
Specific NOX4 Inhibition Preserves Mitochondrial Function and Dampens Kidney Dysfunction Following Ischemia–Reperfusion-Induced Kidney Injury
 , , and
, , and 
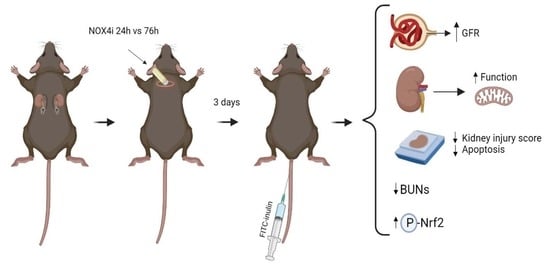
Abstract
Highlights
- NADPH oxidases (NOXs) are induced following ischemia-reperfusion (IR), which aggravates acute kidney injury
- NOX4 inhibition improves kidney and mitochondria function in IR, and dampens renal injury
- These findings can facilitate development of new preventive strategies of IR-induced AKI, which is a global medical problem.
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Ethics Statement
2.2. NOX4 Inhibitor
2.3. Animals and Renal Ischemia–Reperfusion Model
2.4. Glomerular Filtration Rate
2.5. Histological Evaluation
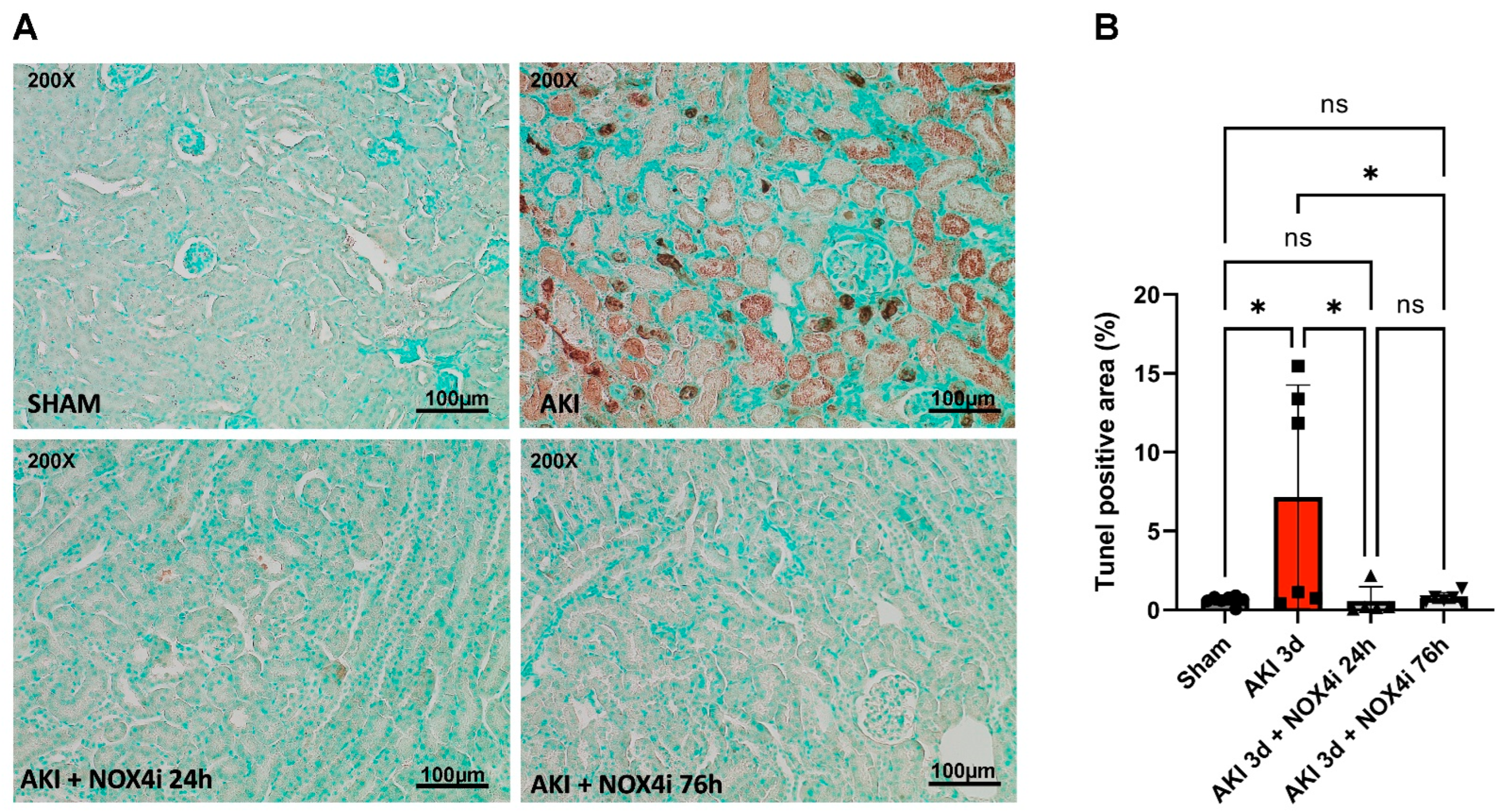
2.6. Evaluation of Apoptosis in Kidney Tissue
2.7. Mitochondrial Isolation
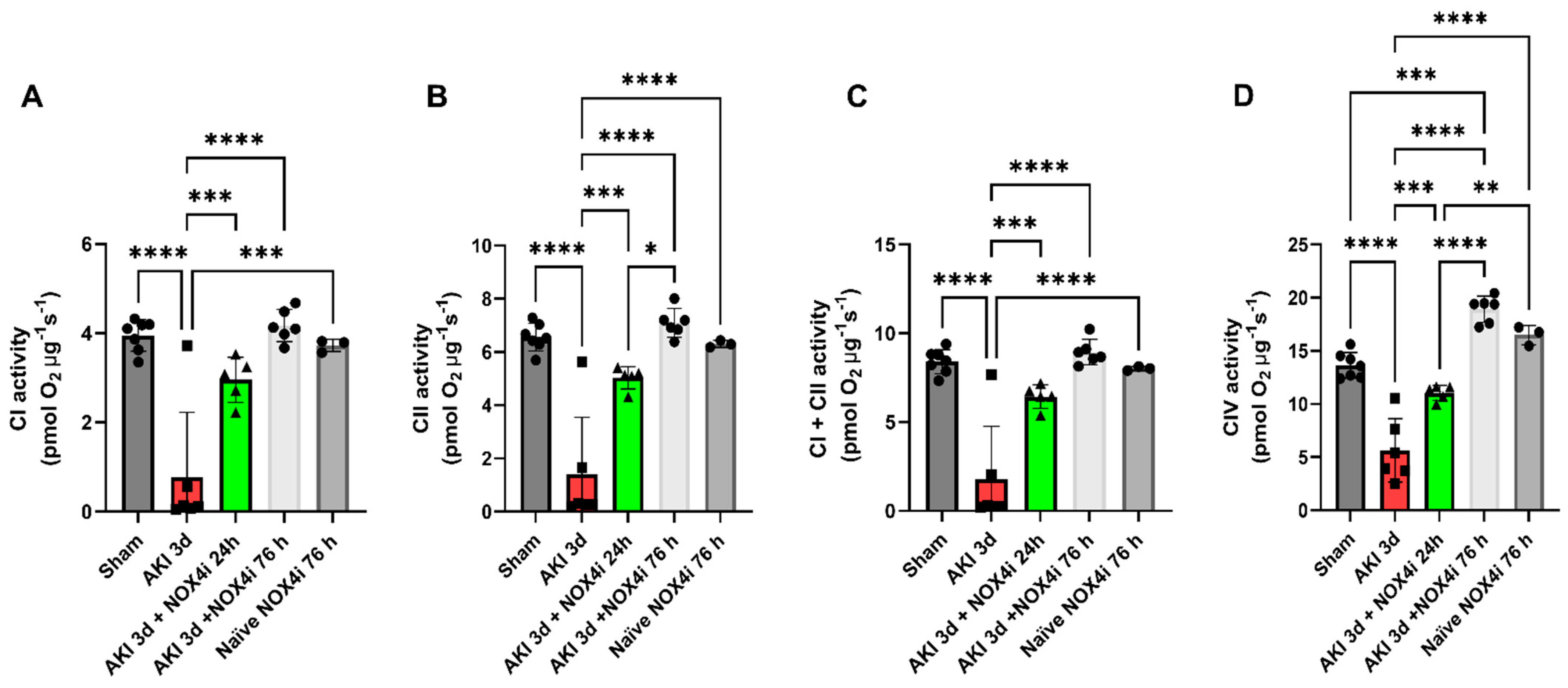
2.8. Mitochondrial Function
2.9. Kidney Tissue Citrate Synthase Activity
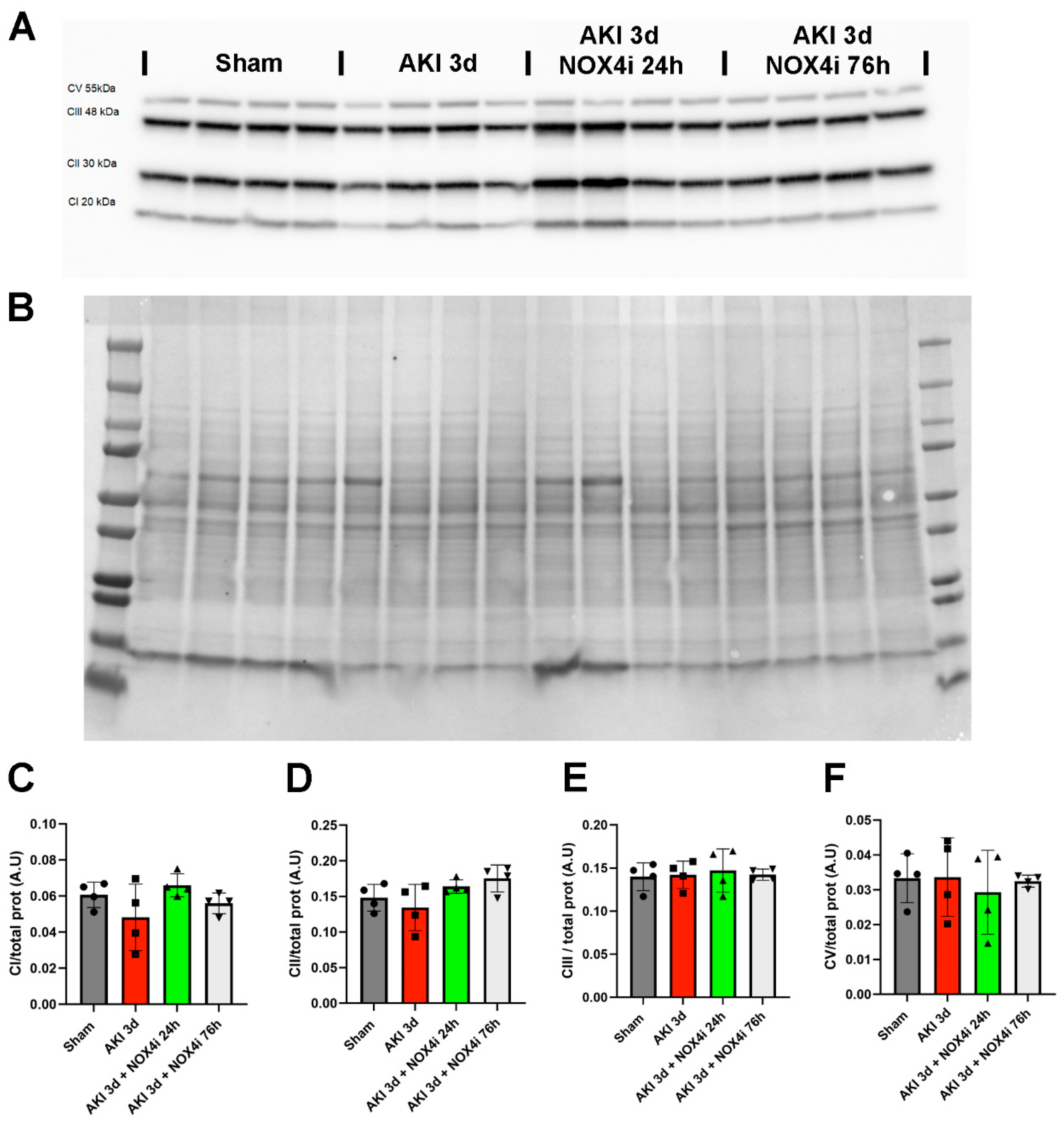
2.10. Immunoblotting
2.11. Cell Culture
2.12. Hypoxia Reoxygenation Protocol in HK-2 Cells
2.13. Mitochondrial ROS Production in HK-2 Cells
2.14. NOX Activity in HK-2 Cells
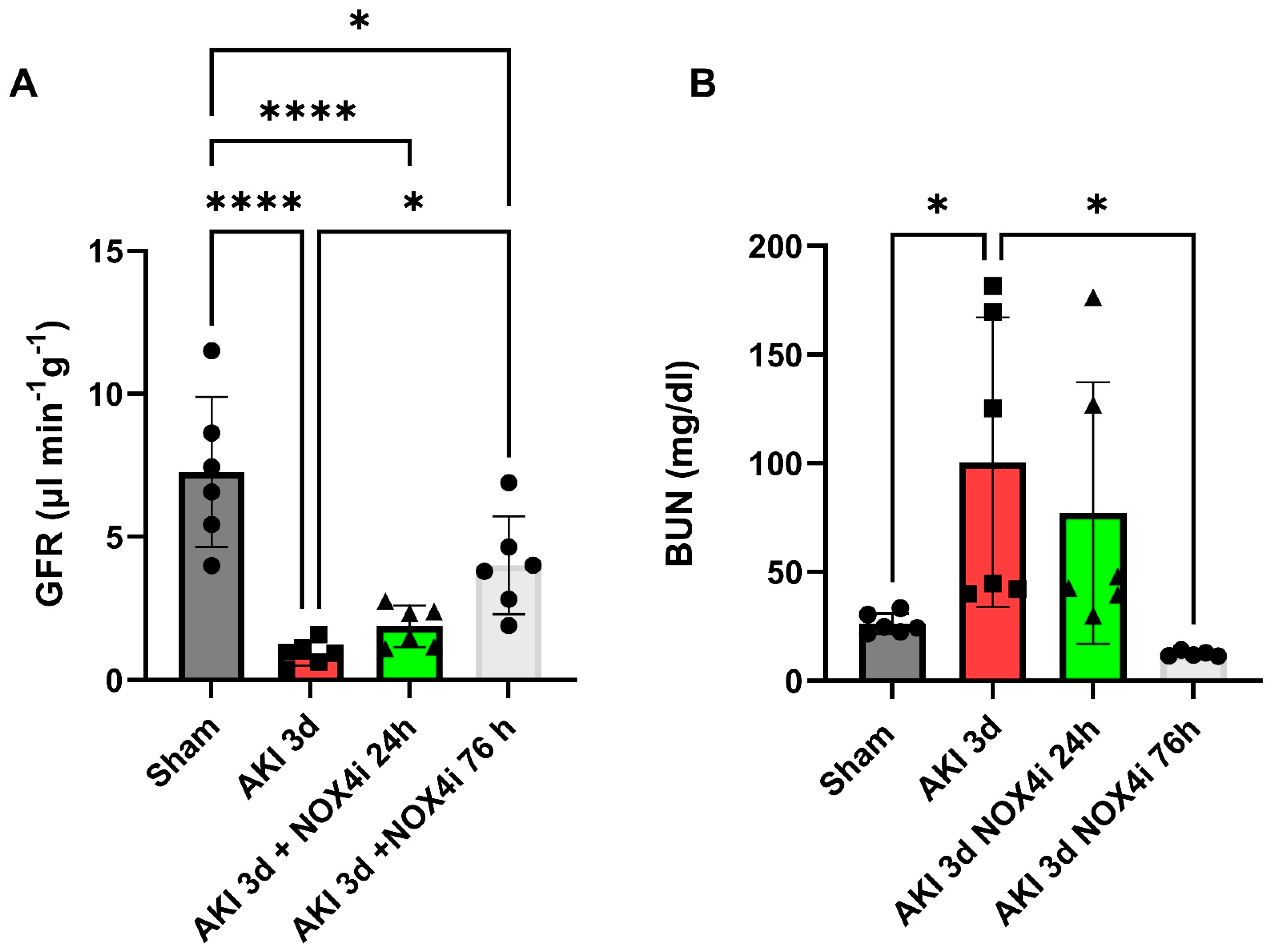
2.15. Blood Urea Nitrogen
2.16. Statistical Analysis
3. Results
3.1. Kidney Function
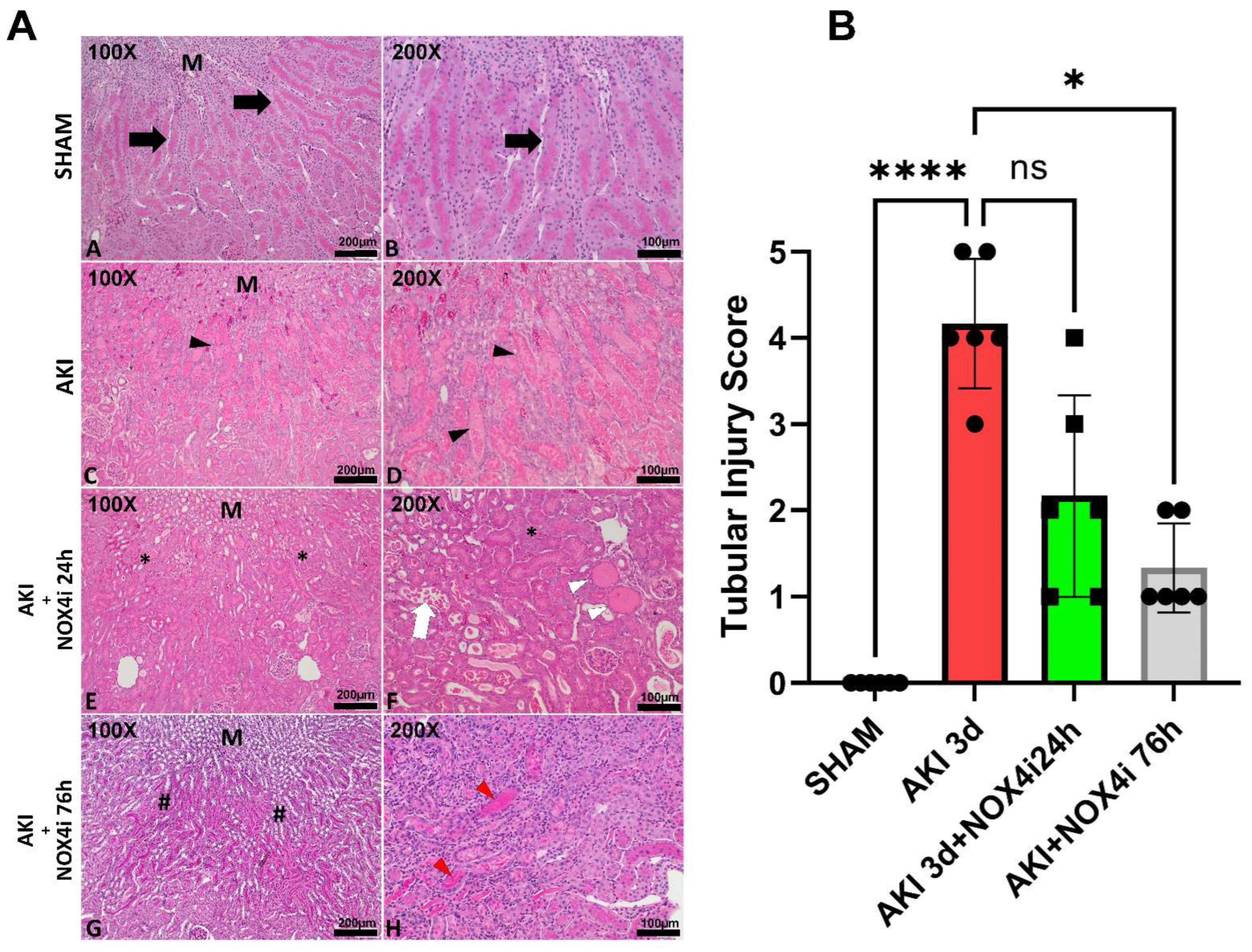
3.2. Histopathological Evaluation
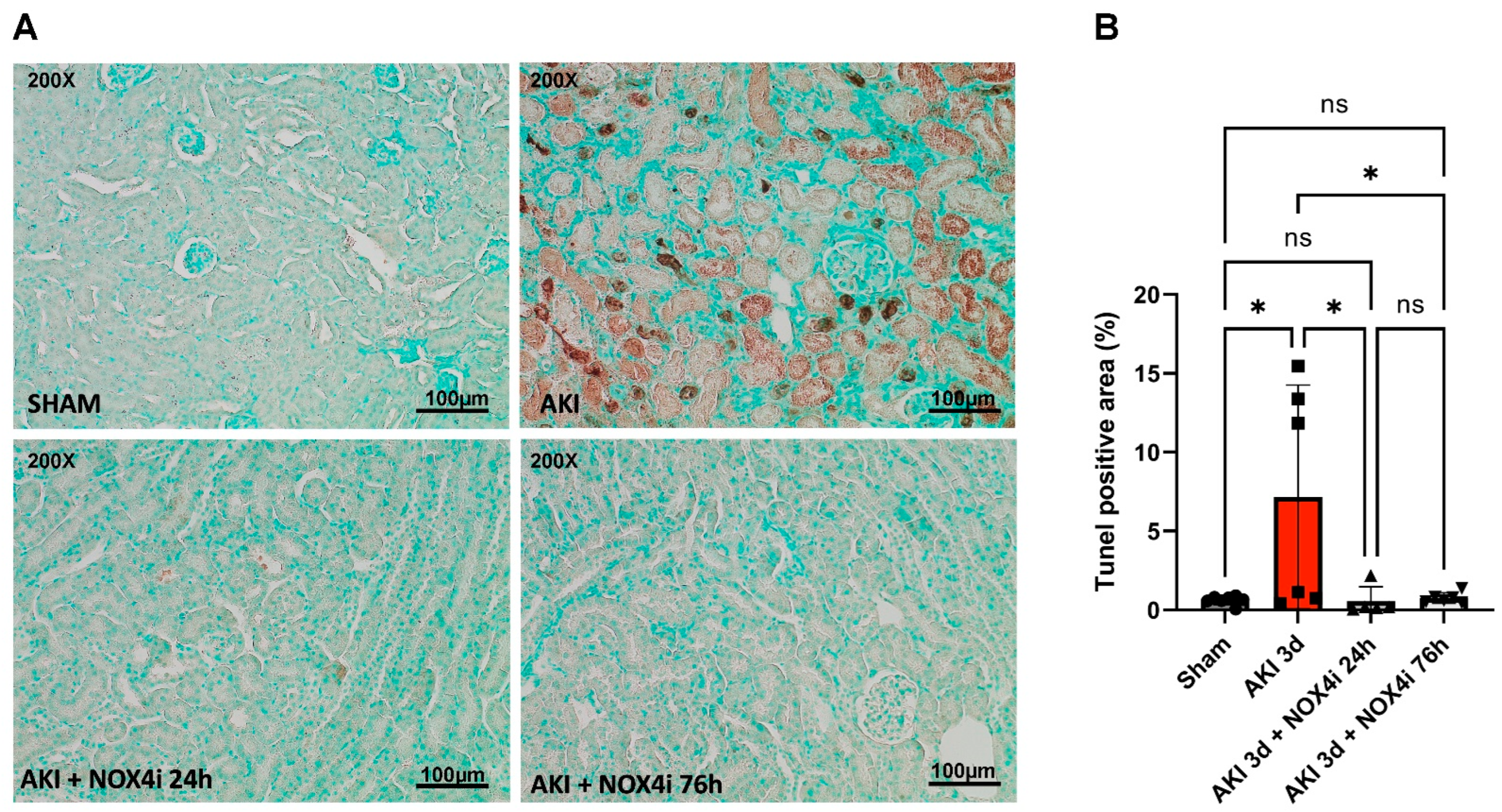
3.3. Evaluation of Apoptotic Tissue
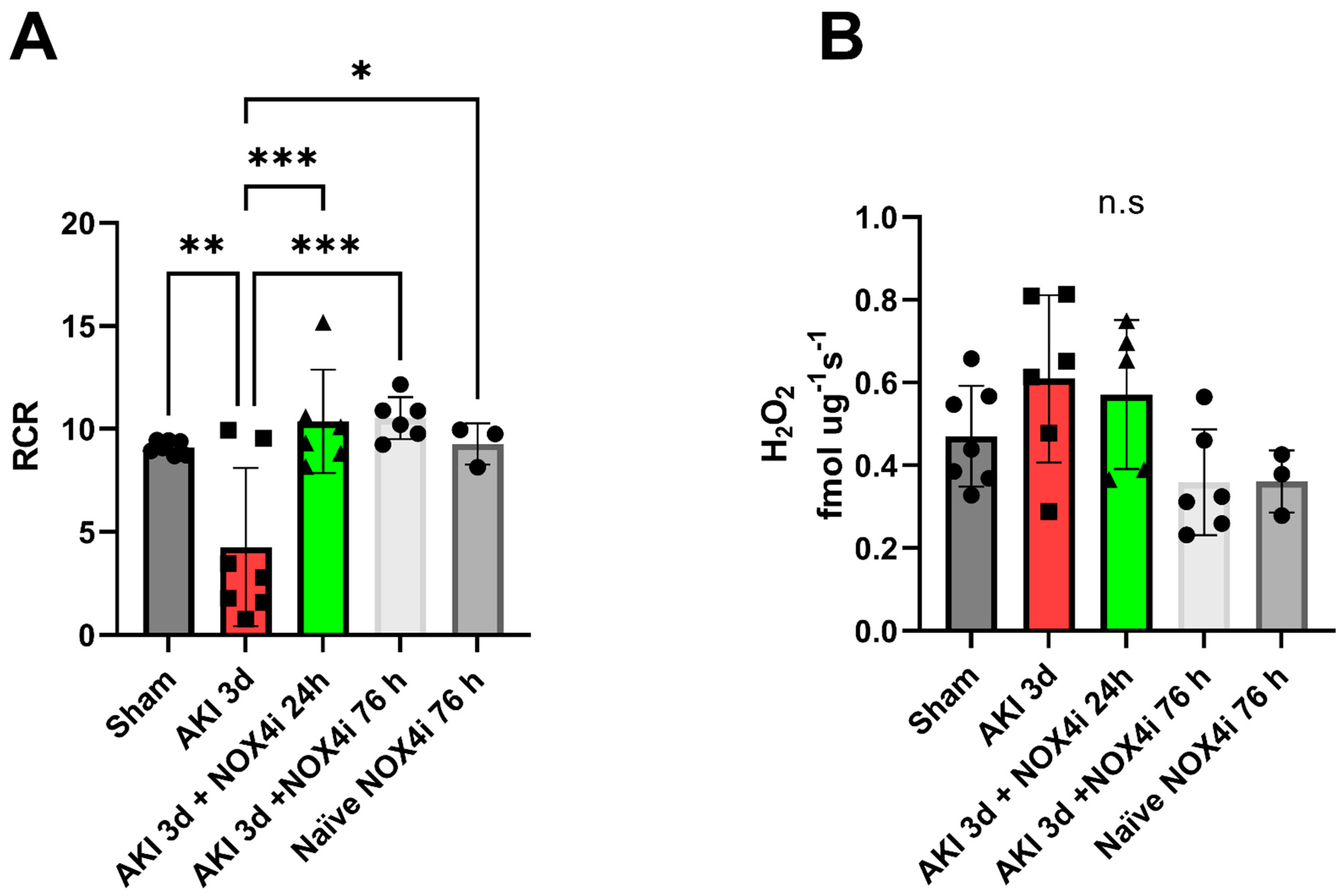
3.4. Mitochondrial Function
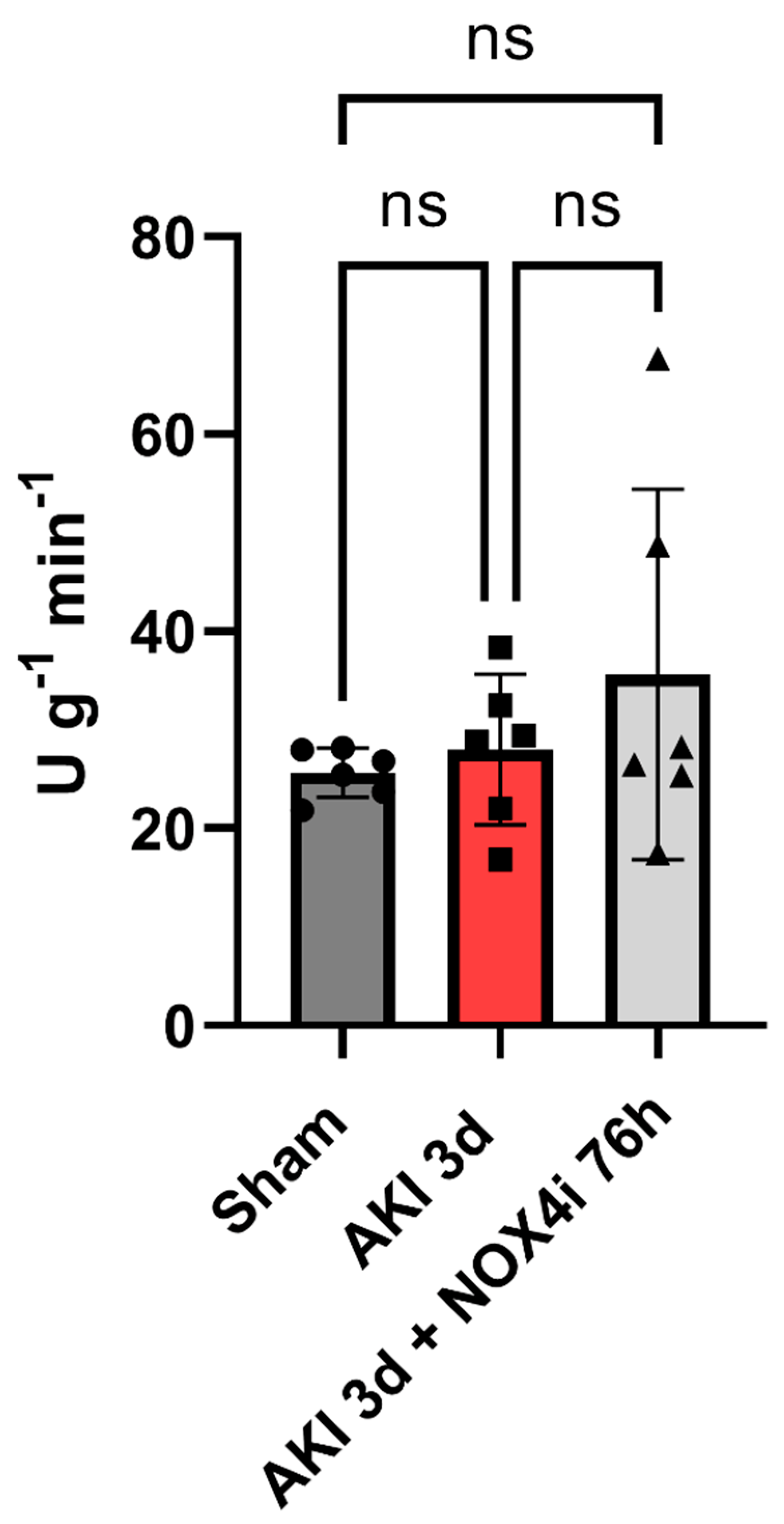
3.5. Tissue Citrate Synthase Activity
3.6. Protein Levels of Components of the Mitochondrial Respiratory Chain
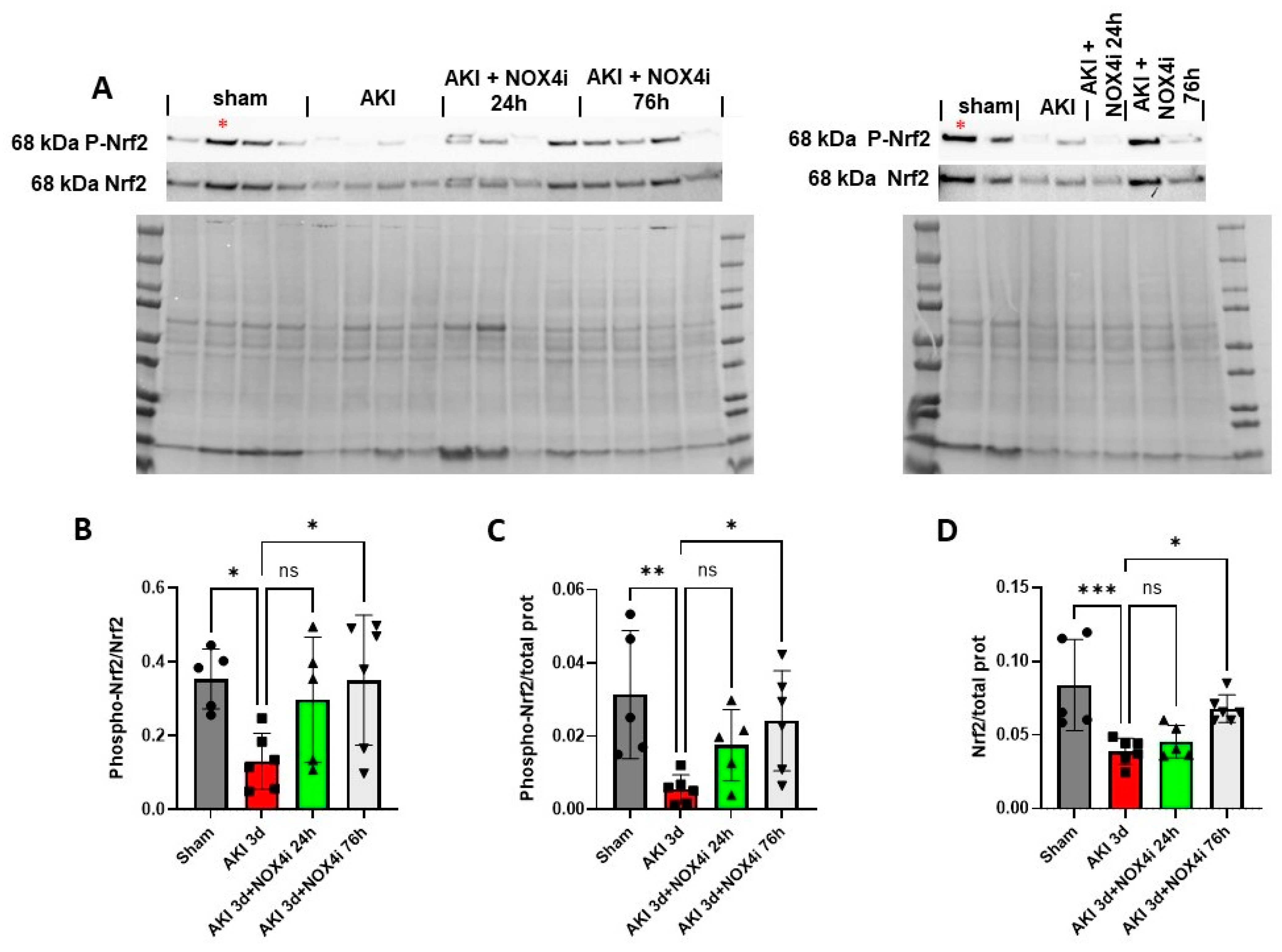
3.7. Nrf2 Serine Residue 40 Phosphorylation
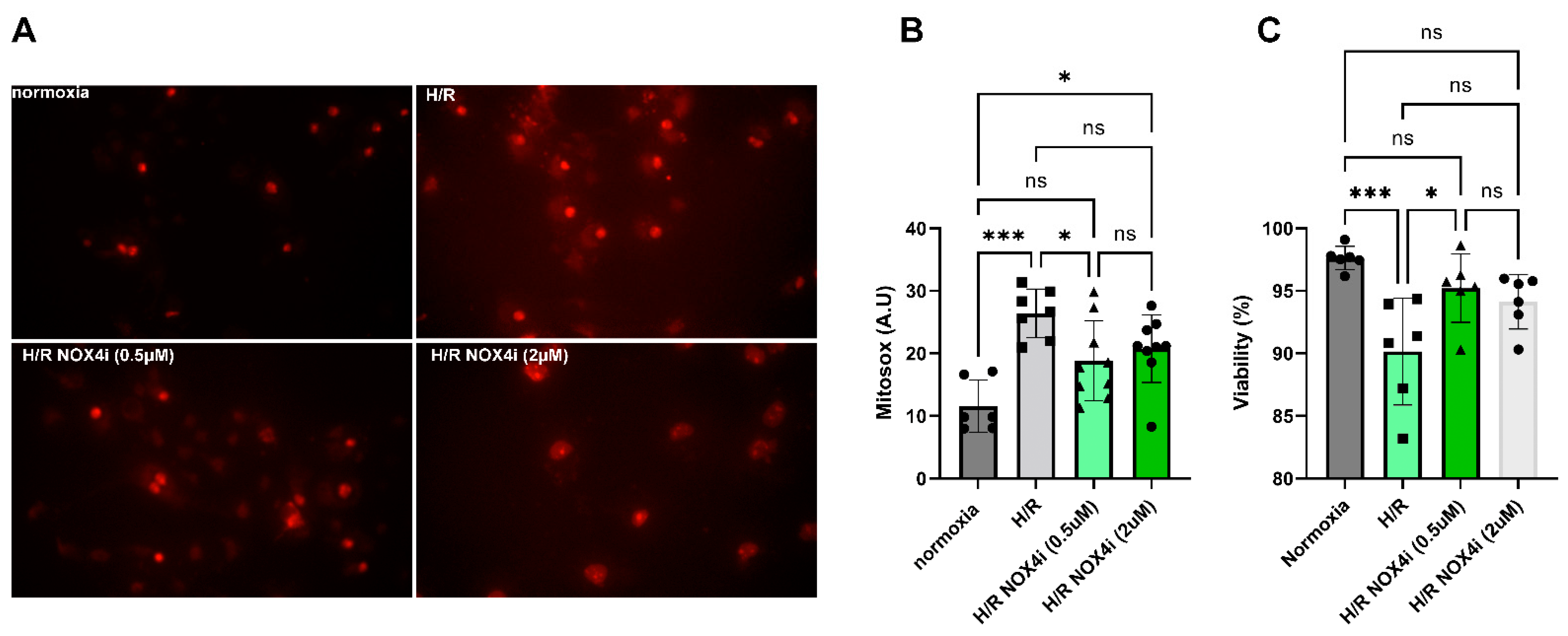
3.8. Mitochondrial ROS Production in HK-2 Cells Exposed to Hypoxia Reoxygenation
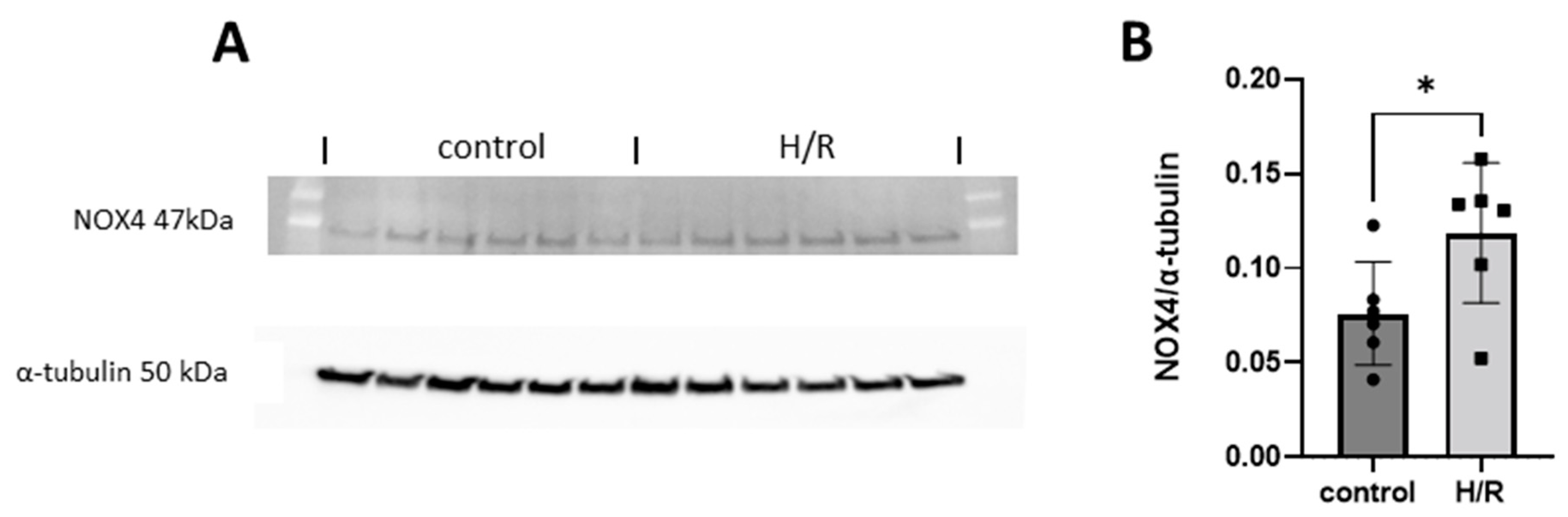
3.9. NOX4 Expression in HK-2 Cells Exposed to Hypoxia Reoxygenation
3.10. The Relative Contribution of H2O2 Production from NOX4 in HK-2 Cells Exposed to Hypoxia Reoxygenation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lewington, A.J.; Cerda, J.; Mehta, R.L. Raising awareness of acute kidney injury: A global perspective of a silent killer. Kidney Int. 2013, 84, 457–467. [Google Scholar] [CrossRef]
- Kellum, J.A.; Romagnani, P.; Ashuntantang, G.; Ronco, C.; Zarbock, A.; Anders, H.-J. Acute kidney injury. Nat. Rev. Dis. Primers 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef]
- Martin-Cleary, C.; Molinero-Casares, L.M.; Ortiz, A.; Arce-Obieta, J.M. Development and internal validation of a prediction model for hospital-acquired acute kidney injury. Clin. Kidney J. 2021, 14, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.J.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerda, J.; Chawla, L.S. Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Kounatidis, D.; Vallianou, N.G.; Psallida, S.; Panagopoulos, F.; Margellou, E.; Tsilingiris, D.; Karampela, I.; Stratigou, T.; Dalamaga, M. Sepsis-Associated Acute Kidney Injury: Where Are We Now? Medicina 2024, 60, 434. [Google Scholar] [CrossRef]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Sadoshima, J. Yin and Yang of NADPH Oxidases in Myocardial Ischemia-Reperfusion. Antioxidants 2022, 11, 1069. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Maulik, G.; Cordis, G.A.; Das, D.K. Oxidative damage to myocardial proteins and DNA during ischemia and reperfusion. Ann. N. Y. Acad. Sci. 1996, 793, 431–436. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Kezic, A.; Stajic, N.; Thaiss, F. Innate Immune Response in Kidney Ischemia/Reperfusion Injury: Potential Target for Therapy. J. Immunol. Res. 2017, 2017, 6305439. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Forro, L.; Schlegel, W.; Krause, K.H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef]
- Koziel, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen-Durr, P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013, 452, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Logsdon, N.J.; Miguel, V.; Benavides, G.A.; Zhang, J.; Carter, A.B.; Darley-Usmar, V.M.; Thannickal, V.J. NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-derived 2-like 2 (Nrf2)-dependent Pathway. J. Biol. Chem. 2017, 292, 3029–3038. [Google Scholar] [CrossRef]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Ikeda, Y.; Oka, S.; Fong, G.H.; Tian, R.; Sadoshima, J. Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1alpha and upregulation of peroxisome proliferator-activated receptor-alpha. Circ. Res. 2013, 112, 1135–1149. [Google Scholar] [CrossRef]
- Matsushima, S.; Tsutsui, H.; Sadoshima, J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends Cardiovasc. Med. 2014, 24, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.L.; Lotz, C.; Ping, P.; Cai, H. Netrin-1 abrogates ischemia/reperfusion-induced cardiac mitochondrial dysfunction via nitric oxide-dependent attenuation of NOX4 activation and recoupling of NOS. J. Mol. Cell. Cardiol. 2015, 78, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, F.L.M.; Walum, E.; Wikstrom, P.; Arner, A. A small molecule inhibitor of Nox2 and Nox4 improves contractile function after ischemia-reperfusion in the mouse heart. Sci. Rep. 2021, 11, 11970. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef] [PubMed]
- Casas, A.I.; Geuss, E.; Kleikers, P.W.M.; Mencl, S.; Herrmann, A.M.; Buendia, I.; Egea, J.; Meuth, S.G.; Lopez, M.G.; Kleinschnitz, C.; et al. NOX4-dependent neuronal autotoxicity and BBB breakdown explain the superior sensitivity of the brain to ischemic damage. Proc. Natl. Acad. Sci. USA 2017, 114, 12315–12320. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, L.; Wang, B.; Zhang, Z.; Jiang, L.; Qin, Z.; Zhao, Y.; Su, B. NOX4 is a potential therapeutic target in septic acute kidney injury by inhibiting mitochondrial dysfunction and inflammation. Theranostics 2023, 13, 2863–2878. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Lee, C.F.; Wang, W.; Karamanlidis, G.; Kuroda, J.; Matsushima, S.; Sadoshima, J.; Tian, R. Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury. J. Am. Heart Assoc. 2014, 3, e000555. [Google Scholar] [CrossRef]
- Nlandu-Khodo, S.; Dissard, R.; Hasler, U.; Schafer, M.; Pircher, H.; Jansen-Durr, P.; Krause, K.H.; Martin, P.Y.; de Seigneux, S. NADPH oxidase 4 deficiency increases tubular cell death during acute ischemic reperfusion injury. Sci. Rep. 2016, 6, 38598. [Google Scholar] [CrossRef]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef]
- Wang, X.; Elksnis, A.; Wikstrom, P.; Walum, E.; Welsh, N.; Carlsson, P.O. The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro. PLoS ONE 2018, 13, e0204271. [Google Scholar] [CrossRef] [PubMed]
- Carlstrom, M.; Rannier Ribeiro Antonino Carvalho, L.; Guimaraes, D.; Boeder, A.; Schiffer, T.A. Dimethyl malonate preserves renal and mitochondrial functions following ischemia-reperfusion via inhibition of succinate dehydrogenase. Redox Biol. 2023, 69, 102984. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsson, J.; Weiner, D. Non-compartmental Analysis. In Pharmacokinetic and Pharmacodynamic Data Analysis; Concepts and Applications; Swedish Pharmaceutical Press: Stockholm, Sweden, 2006; pp. 161–180. [Google Scholar]
- Dong, Y.; Zhang, Q.; Wen, J.; Chen, T.; He, L.; Wang, Y.; Yin, J.; Wu, R.; Xue, R.; Li, S.; et al. Ischemic Duration and Frequency Determines AKI-to-CKD Progression Monitored by Dynamic Changes of Tubular Biomarkers in IRI Mice. Front. Physiol. 2019, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, T.A.; Gustafsson, H.; Palm, F. Kidney outer medulla mitochondria are more efficient compared with cortex mitochondria as a strategy to sustain ATP production in a suboptimal environment. Am. J. Physiol. Renal Physiol. 2018, 315, F677–F681. [Google Scholar] [CrossRef] [PubMed]
- Iommarini, L.; Ghelli, A.; Leone, G.; Tropeano, C.V.; Kurelac, I.; Amato, L.B.; Gasparre, G.; Porcelli, A.M. Mild phenotypes and proper supercomplex assembly in human cells carrying the homoplasmic m.15557G > A mutation in cytochrome b gene. Hum. Mutat. 2018, 39, 92–102. [Google Scholar] [CrossRef]
- Tang, T.T.; Lv, L.L.; Pan, M.M.; Wen, Y.; Wang, B.; Li, Z.L.; Wu, M.; Wang, F.M.; Crowley, S.D.; Liu, B.C. Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis. 2018, 9, 351. [Google Scholar] [CrossRef]
- Havasi, A.; Borkan, S.C. Apoptosis and acute kidney injury. Kidney Int. 2011, 80, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88 Pt B, 179–188. [Google Scholar] [CrossRef]
- Merry, T.L.; Ristow, M. Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J. Physiol. 2016, 594, 5195–5207. [Google Scholar] [CrossRef]
- Elksnis, A.; Cen, J.; Wikstrom, P.; Carlsson, P.O.; Welsh, N. Pharmacological Inhibition of NOX4 Improves Mitochondrial Function and Survival in Human Beta-Cells. Biomedicines 2021, 9, 1865. [Google Scholar] [CrossRef]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef] [PubMed]
- Granado, R.C.; Neyra, J.A.; Basu, R.K. Acute Kidney Injury: Gaps and Opportunities for Knowledge and Growth. Semin. Nephrol. 2023, 43, 151439. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, T.A.; Lundberg, J.O.; Weitzberg, E.; Carlstrom, M. Modulation of mitochondria and NADPH oxidase function by the nitrate-nitrite-NO pathway in metabolic disease with focus on type 2 diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165811. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, A.; Viviano, M.; Feoli, A.; Milite, C.; Sarno, G.; Castellano, S.; Sbardella, G. NADPH Oxidases: From Molecular Mechanisms to Current Inhibitors. J. Med. Chem. 2023, 66, 11632–11655. [Google Scholar] [CrossRef] [PubMed]
- Molitoris, B.A. Therapeutic translation in acute kidney injury: The epithelial/endothelial axis. J. Clin. Investig. 2014, 124, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V. Tubular Transport in Acute Kidney Injury: Relevance for Diagnosis, Prognosis and Intervention. Nephron 2016, 134, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Xiong, Y.; Lei, Y.; Huang, Q.; Liu, H.; Zhao, X.; Chen, Z.; Chen, H.; Liu, X.; Wang, L.; et al. Lysine-specific demethylase 1 aggravated oxidative stress and ferroptosis induced by renal ischemia and reperfusion injury through activation of TLR4/NOX4 pathway in mice. J. Cell. Mol. Med. 2022, 26, 4254–4267. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.Y.; Oh, S.H.; Ahn, J.S.; Oh, E.J.; Kim, Y.J.; Kim, C.D.; Park, S.H.; Kim, Y.L.; Cho, J.H. NOX1 Inhibition Attenuates Kidney Ischemia-Reperfusion Injury via Inhibition of ROS-Mediated ERK Signaling. Int. J. Mol. Sci. 2020, 21, 6911. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis*. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 2012, 52, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Hancock, M.; Hafstad, A.D.; Nabeebaccus, A.A.; Catibog, N.; Logan, A.; Smyrnias, I.; Hansen, S.S.; Lanner, J.; Schroder, K.; Murphy, M.P.; et al. Myocardial NADPH oxidase-4 regulates the physiological response to acute exercise. eLife 2018, 7, e41044. [Google Scholar] [CrossRef] [PubMed]
- Gou, H.; Chen, X.; Zhu, X.; Li, L.; Hou, L.; Zhou, Y.; Xu, Y. Sequestered SQSTM1/p62 crosstalk with Keap1/NRF2 axis in hPDLCs promotes oxidative stress injury induced by periodontitis. Free Radic. Biol. Med. 2022, 190, 62–74. [Google Scholar] [CrossRef]
- Kageyama, S.; Saito, T.; Obata, M.; Koide, R.H.; Ichimura, Y.; Komatsu, M. Negative Regulation of the Keap1-Nrf2 Pathway by a p62/Sqstm1 Splicing Variant. Mol. Cell. Biol. 2018, 38, e00642-17. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Activation of Nrf2 by Natural Bioactive Compounds: A Promising Approach for Stroke? Int. J. Mol. Sci. 2020, 21, 4875. [Google Scholar] [CrossRef] [PubMed]
- Strom, J.; Xu, B.; Tian, X.; Chen, Q.M. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016, 30, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Hennig, P.; Fenini, G.; Di Filippo, M.; Karakaya, T.; Beer, H.D. The Pathways Underlying the Multiple Roles of p62 in Inflammation and Cancer. Biomedicines 2021, 9, 707. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Q.; Kim, M.Y.; Godoy, L.C.; Thiantanawat, A.; Trudel, L.J.; Wogan, G.N. Nitric oxide activation of Keap1/Nrf2 signaling in human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14547–14551. [Google Scholar] [CrossRef]
- McLaughlin, K.L.; Hagen, J.T.; Coalson, H.S.; Nelson, M.A.M.; Kew, K.A.; Wooten, A.R.; Fisher-Wellman, K.H. Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci. Rep. 2020, 10, 17599. [Google Scholar] [CrossRef]
- Mogensen, M.; Sahlin, K.; Fernstrom, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Hojlund, K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Yu, S.L.; Kang, J.; Jeong, B.Y.; Lee, H.Y.; Park, C.G.; Yu, Y.B.; Jin, D.C.; Hwang, W.M.; Yun, S.R.; et al. NADPH oxidase 4 mediates TGF-beta1/Smad signaling pathway induced acute kidney injury in hypoxia. PLoS ONE 2019, 14, e0219483. [Google Scholar] [CrossRef]
- Allen, K.L.; Almeida, A.; Bates, T.E.; Clark, J.B. Changes of respiratory chain activity in mitochondrial and synaptosomal fractions isolated from the gerbil brain after graded ischaemia. J. Neurochem. 1995, 64, 2222–2229. [Google Scholar] [CrossRef]
- Almeida, A.; Allen, K.L.; Bates, T.E.; Clark, J.B. Effect of reperfusion following cerebral ischaemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J. Neurochem. 1995, 65, 1698–1703. [Google Scholar] [CrossRef]
- Ginsberg, M.D.; Mela, L.; Wrobel-Kuhl, K.; Reivich, M. Mitochondrial metabolism following bilateral cerebral ischemia in the gerbil. Ann. Neurol. 1977, 1, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Hillered, L.; Siesjo, B.K.; Arfors, K.E. Mitochondrial response to transient forebrain ischemia and recirculation in the rat. J. Cereb. Blood Flow Metab. 1984, 4, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.R. Selective impairment of respiration in mitochondria isolated from brain subregions following transient forebrain ischemia in the rat. J. Neurochem. 1991, 56, 1836–1844. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.R.; Pulsinelli, W.A. Altered mitochondrial respiration in selectively vulnerable brain subregions following transient forebrain ischemia in the rat. J. Neurochem. 1987, 49, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, A.; Sosunov, S.; Niatsetskaya, Z.; Konrad, C.; Starkov, A.A.; Manfredi, G.; Wittig, I.; Ten, V.; Galkin, A. Redox-Dependent Loss of Flavin by Mitochondrial Complex I in Brain Ischemia/Reperfusion Injury. Antioxid. Redox Signal. 2019, 31, 608–622. [Google Scholar] [CrossRef]
- Kahl, A.; Stepanova, A.; Konrad, C.; Anderson, C.; Manfredi, G.; Zhou, P.; Iadecola, C.; Galkin, A. Critical Role of Flavin and Glutathione in Complex I-Mediated Bioenergetic Failure in Brain Ischemia/Reperfusion Injury. Stroke 2018, 49, 1223–1231. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiffer, T.A.; Carvalho, L.R.R.A.; Guimaraes, D.; Boeder, A.; Wikström, P.; Carlström, M. Specific NOX4 Inhibition Preserves Mitochondrial Function and Dampens Kidney Dysfunction Following Ischemia–Reperfusion-Induced Kidney Injury. Antioxidants 2024, 13, 489. https://doi.org/10.3390/antiox13040489
Schiffer TA, Carvalho LRRA, Guimaraes D, Boeder A, Wikström P, Carlström M. Specific NOX4 Inhibition Preserves Mitochondrial Function and Dampens Kidney Dysfunction Following Ischemia–Reperfusion-Induced Kidney Injury. Antioxidants. 2024; 13(4):489. https://doi.org/10.3390/antiox13040489
Chicago/Turabian StyleSchiffer, Tomas A., Lucas Rannier Ribeiro Antonino Carvalho, Drielle Guimaraes, Ariela Boeder, Per Wikström, and Mattias Carlström. 2024. "Specific NOX4 Inhibition Preserves Mitochondrial Function and Dampens Kidney Dysfunction Following Ischemia–Reperfusion-Induced Kidney Injury" Antioxidants 13, no. 4: 489. https://doi.org/10.3390/antiox13040489
APA StyleSchiffer, T. A., Carvalho, L. R. R. A., Guimaraes, D., Boeder, A., Wikström, P., & Carlström, M. (2024). Specific NOX4 Inhibition Preserves Mitochondrial Function and Dampens Kidney Dysfunction Following Ischemia–Reperfusion-Induced Kidney Injury. Antioxidants, 13(4), 489. https://doi.org/10.3390/antiox13040489