek, L.M.; Narayanan, V.L.; Adjei, A.A.; Kay, N.E.; Tefferi, A.; et al. Involvement of reactive oxygen species in adaphostin-induced cytotoxicity in human leukemia cells. Blood 2003, 102, 4512–4519. [Google Scholar] [CrossRef] [PubMed]
- Avramis, I.A.; Laug, W.E.; Sausville, E.A.; Avramis, V.I. Determination of drug synergism between the tyrosine kinase inhibitors NSC 680410 (adaphostin) and/or STI571 (imatinib mesylate, Gleevec) with cytotoxic drugs against human leukemia cell lines. Cancer Chemother. Pharmacol. 2003, 52, 307–318. [Google Scholar] [CrossRef]
- Avramis, I.; Christodoulopoulos, G.; Suzuki, A.; Laug, W.E.; Gonzalez-Gomez, I.; McNamara, G.; Sausville, E.A.; Avramis, V.I. In vitro and in vivo anticancer evaluations of the novel tyrosine kinase inhibitor NSC 680410. Cancer Chemother. Pharmacol. 2002, 50, 479–489. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Lee, Y.K.; Bone, N.D.; Strege, A.K.; Narayanan, V.L.; Sausville, E.A.; Geyer, S.M.; Kaufmann, S.H.; Kay, N.E. Adaphostin-induced apoptosis in CLL B cells is associated with induction of oxidative stress and exhibits synergy with fludarabine. Blood 2005, 105, 2099–2106. [Google Scholar] [CrossRef][Green Version]
- Knop, S.; Straka, C.; Haen, M.; Schwedes, R.; Hebart, H.; Einsele, H. The efficacy and toxicity of bendamustine in recurrent multiple myeloma after high-dose chemotherapy. Haematologica 2005, 90, 1287–1288. [Google Scholar]
- von Minckwitz, G.; Chernozemsky, I.; Sirakova, L.; Chilingirov, P.; Souchon, R.; Marschner, N.; Kleeberg, U.; Tsekov, C.; Fritze, D.; Thomssen, C.; et al. Bendamustine prolongs progression-free survival in metastatic breast cancer (MBC): A phase III prospective, randomized, multicenter trial of bendamustine hydrochloride, methotrexate and 5-fluorouracil (BMF) versus cyclophosphamide, methotrexate and 5-fluorouracil (CMF) as first-line treatment of MBC. Anti-Cancer Drugs 2005, 16, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, M.A.; Goebeler, M.E.; Herold, M.; Emmerich, B.; Wilhelm, M.; Ruelfs, C.; Boening, L.; Hallek, M.J.; German CLL Study Group. Efficacy of bendamustine in patients with relapsed or refractory chronic lymphocytic leukemia: Results of a phase I/II study of the German CLL Study Group. Haematologica 2005, 90, 1357–1364. [Google Scholar]
- Bottke, D.; Bathe, K.; Wiegel, T.; Hinkelbein, W. Phase I trial of radiochemotherapy with bendamustine in patients with recurrent squamous cell carcinoma of the head and neck. Strahlenther. Onkol. 2007, 183, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Rummel, M.J.; Mitrou, P.S.; Hoelzer, D. Bendamustine in the treatment of non-Hodgkin’s lymphoma: Results and future perspectives. Semin. Oncol. 2002, 29, 27–32. [Google Scholar] [CrossRef]
- Teichert, J.; Baumann, F.; Chao, Q.; Franklin, C.; Bailey, B.; Hennig, L.; Caca, K.; Schoppmeyer, K.; Patzak, U.; Preiss, R. Characterization of two phase I metabolites of bendamustine in human liver microsomes and in cancer patients treated with bendamustine hydrochloride. Cancer Chemother. Pharmacol. 2007, 59, 759–770. [Google Scholar] [CrossRef]
- Schwanen, C.; Hecker, T.; Hubinger, G.; Wölfle, M.; Rittgen, W.; Bergmann, L.; Karakas, T. In vitro evaluation of bendamustine induced apoptosis in Bchronic lymphocytic leukemia. Leukemia 2002, 16, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Erlacher, M.; Labi, V.; Manzl, C.; Böck, G.; Tzankov, A.; Häcker, G.; Michalak, E.; Strasser, A.; Villunger, A. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J. Exp. Med. 2006, 203, 2939–2951. [Google Scholar] [CrossRef]
- Perez-Galan, P.; Roue, G.; Villamor, N.; Montserrat, E.; Campo, E.; Colomer, D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood 2006, 107, 257–264. [Google Scholar] [CrossRef]
- Vousden, K.H. Apoptosis. p53 and PUMA: A deadly duo. Science 2005, 309, 1685–1686. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, Y.; Verhaegen, M.; Miller, T.P.; Rush, J.L.; Steiner, P.; Opipari, A.W., Jr.; Lowe, S.W.; Soengas, M.S. Differential regulation of noxa in normalmelanocytes and melanoma cells by proteasome inhibition: Therapeutic implications. Cancer Res. 2005, 65, 6294–6304. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.Z.; Ziffra, J.; Stennett, L.; Bodner, B.; Bonish, B.K.; Chaturvedi, V.; Bennett, F.; Pollock, P.M.; Trent, J.M.; Hendrix, M.J.C.; et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005, 65, 6282–6293. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Veal, G.J.; Redfern, C.P.; Lovat, P.E. Role of Noxa in p53-independent fenretinide-induced apoptosis of neuroectodermal tumours. Apoptosis 2007, 12, 613–622. [Google Scholar] [CrossRef]
- Roué, G.; López-Guerra, M.; Milpied, P.; Pérez-Galán, P.; Villamor, N.; Montserrat, E.; Campo, E.; Colomer, D. Bendamustine is effective in p53-deficient B-cell neoplasms and requires oxidative stress and caspase-independent signaling. Clin. Cancer Res. 2008, 14, 6907–6915. [Google Scholar] [CrossRef] [PubMed]
- Li, G.X.; Hu, H.; Jiang, C.; Schuster, T.; Lu, J. Differential involvement of reactive oxygen species in apoptosis induced by two classes of selenium compounds in human prostate cancer cells. Int. J. Cancer 2007, 120, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- May, M.J.; Madge, L.A. Caspase inhibition sensitizes inhibitor of NF-nB kinase h-deficient fibro-blasts to caspase-independent cell death via the generation of reactive oxygen species. J. Biol. Chem. 2007, 282, 16105–16116. [Google Scholar] [CrossRef]
- Sutton, B.M.; McGusty, E.; Walz, D.T.; DiMartino, M.J. Oral gold. Antiarthritic properties of alkylphosphinegold coordination complexes. J. Med. Chem. 1972, 15, 1095–1098. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs R D 2015, 15, 13–20. [Google Scholar] [CrossRef]
- Meuillet, E.J.; Mahadevan, D.; Berggren, M.; Coon, A.; Powis, G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN’s lipid phosphatase activity and membrane binding: A mechanism for the functional loss of PTEN’s tumor suppressor activity. Arch. Biochem. Biophys. 2004, 429, 123–133. [Google Scholar] [CrossRef]
- Mohammadi, F.; Soltani, A.; Ghahremanloo, A.; Javid, H.; Hashemy, S.I. The thioredoxin system and cancer therapy: A review. Cancer Chemother. Pharmacol. 2019, 84, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Abdalbari, F.H.; Telleria, C.M. The gold complex auranofin: New perspectives for cancer therapy. Discov. Oncol. 2021, 12, 42. [Google Scholar] [CrossRef]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S.D.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schor-no, K.; et al. Auranofin induces lethal oxidative and endoplasmic reticulum stress and exerts potent preclinical activity against chronic lymphocytic leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Goldstein, J.C.; Kluck, R.M.; Newmeyer, D.D.; Green, D.R. The (Holey) study of mitochondria in apoptosis. Methods Cell Biol. 2001, 66, 365–391. [Google Scholar]
- Lu, W.; Ogasawara, M.A.; Huang, P. Models of reactive oxygen species in cancer. Drug Discov. Today Dis. Models 2007, 4, 67–73. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef]
- Kim, H.R.; Park, C.G.; Jung, J.Y. Acacetin (5, 7-dihydroxy-4′-methoxyflavone) exhibits in vitro and in vivo anticancer activity through the suppression of NF-kB/Akt signaling in prostate cancer cells. Int. J. Mol. Med. 2014, 33, 317–324. [Google Scholar] [CrossRef]
- Wang, S.; Lin, B.; Liu, W.; Wei, G.; Li, Z.; Yu, N.; Xue, X.; Ji, G. Acacetin Induces Apoptosis in Human Osteosarcoma Cells by Modulation of ROS/JNK Activation. Drug Des. Devel Ther. 2020, 14, 5077–5085. [Google Scholar] [CrossRef] [PubMed]
- Salimi, A.; Roudkenar, M.H.; Sadeghi, L.; Mohseni, A.; Seydi, E.; Pirahmadi, N.; Pourahmad, J. Selective Anticancer Activity of Acacetin Against Chronic Lymphocytic Leukemia Using Both In Vivo and In Vitro Methods: Key Role of Oxidative Stress and Cancerous Mitochondria. Nutr. Cancer 2016, 68, 1404–1416. [Google Scholar] [CrossRef]
- Gao, B.; Peng, C.; Lin, B.; Chen, Q.; Zhang, J.; Shi, Q. Screening and validation of highly-efficient insecticidal conotoxins from a transcriptome-based dataset of Chinese tubular cone snail. Toxins 2017, 9, 214. [Google Scholar] [CrossRef]
- Gao, B.; Peng, C.; Yang, J.; Yi, Y.; Zhang, J.; Shi, Q. Cone snails: A big store of conotoxins for novel drug discovery. Toxins 2017, 9, 397. [Google Scholar] [CrossRef]
- Fu, Y.; Li, C.; Dong, S.; Wu, Y.; Zhangsun, D.; Luo, S. Discovery methodology of novel conotoxins from Conus species. Mar. Drugs 2018, 16, 417. [Google Scholar] [CrossRef]
- Salimi, A.; Rahimitabar, N.; Vazirizadeh, A.; Adhami, V.; Pourahmad, J. Persian Gulf Snail Crude Venom (Conus textile): A potential source of anti-cancer therapeutic agents for glioblastoma through mitochondrial-mediated apoptosis. Asian Pac. J. Cancer Prev. 2020, 21, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Salimi, A.; Salehian, S.; Aboutorabi, A.; Vazirizadeh, A.; Adhami, V.; Sajjadi Alehashem, S.H.; Seydi, E.; Pourahmad, J. Cytotoxicity Studies of the Crude venom and Fractions of Persian Gulf Snail (Conus textile) on Chronic Lymphocytic Leukemia and Normal Lymphocytes. Asian Pac. J. Cancer Prev. 2021, 22, 1523–1529. [Google Scholar] [CrossRef]
- Evens, A.M.; Balasubramanian, L.; Gordon, L.I. Motexafin gadolinium induces oxidative stress and apoptosis in hematologic malignancies. Curr. Treat. Options Oncol. 2005, 6, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood 2009, 114, 3367–3375. [Google Scholar] [CrossRef]
- Cuní, S.; Pérez-Aciego, P.; Pérez-Chacón, G.; Vargas, J.A.; Sánchez, A.; Martín-Saavedra, F.M.; Ballester, S.; García-Marco, J.; Jordá, J.; Durántez, A. A sustained activation of PI3K/NF-kappaB pathway is critical for the survival of chronic lymphocytic leukemia B cells. Leukemia 2004, 18, 1391–1400. [Google Scholar] [CrossRef]
- Herishanu, Y.; Gibellini, F.; Njuguna, N.; Hazan-Halevy, I.; Farooqui, M.; Bern, S.; Keyvanfar, K.; Lee, E.; Wilson, W.; Wiestner, A. Activation of CD44, a receptor for extracellular matrix components, protects chronic lymphocytic leukemia cells from spontaneous and drug induced apoptosis through MCL-1. Leuk. Lymphoma 2011, 52, 1758–1769. [Google Scholar] [CrossRef] [PubMed]
- Raffoux, E.; Rousselot, P.; Poupon, J.; Daniel, M.T.; Cassinat, B.; Delarue, E.; Taksin, A.L.; Réa, D.; Buzyn, A.; Tibi, A.; et al. Combined treatment with arsenic trioxide and all-trans-retinoic acid in patients with relapsed acute promyelocytic leukemia. J. Clin. Oncol. 2003, 21, 2326–2334. [Google Scholar] [CrossRef]
- Shen, Z.Y.; Shen, J.; Cai, W.J.; Hong, C.; Zheng, M.H. The alteration of mitochondria is an early event of arsenic trioxide induced apoptosis in esophageal carcinoma cells. Int. J. Mol. Med. 2000, 5, 155–158. [Google Scholar] [CrossRef]
- Park, W.H.; Seol, J.G.; Kim, E.S.; Hyun, J.M.; Jung, C.W.; Lee, C.C.; Kim, B.K.; Lee, Y.Y. Arsenic trioxide-mediated growth inhibition in MC/CAR myeloma cells via cell cycle arrest in association with induction of cyclindependent kinase inhibitor, p21, and apoptosis. Cancer Res. 2000, 60, 3065–3071. [Google Scholar]
- Zhou, Y.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003, 101, 4098–4104. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Minami, Y.; Yamamoto, K.; Akao, Y.; Kiyoi, H.; Saito, H.; Naoe, T. Involvement of CD95-independent caspase 8 activation in arsenic trioxide-induced apoptosis. Leukemia 2000, 14, 1743–1750. [Google Scholar] [CrossRef]
- Scholz, C.; Richter, A.; Lehmann, M.; Schulze-Osthoff, K.; Dörken, B.; Daniel, P.T. Arsenic trioxide induces regulated, death receptor-independent cell death through a Bcl-2-controlled pathway. Oncogene 2005, 24, 7031–7042. [Google Scholar] [CrossRef]
- Scholz, C.; Wieder, T.; Starck, L.; Essmann, F.; Schulze-Osthoff, K.; Dörken, B.; Daniel, P.T. Arsenic trioxide triggers a regulated form of caspase-independent necrotic cell death via the mitochondrial death pathway. Oncogene 2005, 24, 1904–1913. [Google Scholar] [CrossRef]
- Liu, Q.; Hilsenbeck, S.; Gazitt, Y. Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood 2003, 101, 4078–4087. [Google Scholar] [CrossRef] [PubMed]
- Merkel, O.; Heyder, C.; Asslaber, D.; Hamacher, F.; Tinhofer, I.; Holler, C.; Stöcher, M.; Prokesch, A.; Papak, C.; Scheideler, M.; et al. Arsenic trioxide induces apoptosis preferentially in B-CLL cells of patients with unfavourable prognostic factors including del17p13. J. Mol. Med. 2008, 86, 541–552. [Google Scholar] [CrossRef]
- Fayad, L.; Keating, M.J.; Reuben, J.M.; O’Brien, S.; Lee, B.N.; Lerner, S.; Kurzrock, R. Interleukin-6 and interleukin-10 levels in chronic lymphocytic leukemia: Correlation with phenotypic characteristics and outcome. Blood 2001, 97, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Kohda, H.; Kawaguchi, T.; Ohhira, M.; Sekiya, C.; Namiki, M.; Takeyasu, A.; Taniguchi, N. Induction of Mn-superoxide dismutase by tumor necrosis factor, interleukin-1 and interleukin-6 in human hepatoma cells. Biochem. Biophys. Res. Commun. 1992, 182, 1100–1107. [Google Scholar] [CrossRef]
- Cooper, K.L.; Liu, K.J.; Hudson, L.G. Contributions of reactive oxygen species and mitogen-activated protein kinase signaling in arsenite-stimulated hemeoxygenase-1 production. Toxicol. Appl. Pharmacol. 2007, 218, 119–127. [Google Scholar] [CrossRef]
- Was, H.; Dulak, J.; Jozkowicz, A. Heme oxygenase-1 in tumor biology and therapy. Curr. Drug Targets. 2010, 11, 1551–1570. [Google Scholar] [CrossRef] [PubMed]
- Menzel, D.B.; Rasmussen, R.E.; Lee, E.; Meacher, D.M.; Said, B.; Hamadeh, H.; Vargas, M.; Greene, H.; Roth, R.N. Human lymphocyte heme oxygenase 1 as a response biomarker to inorganic arsenic. Biochem. Biophys. Res. Commun. 1998, 250, 653–656. [Google Scholar] [CrossRef]
- Bourdonnay, E.; Morzadec, C.; Fardel, O.; Vernhet, L. Redox-sensitive regulation of gene expression in human primary macrophages exposed to inorganic arsenic. J. Cell Biochem. 2009, 107, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Akagi, R.; Takahashi, T.; Sassa, S. Cytoprotective effects of heme oxygenase in acute renal failure. Contrib. Nephrol. 2005, 148, 70–85. [Google Scholar] [PubMed]
- Yu, H.P.; Choudhry, M.A.; Shimizu, T.; Hsieh, Y.C.; Schwacha, M.G.; Yang, S.; Chaudry, I.H. Mechanism of the salutary effects of flutamide on intestinal myeloperoxidase activity following trauma-hemorrhage: Up-regulation of estrogen receptor-{beta}-dependent HO-1. J. Leukoc. Biol. 2006, 79, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Amigo-Jiménez, I.; Bailón, E.; Aguilera-Montilla, N.; García-Marco, J.A.; García-Pardo, A. Gene expression profile induced by arsenic trioxide in chronic lymphocytic leukemia cells reveals a central role for heme oxygenase-1 in apoptosis and regulation of matrix metalloproteinase-9. Oncotarget 2016, 7, 83359–83377. [Google Scholar] [CrossRef] [PubMed][Green Version]
- ‘Plenty of room’ revisited. Nat. Nanotechnol. 2009, 4, 781. [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 23. [Google Scholar] [CrossRef]
- Singh, A.P.; Biswas, A.; Shukla, A.; Maiti, P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal. Transduct. Target. Ther. 2019, 4, 33. [Google Scholar] [CrossRef]
- Chasara, R.S.; Ajayi, T.O.; Leshilo, D.M.; Poka, M.S.; Witika, B.A. Exploring novel strategies to improve anti-tumour efficiency: The potential for targeting reactive oxygen species. Heliyon 2023, 9, e19896. [Google Scholar] [CrossRef]
- Allegra, A.; Gioacchino, M.D.; Tonacci, A.; Petrarca, C.; Gangemi, S. Nanomedicine for Immunotherapy Targeting Hematological Malignancies: Current Approaches and Perspective. Nanomaterials 2021, 11, 2792. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Penna, G.; Alonci, A.; Rizzo, V.; Russo, S.; Musolino, C. Nanoparticles in oncology: The new theragnostic molecules. Anticancer Agents Med. Chem. 2011, 11, 669–686. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, J.; Sun, X. Reactive oxygen species-based nanomaterials for cancer therapy. Front. Chem. 2021, 9, 650587. [Google Scholar] [CrossRef]
- Koziner, B. Potential therapeutic applications of oblimersen in CLL. Oncology 2004, 18, 32–38. [Google Scholar]
- Yu, B.; Mao, Y.; Bai, L.Y.; Herman, S.E.; Wang, X.; Ramanunni, A.; Jin, Y.; Mo, X.; Cheney, C.; Chan, K.K.; et al. Targeted nanoparticle delivery overcomes off-target immunostimulatory effects of oligonucleotides and improves therapeutic efficacy in chronic lymphocytic leukemia. Blood 2013, 121, 136–147. [Google Scholar] [CrossRef][Green Version]
- Chiang, C.L.; Goswami, S.; Frissora, F.W.; Xie, Z.; Yan, P.S.; Bundschuh, R.; Walker, L.A.; Huang, X.; Mani, R.; Mo, X.M.; et al. ROR1-targeted delivery of miR-29b induces cell cycle arrest and therapeutic benefit in vivo in a CLL mouse model. Blood 2019, 134, 432–444. [Google Scholar] [CrossRef]
- Luo, Z.T.; Zheng, K.Y.; Xie, J.P. Engineering ultrasmall water soluble gold and silver nanoclusters for biomedical applications. Chem. Commun. 2014, 50, 5143–5155. [Google Scholar] [CrossRef]
- Li, J.G.; Zhao, T.T.; Chen, T.K.; Liu, Y.B.; Ong, C.N.; Xie, J.P. Engineering noble metal nanomaterials for environmental applications. Nanoscale 2015, 7, 7502–7519. [Google Scholar] [CrossRef]
- Yuan, Q.; Wang, Y.; Zhao, L.; Liu, R.; Gao, F.; Gao, L.; Gao, X. Peptide protected gold clusters: Chemical synthesis and biomedical applications. Nanoscale 2016, 8, 12095–12104. [Google Scholar] [CrossRef]
- Yao, Y.; Lu, C.; Gao, L.; Cao, K.; Yuan, H.; Zhang, X.; Gao, X.; Yuan, Q. Gold Cluster Capped with a BCL-2 Antagonistic Peptide Exerts Synergistic Antitumor Activity in Chronic Lymphocytic Leukemia Cells. ACS Appl. Mater. Interfaces 2021, 13, 21108–21118. [Google Scholar] [CrossRef]
- Maniaci, C.; Hughes, S.J.; Testa, A.; Chen, W.; Lamont, D.J.; Rocha, S.; Alessi, D.R.; Romeo, R.; Ciulli, A. Homo-PROTACs: Bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 2017, 8, 830. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Paiva, S.L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Fu, L. Small-molecule PROTACs: An Emerging and Promising Approach for the Development of Targeted Therapy Drugs. eBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef]
- Li, X.; Yao, Y.; Wu, F.; Song, Y. A proteolysis-targeting chimera molecule selectively degrades ENL and inhibits malignant gene expression and tumor growth. J. Hematol. Oncol. 2022, 15, 41. [Google Scholar] [CrossRef]
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.A.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homo-log-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306.e9. [Google Scholar] [CrossRef] [PubMed]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.-H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Woyach, J.A.; Bojnik, E.; Ruppert, A.S.; Stefanovski, M.R.; Goettl, V.M.; Smucker, K.A.; Smith, L.L.; Dubovsky, J.A.; Towns, W.H.; MacMurray, J.; et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood 2014, 123, 1207–1213. [Google Scholar] [CrossRef]
- Burger, J.A. BTK Inhibitors: Present and Future. Cancer J. 2019, 25, 386–393. [Google Scholar] [CrossRef]
- Arthur, R.; Valle-Argos, B.; Steele, A.J.; Packham, G. Development of PROTACs to address clinical limitations associated with BTK-targeted kinase inhibitors. Explor. Target. Anti-Tumor Ther. 2020, 1, 131–152. [Google Scholar] [CrossRef] [PubMed]
- Jaime-Figueroa, S.; Buhimschi, A.D.; Toure, M.; Hines, J.; Crews, C.M. Design, synthesis and biological evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2020, 30, 126877. [Google Scholar] [CrossRef] [PubMed]
- Anwar, Z.; Ali, M.S.; Galvano, A.; Perez, A.; La Mantia, M.; Bukhari, I.; Swiatczak, B. Protacs: The future of leukemia therapeutics. Front. Cell Dev. Biol. 2022, 10, 851087. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, R.; Shraga, A.; Gehrtz, P.; Livnah, E.; Shorer, Y.; Gurwicz, N.; Avram, L.; Unger, T.; Aharoni, H.; Albeck, S. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J. Am. Chem. Soc. 2020, 142, 11734–11742. [Google Scholar] [CrossRef] [PubMed]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, X.; Ding, N.; Gao, H.; Wu, Y.; Yang, Y.; Zhao, M.; Hwang, J.; Song, Y.; Liu, W.; et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Smith, B.E.; Burslem, G.M.; Buhimschi, A.D.; Hines, J.; Jaime-Figueroa, S.; Wang, J.; Hamman, B.D.; Ishchenko, A.; Crews, C.M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018, 25, 78–87.e5. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.T.; Dobrovolsky, D.; Paulk, J.; Yang, G.; Weisberg, E.L.; Doctor, Z.M.; Buckley, D.L.; Cho, J.H.; Ko, E.; Jang, J.; et al. A chemo-proteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 2018, 25, 88–99.e6. [Google Scholar] [CrossRef]




| Types of Recurrent Gene Alterations | Genes |
|---|---|
| Signaling pathways of B-cell receptor and Toll-like receptor | PAX5, MYD88, BCOR, IKZF3 |
| Splicing of RNA and metabolic processes | U1, DDX3X, RPS15, SF3B1 |
| Cell cycle regulators | CDKN1B, CDKN2A |
| MAPK-ERK | BRAF, NRAS, KRAS |
| Changes in chromatin structure | SETD2, KMT2D, CHD2, ASXL1 |
| NF-KB pathway | TRAF2, TRAF3, BIRC3 |
| Criteria for Initiating Treatment: “Active Disease” |
|---|
| Indications indicate gradual decline in bone marrow function, characterized by the occurrence or deterioration of anemia and/or thrombocytopenia. When the amount of Hb is less than 10 g/dL or the platelet count is less than 100 × 109/L, it is typically seen as an indication for therapy. Nevertheless, in certain individuals, platelet counts below 100 × 109/L might persist without significant changes for an extended duration, and in such cases, there is no immediate need for therapeutic action. |
| Splenomegaly that is either large (≥6 cm below the left costal border), progressing, or causing symptoms. |
| Lymph nodes that are large (≥10 cm in longest diameter) or lymphadenopathy that is worsening or causing symptoms. |
| Increasing lymphocytosis is characterized by a significant rise of at least 50% during a period of two months, or a lymphocyte doubling time (LDT) of less than six months. The LDT may be calculated using linear regression extrapolation of absolute lymphocyte counts recorded every 2 weeks during a 2–3 month observation period. However, patients with baseline blood lymphocyte counts below 30 × 109/L may need a longer observation time to accurately establish the LDT. When diagnosing lymphocytosis, it is important to rule out factors other than CLL, such as infections or the use of steroids. |
| Anemia and thrombocytopenia are examples of autoimmune complications that exhibit a diminished response to corticosteroids. |
| Extranodal involvement, whether symptomatic or functional, can occur in several parts of the body such as the skin, kidney, lung, and spine. |
| Signs and symptoms associated with the disease, as defined by any of the following sources: Unintentional weight loss of at least 10% within the preceding six months, significant fatigue (defined as ECOG performance scale 2 or worse; inability to work or execute typical activities), fevers of at least 100.5 °F or 38.0 °C for a period of two weeks or longer without any indication of infection, and night sweats for a period of at least one month without any indication of infection are all indicators of a possible infection. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sciaccotta, R.; Gangemi, S.; Penna, G.; Giordano, L.; Pioggia, G.; Allegra, A. Potential New Therapies “ROS-Based” in CLL: An Innovative Paradigm in the Induction of Tumor Cell Apoptosis. Antioxidants 2024, 13, 475. https://doi.org/10.3390/antiox13040475
Sciaccotta R, Gangemi S, Penna G, Giordano L, Pioggia G, Allegra A. Potential New Therapies “ROS-Based” in CLL: An Innovative Paradigm in the Induction of Tumor Cell Apoptosis. Antioxidants. 2024; 13(4):475. https://doi.org/10.3390/antiox13040475
Chicago/Turabian StyleSciaccotta, Raffaele, Sebastiano Gangemi, Giuseppa Penna, Laura Giordano, Giovanni Pioggia, and Alessandro Allegra. 2024. "Potential New Therapies “ROS-Based” in CLL: An Innovative Paradigm in the Induction of Tumor Cell Apoptosis" Antioxidants 13, no. 4: 475. https://doi.org/10.3390/antiox13040475
APA StyleSciaccotta, R., Gangemi, S., Penna, G., Giordano, L., Pioggia, G., & Allegra, A. (2024). Potential New Therapies “ROS-Based” in CLL: An Innovative Paradigm in the Induction of Tumor Cell Apoptosis. Antioxidants, 13(4), 475. https://doi.org/10.3390/antiox13040475