

Oxidative Stress: A Culprit in the Progression of Diabetic Kidney Disease

Abstract
1. Introduction
2. Sources of Endogenous ROS
2.1. Mitochondrial Superoxide Production
2.2. NADPH Oxidases (Noxs)
2.3. Uncoupled NOS
2.4. Xanthine Oxidase (XO)
2.5. Cytochrome P450
2.6. Lipoxygenase
3. The Pathogenesis of Diabetic Kidney Disease (DKD) with Oxidative Stress
3.1. Immune Cells
3.2. Complement System
3.3. The Upstream Signaling Cascades That Trigger Oxidative Stress Response
3.3.1. AGEs/RAGE Pathway
3.3.2. Hexosamine Pathway
3.3.3. Polyol Pathway
3.3.4. Activation of the Protein Kinase C (PKC) Pathway
3.4. The Downstream Signaling of Oxidative Stress
3.4.1. NF-κB Pathway
3.4.2. TGF-β Pathway
3.4.3. Phosphoinositide 3-Kinase (PI3K)/Akt Pathway
3.4.4. Nrf2/ARE Signaling Pathway
3.4.5. Janus Kinase 2-Signal Transducer/Activator of Transcription 3 (JAK2/STAT3) Signaling Pathway
3.4.6. Adenosine Monophosphate-Activated Protein Kinase (AMPK) Signaling Pathway
4. Oxidative Stress and Epigenetic Modifications
4.1. DNA Methylation
4.2. Histone Modifications
4.3. Non-Coding RNAs
5. Antioxidative Therapies
5.1. Glucose-Lowering Drugs
5.1.1. Metformin
5.1.2. Thiazolidinedione
5.1.3. SGLT2 Inhibitors
5.1.4. Glucagon-like Peptide 1 Receptor Agonists (GLP-1 RAs)
5.2. NRF2 Activation
5.3. Other Novel Antioxidative Therapies
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Jandeleit-Dahm, K. The pathobiology of diabetic vascular complications—Cardiovascular and kidney disease. J. Mol. Med. 2014, 92, 441–452. [Google Scholar] [CrossRef]
- Zhang, P.N.; Zhou, M.Q.; Guo, J.; Zheng, H.J.; Tang, J.; Zhang, C.; Liu, Y.N.; Liu, W.J.; Wang, Y.X. Mitochondrial Dysfunction and Diabetic Nephropathy: Nontraditional Therapeutic Opportunities. J. Diabetes Res. 2021, 2021, 1010268. [Google Scholar] [CrossRef]
- American Diabetes Association. Addendum. 11. Microvascular Complications and Foot Care: Standards of Medical Care in Diabetes—2021: Diabetes Care 2021;44(Suppl. 1):S151–S167. Diabetes Care 2021, 44, 2186–2187. [Google Scholar] [CrossRef]
- Viigimaa, M.; Sachinidis, A.; Toumpourleka, M.; Koutsampasopoulos, K.; Alliksoo, S.; Titma, T. Macrovascular Complications of Type 2 Diabetes Mellitus. Curr. Vasc. Pharmacol. 2020, 18, 110–116. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes Diabetes Work Group. KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2022, 102, S1–S127. [Google Scholar] [CrossRef]
- Pugliese, G.; Penno, G.; Natali, A.; Barutta, F.; Di Paolo, S.; Reboldi, G.; Gesualdo, L.; De Nicola, L.; Italian Diabetes Society; Italian Society of Nephrology. Diabetic kidney disease: New clinical and therapeutic issues. Joint position statement of the Italian Diabetes Society and the Italian Society of Nephrology on “The natural history of diabetic kidney disease and treatment of hyperglycemia in patients with type 2 diabetes and impaired renal function”. J. Nephrol. 2020, 33, 9–35. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Pak, W.L.W.; Tanaka, T.; Tang, S.C.W.; Nangaku, M. Update on diagnosis, pathophysiology, and management of diabetic kidney disease. Nephrology 2021, 26, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Shimizu, M.; Yamanouchi, M.; Toyama, T.; Hara, A.; Furuichi, K.; Wada, T. Trajectories of kidney function in diabetes: A clinicopathological update. Nat. Rev. Nephrol. 2021, 17, 740–750. [Google Scholar] [CrossRef]
- Aboolian, A.; Urner, S.; Roden, M.; Jha, J.C.; Jandeleit-Dahm, K. Diabetic Kidney Disease: From Pathogenesis to Novel Treatment Possibilities. Handb. Exp. Pharmacol. 2022, 274, 269–307. [Google Scholar] [CrossRef]
- Shim, K.; Begum, R.; Yang, C.; Wang, H. Complement activation in obesity, insulin resistance, and type 2 diabetes mellitus. World J. Diabetes 2020, 11, 1–12. [Google Scholar] [CrossRef]
- de Boer, I.H.; Khunti, K.; Sadusky, T.; Tuttle, K.R.; Neumiller, J.J.; Rhee, C.M.; Rosas, S.E.; Rossing, P.; Bakris, G. Diabetes Management in Chronic Kidney Disease: A Consensus Report by the American Diabetes Association (ADA) and Kidney Disease: Improving Global Outcomes (KDIGO). Diabetes Care 2022, 45, 3075–3090. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Morii, T.; Fujishima, H.; Sato, T.; Shimizu, T.; Hosoba, M.; Tsukiyama, K.; Narita, T.; Takahashi, T.; Drucker, D.J.; et al. The protective roles of GLP-1R signaling in diabetic nephropathy: Possible mechanism and therapeutic potential. Kidney Int. 2014, 85, 579–589. [Google Scholar] [CrossRef]
- Kodera, R.; Shikata, K.; Kataoka, H.U.; Takatsuka, T.; Miyamoto, S.; Sasaki, M.; Kajitani, N.; Nishishita, S.; Sarai, K.; Hirota, D.; et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia 2011, 54, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Sourris, K.C.; Yao, H.; Jerums, G.; Cooper, M.E.; Ekinci, E.I.; Coughlan, M.T. Can Targeting the Incretin Pathway Dampen RAGE-Mediated Events in Diabetic Nephropathy? Curr. Drug Targets 2016, 17, 1252–1264. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Yamagishi, S. Tofogliflozin, A Highly Selective Inhibitor of SGLT2 Blocks Proinflammatory and Proapoptotic Effects of Glucose Overload on Proximal Tubular Cells Partly by Suppressing Oxidative Stress Generation. Horm. Metab. Res. 2016, 48, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Kleibert, M.; Zygmunciak, P.; Lakomska, K.; Mila, K.; Zgliczynski, W.; Mrozikiewicz-Rakowska, B. Insight into the Molecular Mechanism of Diabetic Kidney Disease and the Role of Metformin in Its Pathogenesis. Int. J. Mol. Sci. 2023, 24, 13038. [Google Scholar] [CrossRef]
- Giardino, I.; Edelstein, D.; Brownlee, M. BCL-2 expression or antioxidants prevent hyperglycemia-induced formation of intracellular advanced glycation endproducts in bovine endothelial cells. J. Clin. Investig. 1996, 97, 1422–1428. [Google Scholar] [CrossRef]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Zhang, X.G.; Liu, A.X.; Zhang, Y.X.; Zhou, M.Y.; Li, X.Y.; Fu, M.H.; Pan, Y.P.; Xu, J.; Zhang, J.Q. A diarylheptanoid compound from Alpinia officinarum Hance ameliorates high glucose-induced insulin resistance by regulating PI3K/AKT-Nrf2-GSK3beta signaling pathways in HepG2 cells. J. Ethnopharmacol. 2022, 295, 115397. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Whiteman, M. Measuring reactive species and oxidative damage in vivo and in cell culture: How should you do it and what do the results mean? Br. J. Pharmacol. 2004, 142, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Shang, W.; Liu, J.; Cheang, W.S.; Wang, Y.; Xiang, L.; Lau, C.W.; Luo, J.Y.; Ng, C.F.; Huang, Y.; et al. Activation of AMPK/miR-181b Axis Alleviates Endothelial Dysfunction and Vascular Inflammation in Diabetic Mice. Antioxidants 2022, 11, 1137. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Shen, P.; Song, Y.; Huang, Y.; Tu, S. Reactive Oxygen Species in Autoimmune Cells: Function, Differentiation, and Metabolism. Front. Immunol. 2021, 12, 635021. [Google Scholar] [CrossRef] [PubMed]
- Flemming, N.B.; Gallo, L.A.; Forbes, J.M. Mitochondrial Dysfunction and Signaling in Diabetic Kidney Disease: Oxidative Stress and Beyond. Semin. Nephrol. 2018, 38, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Herlein, J.A.; Fink, B.D.; O’Malley, Y.; Sivitz, W.I. Superoxide and respiratory coupling in mitochondria of insulin-deficient diabetic rats. Endocrinology 2009, 150, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Nguyen, T.V.; Penfold, S.A.; Higgins, G.C.; Thallas-Bonke, V.; Tan, S.M.; Van Bergen, N.J.; Sourris, K.C.; Harcourt, B.E.; Thorburn, D.R.; et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin. Sci. 2016, 130, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, S.; Yu, X.; Chen, W.; Ma, H.; Shao, C.; Zhang, Y.; Zhang, A.; Huang, S.; Jia, Z. Mitochondrial activity contributes to impaired renal metabolic homeostasis and renal pathology in STZ-induced diabetic mice. Am. J. Physiol. Ren. Physiol. 2019, 317, F593–F605. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Li, H.; Dai, Q.; Lu, C.; Xu, M.; Zhang, J.; Feng, J. DUSP1 recuses diabetic nephropathy via repressing JNK-Mff-mitochondrial fission pathways. J. Cell Physiol. 2019, 234, 3043–3057. [Google Scholar] [CrossRef]
- Cleveland, K.H.; Schnellmann, R.G. Pharmacological Targeting of Mitochondria in Diabetic Kidney Disease. Pharmacol. Rev. 2023, 75, 250–262. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef]
- Mohri, H.; Ninoyu, Y.; Sakaguchi, H.; Hirano, S.; Saito, N.; Ueyama, T. Nox3-Derived Superoxide in Cochleae Induces Sensorineural Hearing Loss. J. Neurosci. 2021, 41, 4716–4731. [Google Scholar] [CrossRef]
- Banfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Adeyemo, A.A.; Zhou, J.; Chen, Y.; Doumatey, A.; Lashley, K.; Huang, H.; Amoah, A.; Agyenim-Boateng, K.; Eghan, B.A., Jr.; et al. A genome-wide search for linkage to renal function phenotypes in West Africans with type 2 diabetes. Am. J. Kidney Dis. 2007, 49, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Gray, S.P.; Barit, D.; Okabe, J.; El-Osta, A.; Namikoshi, T.; Thallas-Bonke, V.; Wingler, K.; Szyndralewiez, C.; Heitz, F.; et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1237–1254. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Thallas-Bonke, V.; Banal, C.; Gray, S.P.; Chow, B.S.; Ramm, G.; Quaggin, S.E.; Cooper, M.E.; Schmidt, H.H.; Jandeleit-Dahm, K.A. Podocyte-specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia 2016, 59, 379–389. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Kennedy, K.; Chew, P.; Okabe, J.; El-Osta, A.; Calkin, A.C.; Biessen, E.A.; Touyz, R.M.; Cooper, M.E.; et al. Reactive Oxygen Species Can Provide Atheroprotection via NOX4-Dependent Inhibition of Inflammation and Vascular Remodeling. Arter. Thromb. Vasc. Biol. 2016, 36, 295–307. [Google Scholar] [CrossRef]
- Qin, H.; Zhang, L.; Li, M.; Liu, Y.; Sun, S.; Nie, W.; Bai, B.; Li, G.; Zhang, G. EGR1/NOX4 pathway regulates oxidative stress and further facilitates fibrosis progression in keloids responses to TGF-beta1. J. Dermatol. Sci. 2022, 108, 138–145. [Google Scholar] [CrossRef]
- Holterman, C.E.; Thibodeau, J.F.; Towaij, C.; Gutsol, A.; Montezano, A.C.; Parks, R.J.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R. Nephropathy and elevated BP in mice with podocyte-specific NADPH oxidase 5 expression. J. Am. Soc. Nephrol. 2014, 25, 784–797. [Google Scholar] [CrossRef]
- Jha, J.C.; Dai, A.; Holterman, C.E.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.; Jandeleit-Dahm, K.A.M. Endothelial or vascular smooth muscle cell-specific expression of human NOX5 exacerbates renal inflammation, fibrosis and albuminuria in the Akita mouse. Diabetologia 2019, 62, 1712–1726. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Okabe, J.; Gray, S.P.; Hettige, T.; Chow, B.S.M.; Thallas-Bonke, V.; De Vos, L.; Holterman, C.E.; Coughlan, M.T.; et al. NADPH Oxidase Nox5 Accelerates Renal Injury in Diabetic Nephropathy. Diabetes 2017, 66, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Deliyanti, D.; Alrashdi, S.F.; Touyz, R.M.; Kennedy, C.R.; Jha, J.C.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Wilkinson-Berka, J.L. Nox (NADPH Oxidase) 1, Nox4, and Nox5 Promote Vascular Permeability and Neovascularization in Retinopathy. Hypertension 2020, 75, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Eid, S.A.; Savelieff, M.G.; Eid, A.A.; Feldman, E.L. Nox, Nox, Are You There? The Role of NADPH Oxidases in the Peripheral Nervous System. Antioxid. Redox Signal. 2022, 37, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, S.S. Role of nitric oxide in diabetic nephropathy. Semin. Nephrol. 2004, 24, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Noiri, E.; Satoh, H.; Taguchi, J.; Brodsky, S.V.; Nakao, A.; Ogawa, Y.; Nishijima, S.; Yokomizo, T.; Tokunaga, K.; Fujita, T. Association of eNOS Glu298Asp polymorphism with end-stage renal disease. Hypertension 2002, 40, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Choi, J.; Sim, L.; Dey, A.; Mohan, M.; Kantharidis, P.; Dietz, L.; Sandner, P.; de Haan, J.B. Ameliorating diabetes-associated atherosclerosis and diabetic nephropathy through modulation of soluble guanylate cyclase. Front. Cardiovasc. Med. 2023, 10, 1220095. [Google Scholar] [CrossRef] [PubMed]
- Czirok, S.; Fang, L.; Radovits, T.; Szabo, G.; Szenasi, G.; Rosivall, L.; Merkely, B.; Kokeny, G. Cinaciguat ameliorates glomerular damage by reducing ERK1/2 activity and TGF-ss expression in type-1 diabetic rats. Sci. Rep. 2017, 7, 11218. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.J.; Wang, S.; Cheng, H.; Zhang, M.Z.; Takahashi, T.; Fogo, A.B.; Breyer, M.D.; Harris, R.C. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J. Am. Soc. Nephrol. 2006, 17, 2664–2669. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Fujimoto, S.; Haruna, Y.; Arakawa, S.; Horike, H.; Komai, N.; Sasaki, T.; Tsujioka, K.; Makino, H.; Kashihara, N. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2005, 288, F1144–F1152. [Google Scholar] [CrossRef]
- Wenzel, P.; Schulz, E.; Oelze, M.; Muller, J.; Schuhmacher, S.; Alhamdani, M.S.; Debrezion, J.; Hortmann, M.; Reifenberg, K.; Fleming, I.; et al. AT1-receptor blockade by telmisartan upregulates GTP-cyclohydrolase I and protects eNOS in diabetic rats. Free Radic. Biol. Med. 2008, 45, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Daiber, A.; Oelze, M.; Brandt, M.; Closs, E.; Xu, J.; Thum, T.; Bauersachs, J.; Ertl, G.; Zou, M.H.; et al. Mechanisms underlying recoupling of eNOS by HMG-CoA reductase inhibition in a rat model of streptozotocin-induced diabetes mellitus. Atherosclerosis 2008, 198, 65–76. [Google Scholar] [CrossRef]
- Hovind, P.; Rossing, P.; Tarnow, L.; Johnson, R.J.; Parving, H.H. Serum uric acid as a predictor for development of diabetic nephropathy in type 1 diabetes: An inception cohort study. Diabetes 2009, 58, 1668–1671. [Google Scholar] [CrossRef]
- Liu, J.; Wang, C.; Liu, F.; Lu, Y.; Cheng, J. Metabonomics revealed xanthine oxidase-induced oxidative stress and inflammation in the pathogenesis of diabetic nephropathy. Anal. Bioanal. Chem. 2015, 407, 2569–2579. [Google Scholar] [CrossRef] [PubMed]
- Olaniyi, K.S.; Amusa, O.A.; Akinnagbe, N.T.; Ajadi, I.O.; Ajadi, M.B.; Agunbiade, T.B.; Michael, O.S. Acetate ameliorates nephrotoxicity in streptozotocin-nicotinamide-induced diabetic rats: Involvement of xanthine oxidase activity. Cytokine 2021, 142, 155501. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, T.; Nakayama, T.; Heinig, M.; Zhang, L.; Yuzawa, Y.; Sanchez-Lozada, L.G.; Roncal, C.; Johnson, R.J.; Nakagawa, T. Effect of lowering uric acid on renal disease in the type 2 diabetic db/db mice. Am. J. Physiol. Ren. Physiol. 2009, 297, F481–F488. [Google Scholar] [CrossRef]
- Komers, R.; Xu, B.; Schneider, J.; Oyama, T.T. Effects of xanthine oxidase inhibition with febuxostat on the development of nephropathy in experimental type 2 diabetes. Br. J. Pharmacol. 2016, 173, 2573–2588. [Google Scholar] [CrossRef]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, P.; Zhao, G.; Xu, G.; Gruzdev, A.; Zeldin, D.C.; Wang, D.W. Cytochrome P450 epoxygenase CYP2J2 attenuates nephropathy in streptozotocin-induced diabetic mice. Prostaglandins Other Lipid Mediat. 2011, 96, 63–71. [Google Scholar] [CrossRef]
- Njeim, R.; Braych, K.; Ghadieh, H.E.; Azar, N.S.; Azar, W.S.; Dia, B.; Leone, A.; Cappello, F.; Kfoury, H.; Harb, F.; et al. VEGF-A: A Novel Mechanistic Link between CYP2C-Derived EETs and Nox4 in Diabetic Kidney Disease. Diabetes 2023, 72, 947–957. [Google Scholar] [CrossRef]
- Gangadhariah, M.H.; Luther, J.M.; Garcia, V.; Paueksakon, P.; Zhang, M.Z.; Hayward, S.W.; Love, H.D.; Falck, J.R.; Manthati, V.L.; Imig, J.D.; et al. Hypertension is a major contributor to 20-hydroxyeicosatetraenoic acid-mediated kidney injury in diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.A.; Gorin, Y.; Fagg, B.M.; Maalouf, R.; Barnes, J.L.; Block, K.; Abboud, H.E. Mechanisms of podocyte injury in diabetes: Role of cytochrome P450 and NADPH oxidases. Diabetes 2009, 58, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, J.; Pye, C.; Al-Shabrawey, M.; Elmarakby, A.A. Inhibition of 12/15-Lipoxygenase Reduces Renal Inflammation and Injury in Streptozotocin-Induced Diabetic Mice. J. Diabetes Metab. 2015, 6, 555. [Google Scholar] [CrossRef]
- Dong, C.; Liu, S.; Cui, Y.; Guo, Q. 12-Lipoxygenase as a key pharmacological target in the pathogenesis of diabetic nephropathy. Eur. J. Pharmacol. 2020, 879, 173122. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Sun, B.; Liu, Y.; Huang, J.; Chen, G.; Zhang, X.; Chen, C.; Wang, D.; Wang, G. Increased lipoxygenase and decreased cytochrome P450s metabolites correlated with the incidence of diabetic nephropathy: Potential role of eicosanoids from metabolomics in type 2 diabetic patients. Clin. Exp. Pharmacol. Physiol. 2021, 48, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Fernandez-Fernandez, B.; Mora-Fernandez, C.; Marchant, V.; Donate-Correa, J.; Navarro-Gonzalez, J.F.; Ortiz, A.; Ruiz-Ortega, M. Targeting inflammation to treat diabetic kidney disease: The road to 2030. Kidney Int. 2023, 103, 282–296. [Google Scholar] [CrossRef]
- Wilson, P.C.; Wu, H.; Kirita, Y.; Uchimura, K.; Ledru, N.; Rennke, H.G.; Welling, P.A.; Waikar, S.S.; Humphreys, B.D. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 19619–19625. [Google Scholar] [CrossRef]
- Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Rabelink, T.J.; Bajema, I.M.; DHT, I.J. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol. Dial Transpl. 2017, 32, 1322–1329. [Google Scholar] [CrossRef]
- Calle, P.; Hotter, G. Macrophage Phenotype and Fibrosis in Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 2806. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Agarwal, R.; Alpers, C.E.; Bakris, G.L.; Brosius, F.C.; Kolkhof, P.; Uribarri, J. Molecular mechanisms and therapeutic targets for diabetic kidney disease. Kidney Int. 2022, 102, 248–260. [Google Scholar] [CrossRef]
- Jaaban, M.; Zetoune, A.B.; Hesenow, S.; Hessenow, R. Neutrophil-lymphocyte ratio and platelet-lymphocyte ratio as novel risk markers for diabetic nephropathy in patients with type 2 diabetes. Heliyon 2021, 7, e07564. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076. [Google Scholar] [CrossRef] [PubMed]
- Shafqat, A.; Abdul Rab, S.; Ammar, O.; Al Salameh, S.; Alkhudairi, A.; Kashir, J.; Alkattan, K.; Yaqinuddin, A. Emerging role of neutrophil extracellular traps in the complications of diabetes mellitus. Front. Med. 2022, 9, 995993. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Ma, L.; Li, X.; Wang, Z.; Gao, R.; Peng, C.; Kang, B.; Wang, Y.; Luo, T.; Wu, J.; et al. Neutrophil Extracellular Traps Induce Glomerular Endothelial Cell Dysfunction and Pyroptosis in Diabetic Kidney Disease. Diabetes 2022, 71, 2739–2750. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Singh, K.; Fatima, S.; Ambreen, S.; Zimmermann, S.; Younis, R.; Krishnan, S.; Rana, R.; Gadi, I.; Schwab, C.; et al. Neutrophil Extracellular Traps Promote NLRP3 Inflammasome Activation and Glomerular Endothelial Dysfunction in Diabetic Kidney Disease. Nutrients 2022, 14, 2965. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.S.; Brenchley, P.E. Mast cells: The forgotten cells of renal fibrosis. J. Clin. Pathol. 2000, 53, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, P.; Reddy, J.; Singh, M. Do resident renal mast cells play a role in the pathogenesis of diabetic nephropathy? Mol. Cell. Biochem. 2009, 330, 187–192. [Google Scholar] [CrossRef]
- Zheng, J.M.; Yao, G.H.; Cheng, Z.; Wang, R.; Liu, Z.H. Pathogenic role of mast cells in the development of diabetic nephropathy: A study of patients at different stages of the disease. Diabetologia 2012, 55, 801–811. [Google Scholar] [CrossRef]
- Bivona, B.J.; Takai, S.; Seth, D.M.; Satou, R.; Harrison-Bernard, L.M. Chymase inhibition retards albuminuria in type 2 diabetes. Physiol. Rep. 2019, 7, e14302. [Google Scholar] [CrossRef]
- Bradding, P.; Pejler, G. The controversial role of mast cells in fibrosis. Immunol. Rev. 2018, 282, 198–231. [Google Scholar] [CrossRef]
- Moon, J.Y.; Jeong, K.H.; Lee, T.W.; Ihm, C.G.; Lim, S.J.; Lee, S.H. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am. J. Nephrol. 2012, 35, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Chavez, M.D.; Tse, H.M. Targeting Mitochondrial-Derived Reactive Oxygen Species in T Cell-Mediated Autoimmune Diseases. Front. Immunol. 2021, 12, 703972. [Google Scholar] [CrossRef] [PubMed]
- Belikov, A.V.; Schraven, B.; Simeoni, L. T cells and reactive oxygen species. J. Biomed. Sci. 2015, 22, 85. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Sun, L.; Dong, M.; An, G.; Zhang, K.; Zhang, C.; Meng, X. Regulatory T Cells and Diabetes Mellitus. Hum. Gene Ther. 2021, 32, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Z.; Si, Z.; Yang, Y.; Li, S.; Xue, Y. Dapagliflozin reverses the imbalance of T helper 17 and T regulatory cells by inhibiting SGK1 in a mouse model of diabetic kidney disease. FEBS Open Bio 2021, 11, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Andrikopoulos, S.; MacIsaac, R.J.; Mackay, L.K.; Nikolic-Paterson, D.J.; Torkamani, N.; Zafari, N.; Marin, E.C.S.; Ekinci, E.I. Role of the adaptive immune system in diabetic kidney disease. J. Diabetes Investig. 2022, 13, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Pescovitz, M.D.; Greenbaum, C.J.; Bundy, B.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; Moran, A.; Raskin, P.; et al. B-lymphocyte depletion with rituximab and beta-cell function: Two-year results. Diabetes Care 2014, 37, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yu, Z.; Qu, Z.; Zhang, N.; Crew, R.; Jiang, Y. Decreased number of CD19(+)CD24(hi)CD38(hi) regulatory B cells in Diabetic nephropathy. Mol. Immunol. 2019, 112, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Tao, L.; Su, H. The Complement System in Metabolic-Associated Kidney Diseases. Front. Immunol. 2022, 13, 902063. [Google Scholar] [CrossRef]
- Petr, V.; Thurman, J.M. The role of complement in kidney disease. Nat. Rev. Nephrol. 2023, 19, 771–787. [Google Scholar] [CrossRef]
- Flyvbjerg, A. The role of the complement system in diabetic nephropathy. Nat. Rev. Nephrol. 2017, 13, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, D.; Huang, X.; Wang, Y.; Wang, S.; Wang, X.; Yang, X. Association of Complement and Inflammatory Biomarkers with Diabetic Nephropathy. Ann. Clin. Lab. Sci. 2019, 49, 488–495. [Google Scholar] [PubMed]
- Sircar, M.; Rosales, I.A.; Selig, M.K.; Xu, D.; Zsengeller, Z.K.; Stillman, I.E.; Libermann, T.A.; Karumanchi, S.A.; Thadhani, R.I. Complement 7 Is Up-Regulated in Human Early Diabetic Kidney Disease. Am. J. Pathol. 2018, 188, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.J.; Li, X.Q.; Chang, D.Y.; Wang, S.X.; Liu, G.; Chen, M.; Zhao, M.H. Complement deposition on renal histopathology of patients with diabetic nephropathy. Diabetes Metab. 2019, 45, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Di, D.; Jiao, Y.; Zou, G.; Gao, H.; Li, W. Complement Deposition Predicts Worsening Kidney Function and Underlines the Clinical Significance of the 2010 Renal Pathology Society Classification of Diabetic Nephropathy. Front. Immunol. 2022, 13, 868127. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Sun, L.; Nie, G.; Chen, J.; Zhang, C.; Zhu, H.; Huang, Z.; Qian, J.; Zhao, X.; Xing, C.; et al. Association of Glomerular Complement C4c Deposition with the Progression of Diabetic Kidney Disease in Patients with Type 2 Diabetes. Front. Immunol. 2020, 11, 2073. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Corna, D.; Locatelli, M.; Cassis, P.; Carminati, C.E.; Bolognini, S.; Zoja, C.; Remuzzi, G.; Benigni, A.; et al. C3a receptor blockade protects podocytes from injury in diabetic nephropathy. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Chang, D.Y.; Chen, M.; Zhao, M.H. Complement activation in patients with diabetic nephropathy. Diabetes Metab. 2019, 45, 248–253. [Google Scholar] [CrossRef]
- Saraheimo, M.; Forsblom, C.; Hansen, T.K.; Teppo, A.M.; Fagerudd, J.; Pettersson-Fernholm, K.; Thiel, S.; Tarnow, L.; Ebeling, P.; Flyvbjerg, A.; et al. Increased levels of mannan-binding lectin in type 1 diabetic patients with incipient and overt nephropathy. Diabetologia 2005, 48, 198–202. [Google Scholar] [CrossRef]
- Hansen, T.K.; Gall, M.A.; Tarnow, L.; Thiel, S.; Stehouwer, C.D.; Schalkwijk, C.G.; Parving, H.H.; Flyvbjerg, A. Mannose-binding lectin and mortality in type 2 diabetes. Arch. Intern. Med. 2006, 166, 2007–2013. [Google Scholar] [CrossRef]
- Hansen, T.K.; Forsblom, C.; Saraheimo, M.; Thorn, L.; Waden, J.; Hoyem, P.; Ostergaard, J.; Flyvbjerg, A.; Groop, P.H.; FinnDiane Study, G. Association between mannose-binding lectin, high-sensitivity C-reactive protein and the progression of diabetic nephropathy in type 1 diabetes. Diabetologia 2010, 53, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.M.; Ren, X.G.; Jiang, Z.H.; Chen, D.J.; Zhao, W.J.; Li, L.J. Lectin-induced renal local complement activation is involved in tubular interstitial injury in diabetic nephropathy. Clin. Chim. Acta 2018, 482, 65–73. [Google Scholar] [CrossRef]
- Qin, X.; Goldfine, A.; Krumrei, N.; Grubissich, L.; Acosta, J.; Chorev, M.; Hays, A.P.; Halperin, J.A. Glycation inactivation of the complement regulatory protein CD59: A possible role in the pathogenesis of the vascular complications of human diabetes. Diabetes 2004, 53, 2653–2661. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, L.; Zang, J.; Tang, X.; Liu, Y.; Zhang, J.; Bai, L.; Yin, Q.; Lu, Y.; Cheng, J.; et al. C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/beta-catenin signaling pathway in diabetic kidney disease. Metabolism 2015, 64, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.M.; Ziemann, M.; Thallas-Bonke, V.; Snelson, M.; Kumar, V.; Laskowski, A.; Nguyen, T.V.; Huynh, K.; Clarke, M.V.; Libianto, R.; et al. Complement C5a Induces Renal Injury in Diabetic Kidney Disease by Disrupting Mitochondrial Metabolic Agility. Diabetes 2020, 69, 83–98. [Google Scholar] [CrossRef]
- Lim, J.; Iyer, A.; Suen, J.Y.; Seow, V.; Reid, R.C.; Brown, L.; Fairlie, D.P. C5aR and C3aR antagonists each inhibit diet-induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J. 2013, 27, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Peake, P.W.; Charlesworth, J.A.; Timmermans, V.; Gavrilovic, L.; Pussell, B. Does non-enzymatic glycosylation affect complement function in diabetes? Diabetes Res. 1989, 11, 109–114. [Google Scholar] [PubMed]
- Acosta, J.; Hettinga, J.; Fluckiger, R.; Krumrei, N.; Goldfine, A.; Angarita, L.; Halperin, J. Molecular basis for a link between complement and the vascular complications of diabetes. Proc. Natl. Acad. Sci. USA 2000, 97, 5450–5455. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Vaidya, A.; Sahoo, R.; Goldfine, A.; Herring, N.; Bry, L.; Chorev, M.; Halperin, J.A. Glycation of the complement regulatory protein CD59 is a novel biomarker for glucose handling in humans. J. Clin. Endocrinol. Metab. 2014, 99, E999–E1006. [Google Scholar] [CrossRef]
- Xiao, X.; Ma, B.; Dong, B.; Zhao, P.; Tai, N.; Chen, L.; Wong, F.S.; Wen, L. Cellular and humoral immune responses in the early stages of diabetic nephropathy in NOD mice. J. Autoimmun. 2009, 32, 85–93. [Google Scholar] [CrossRef]
- Ostergaard, J.; Thiel, S.; Gadjeva, M.; Hansen, T.K.; Rasch, R.; Flyvbjerg, A. Mannose-binding lectin deficiency attenuates renal changes in a streptozotocin-induced model of type 1 diabetes in mice. Diabetologia 2007, 50, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Lassila, M.; Seah, K.K.; Allen, T.J.; Thallas, V.; Thomas, M.C.; Candido, R.; Burns, W.C.; Forbes, J.M.; Calkin, A.C.; Cooper, M.E.; et al. Accelerated nephropathy in diabetic apolipoprotein e-knockout mouse: Role of advanced glycation end products. J. Am. Soc. Nephrol. 2004, 15, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, T.P.; Alderson, N.L.; Arrington, D.D.; Beattie, R.J.; Basgen, J.M.; Steffes, M.W.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002, 61, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.M.; Gray, S.P.; Jiaze, L.; Soro-Paavonen, A.; Wong, B.; Cooper, M.E.; Bierhaus, A.; Pickering, R.; Tikellis, C.; Tsorotes, D.; et al. Alagebrium reduces glomerular fibrogenesis and inflammation beyond preventing RAGE activation in diabetic apolipoprotein E knockout mice. Diabetes 2012, 61, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Flyvbjerg, A.; Denner, L.; Schrijvers, B.F.; Tilton, R.G.; Mogensen, T.H.; Paludan, S.R.; Rasch, R. Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes 2004, 53, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Gellai, R.; Hodrea, J.; Lenart, L.; Hosszu, A.; Koszegi, S.; Balogh, D.; Ver, A.; Banki, N.F.; Fulop, N.; Molnar, A.; et al. Role of O-linked N-acetylglucosamine modification in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2016, 311, F1172–F1181. [Google Scholar] [CrossRef]
- Singh, L.P.; Cheng, D.W.; Kowluru, R.; Levi, E.; Jiang, Y. Hexosamine induction of oxidative stress, hypertrophy and laminin expression in renal mesangial cells: Effect of the anti-oxidant alpha-lipoic acid. Cell Biochem. Funct. 2007, 25, 537–550. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Forbes, J.M.; Fukami, K.; Cooper, M.E. Diabetic nephropathy: Where hemodynamics meets metabolism. Exp. Clin. Endocrinol. Diabetes 2007, 115, 69–84. [Google Scholar] [CrossRef]
- Tilton, R.G.; Chang, K.; Nyengaard, J.R.; Van den Enden, M.; Ido, Y.; Williamson, J.R. Inhibition of sorbitol dehydrogenase. Effects on vascular and neural dysfunction in streptozocin-induced diabetic rats. Diabetes 1995, 44, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Kasajima, H.; Yamagishi, S.; Sugai, S.; Yagihashi, N.; Yagihashi, S. Enhanced in situ expression of aldose reductase in peripheral nerve and renal glomeruli in diabetic patients. Virchows Arch. 2001, 439, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Z.; Ying, K.; Wang, H.; Liu, P.; Ji, X.; Chi, T.; Zou, L.; Wang, S.; He, Z. WJ-39, an Aldose Reductase Inhibitor, Ameliorates Renal Lesions in Diabetic Nephropathy by Activating Nrf2 Signaling. Oxid. Med. Cell. Longev. 2020, 2020, 7950457. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef]
- Yu, J.; Liu, Y.; Li, H.; Zhang, P. Pathophysiology of diabetic kidney disease and autophagy: A review. Medicine 2023, 102, e33965. [Google Scholar] [CrossRef]
- Jiang, W.; Xiao, T.; Han, W.; Xiong, J.; He, T.; Liu, Y.; Huang, Y.; Yang, K.; Bi, X.; Xu, X.; et al. Klotho inhibits PKCalpha/p66SHC-mediated podocyte injury in diabetic nephropathy. Mol. Cell Endocrinol. 2019, 494, 110490. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Siomek, A. NF-kappaB signaling pathway and free radical impact. Acta Biochim. Pol. 2012, 59, 323–331. [Google Scholar] [CrossRef]
- Zoccali, C.; Mallamaci, F. Nonproteinuric progressive diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2019, 28, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Foresto-Neto, O.; Albino, A.H.; Arias, S.C.A.; Faustino, V.D.; Zambom, F.F.F.; Cenedeze, M.A.; Elias, R.M.; Malheiros, D.; Camara, N.O.S.; Fujihara, C.K.; et al. NF-kappaB System Is Chronically Activated and Promotes Glomerular Injury in Experimental Type 1 Diabetic Kidney Disease. Front. Physiol. 2020, 11, 84. [Google Scholar] [CrossRef]
- Tamada, S.; Asai, T.; Kuwabara, N.; Iwai, T.; Uchida, J.; Teramoto, K.; Kaneda, N.; Yukimura, T.; Komiya, T.; Nakatani, T.; et al. Molecular mechanisms and therapeutic strategies of chronic renal injury: The role of nuclear factor kappaB activation in the development of renal fibrosis. J. Pharmacol. Sci. 2006, 100, 17–21. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.L.; Liu, T.T.; Lan, H.Y. TGF-Beta as a Master Regulator of Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 7881. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Huang, X.R.; Zhu, H.J.; Oldfield, M.; Cooper, M.; Truong, L.D.; Johnson, R.J.; Lan, H.Y. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: Implications for diabetic renal and vascular disease. FASEB J. 2004, 18, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ramos, M.; Mora, I.; de Frutos, S.; Garesse, R.; Rodriguez-Puyol, M.; Olmos, G.; Rodriguez-Puyol, D. Intracellular redox equilibrium is essential for the constitutive expression of AP-1 dependent genes in resting cells: Studies on TGF-beta1 regulation. Int. J. Biochem. Cell Biol. 2012, 44, 963–971. [Google Scholar] [CrossRef]
- Logan, S.K.; Garabedian, M.J.; Campbell, C.E.; Werb, Z. Synergistic transcriptional activation of the tissue inhibitor of metalloproteinases-1 promoter via functional interaction of AP-1 and Ets-1 transcription factors. J. Biol. Chem. 1996, 271, 774–782. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, S.; Pan, X.; Cao, H.; Huang, X.; Xu, Q.; Zhong, H.; Peng, X. TIMP-1 expression induced by IL-32 is mediated through activation of AP-1 signal pathway. Int. Immunopharmacol. 2016, 38, 233–237. [Google Scholar] [CrossRef]
- Zhang, K.; Fan, C.; Cai, D.; Zhang, Y.; Zuo, R.; Zhu, L.; Cao, Y.; Zhang, J.; Liu, C.; Chen, Y.; et al. Contribution of TGF-Beta-Mediated NLRP3-HMGB1 Activation to Tubulointerstitial Fibrosis in Rat With Angiotensin II-Induced Chronic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, X.; Chun, J.; Vilaysane, A.; Clark, S.; French, G.; Bracey, N.A.; Trpkov, K.; Bonni, S.; Duff, H.J.; et al. Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J. Immunol. 2013, 190, 1239–1249. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Han, J. The p38 signal transduction pathway: Activation and function. Cell Signal. 2000, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Gao, K.; Wang, Y.; Wang, X.; Cui, B. Resveratrol ameliorates diabetic nephropathy in rats through negative regulation of the p38 MAPK/TGF-beta1 pathway. Exp. Ther. Med. 2017, 13, 3223–3230. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, L.; Deng, D.; Zhang, Q.; Liu, W. Renalase Protects against Renal Fibrosis by Inhibiting the Activation of the ERK Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 855. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lin, L.; Li, Q.; Ni, Y.; Zhang, C.; Qin, S.; Wei, J. ERK1/2 communicates GPCR and EGFR signaling pathways to promote CTGF-mediated hypertrophic cardiomyopathy upon Ang-II stimulation. BMC Mol. Cell Biol. 2019, 20, 14. [Google Scholar] [CrossRef]
- Huang, X.R.; Chung, A.C.; Wang, X.J.; Lai, K.N.; Lan, H.Y. Mice overexpressing latent TGF-beta1 are protected against renal fibrosis in obstructive kidney disease. Am. J. Physiol. Ren. Physiol. 2008, 295, F118–F127. [Google Scholar] [CrossRef]
- Li, A.G.; Wang, D.; Feng, X.H.; Wang, X.J. Latent TGFbeta1 overexpression in keratinocytes results in a severe psoriasis-like skin disorder. EMBO J. 2004, 23, 1770–1781. [Google Scholar] [CrossRef]
- Wang, R.; Song, F.; Li, S.; Wu, B.; Gu, Y.; Yuan, Y. Salvianolic acid A attenuates CCl(4)-induced liver fibrosis by regulating the PI3K/AKT/mTOR, Bcl-2/Bax and caspase-3/cleaved caspase-3 signaling pathways. Drug Des. Devel. Ther. 2019, 13, 1889–1900. [Google Scholar] [CrossRef]
- Erratum to Associations between the polymorphisms of main components in PI3K/Akt pathway and risk of diabetic kidney disease: A meta-analysis. IUBMB Life 2024, 76, 103. [CrossRef]
- Gui, H.; Chen, X.; Ye, L.; Ma, H. Seven basement membrane-specific expressed genes are considered potential biomarkers for the diagnosis and treatment of diabetic nephropathy. Acta Diabetol. 2023, 60, 493–505. [Google Scholar] [CrossRef]
- Hay, N. Interplay between FOXO, TOR, and Akt. Biochim. Biophys. Acta 2011, 1813, 1965–1970. [Google Scholar] [CrossRef]
- Ying, C.; Mao, Y.; Chen, L.; Wang, S.; Ling, H.; Li, W.; Zhou, X. Bamboo leaf extract ameliorates diabetic nephropathy through activating the AKT signaling pathway in rats. Int. J. Biol. Macromol. 2017, 105, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Ng, J.K.; Fung, W.W.; Chan, G.C.; Chow, K.M.; Szeto, C.C. Intrarenal and Urinary Glycogen Synthase Kinase-3 Beta Levels in Diabetic and Nondiabetic Chronic Kidney Disease. Kidney Blood Press Res. 2023, 48, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, Z.; Gong, R. Long noncoding RNA: An emerging player in diabetes and diabetic kidney disease. Clin. Sci. 2019, 133, 1321–1339. [Google Scholar] [CrossRef]
- Li, X.; Zou, Y.; Xing, J.; Fu, Y.Y.; Wang, K.Y.; Wan, P.Z.; Zhai, X.Y. Pretreatment with Roxadustat (FG-4592) Attenuates Folic Acid-Induced Kidney Injury through Antiferroptosis via Akt/GSK-3beta/Nrf2 Pathway. Oxid. Med. Cell Longev. 2020, 2020, 6286984. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, M.M.; Prasad, S.; D’Silva, K.; Cedillo, E.; Sataranatarajan, K.; Barnes, J.L.; Choudhury, G.G.; Kasinath, B.S. Activation of glycogen synthase kinase 3beta ameliorates diabetes-induced kidney injury. J. Biol. Chem. 2014, 289, 35363–35375. [Google Scholar] [CrossRef]
- Lu, Q.; Wang, W.W.; Zhang, M.Z.; Ma, Z.X.; Qiu, X.R.; Shen, M.; Yin, X.X. ROS induces epithelial-mesenchymal transition via the TGF-beta1/PI3K/Akt/mTOR pathway in diabetic nephropathy. Exp. Ther. Med. 2019, 17, 835–846. [Google Scholar] [CrossRef]
- Yasuda-Yamahara, M.; Kume, S.; Maegawa, H. Roles of mTOR in Diabetic Kidney Disease. Antioxidants 2021, 10, 321. [Google Scholar] [CrossRef]
- Barthel, A.; Schmoll, D.; Unterman, T.G. FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 2005, 16, 183–189. [Google Scholar] [CrossRef]
- Wang, Y.; He, W. Improving the Dysregulation of FoxO1 Activity Is a Potential Therapy for Alleviating Diabetic Kidney Disease. Front. Pharmacol. 2021, 12, 630617. [Google Scholar] [CrossRef]
- Huang, F.; Wang, Q.; Guo, F.; Zhao, Y.; Ji, L.; An, T.; Song, Y.; Liu, Y.; He, Y.; Qin, G. FoxO1-mediated inhibition of STAT1 alleviates tubulointerstitial fibrosis and tubule apoptosis in diabetic kidney disease. EBioMedicine 2019, 48, 491–504. [Google Scholar] [CrossRef]
- Li, W.; Wang, Q.; Du, M.; Ma, X.; Wu, L.; Guo, F.; Zhao, S.; Huang, F.; Wang, H.; Qin, G. Effects of overexpressing FoxO1 on apoptosis in glomeruli of diabetic mice and in podocytes cultured in high glucose medium. Biochem. Biophys. Res. Commun. 2016, 478, 612–617. [Google Scholar] [CrossRef]
- Li, X.; Liao, J.; Guo, Z. Detection value of FOXO1 gene methylation, blood glucose and lipids in patients with type 2 diabetic kidney disease. Medicine 2022, 101, e31663. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.; Yue, G.; Xu, J. Placenta-derived mesenchymal stem cells protect against diabetic kidney disease by upregulating autophagy-mediated SIRT1/FOXO1 pathway. Ren. Fail. 2024, 46, 2303396. [Google Scholar] [CrossRef]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef]
- Ma, L.; Wu, F.; Shao, Q.; Chen, G.; Xu, L.; Lu, F. Baicalin Alleviates Oxidative Stress and Inflammation in Diabetic Nephropathy via Nrf2 and MAPK Signaling Pathway. Drug Des. Devel. Ther. 2021, 15, 3207–3221. [Google Scholar] [CrossRef]
- Sen, Z.; Weida, W.; Jie, M.; Li, S.; Dongming, Z.; Xiaoguang, C. Coumarin glycosides from Hydrangea paniculata slow down the progression of diabetic nephropathy by targeting Nrf2 anti-oxidation and smad2/3-mediated profibrosis. Phytomedicine 2019, 57, 385–395. [Google Scholar] [CrossRef]
- Alshehri, A.S. Kaempferol attenuates diabetic nephropathy in streptozotocin-induced diabetic rats by a hypoglycaemic effect and concomitant activation of the Nrf-2/Ho-1/antioxidants axis. Arch. Physiol. Biochem. 2023, 129, 984–997. [Google Scholar] [CrossRef]
- Huang, K.; Gao, X.; Wei, W. The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-beta1 expressions in rat glomerular mesangial cells. Exp. Cell Res. 2017, 361, 63–72. [Google Scholar] [CrossRef]
- Zhuang, K.; Jiang, X.; Liu, R.; Ye, C.; Wang, Y.; Wang, Y.; Quan, S.; Huang, H. Formononetin Activates the Nrf2/ARE Signaling Pathway Via Sirt1 to Improve Diabetic Renal Fibrosis. Front. Pharmacol. 2020, 11, 616378. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, H.; Chen, J.; Zhu, H. Sinomenine improve diabetic nephropathy by inhibiting fibrosis and regulating the JAK2/STAT3/SOCS1 pathway in streptozotocin-induced diabetic rats. Life Sci. 2021, 265, 118855. [Google Scholar] [CrossRef]
- Liu, F.; Zong, M.; Wen, X.; Li, X.; Wang, J.; Wang, Y.; Jiang, W.; Li, X.; Guo, Z.; Qi, H. Silencing of Histone Deacetylase 9 Expression in Podocytes Attenuates Kidney Injury in Diabetic Nephropathy. Sci. Rep. 2016, 6, 33676. [Google Scholar] [CrossRef]
- Elsherbiny, N.M.; Zaitone, S.A.; Mohammad, H.M.F.; El-Sherbiny, M. Renoprotective effect of nifuroxazide in diabetes-induced nephropathy: Impact on NFkappaB, oxidative stress, and apoptosis. Toxicol. Mech. Methods 2018, 28, 467–473. [Google Scholar] [CrossRef]
- Jo, H.A.; Kim, J.Y.; Yang, S.H.; Han, S.S.; Joo, K.W.; Kim, Y.S.; Kim, D.K. The role of local IL6/JAK2/STAT3 signaling in high glucose-induced podocyte hypertrophy. Kidney Res. Clin. Pract. 2016, 35, 212–218. [Google Scholar] [CrossRef]
- Chow, F.; Ozols, E.; Nikolic-Paterson, D.J.; Atkins, R.C.; Tesch, G.H. Macrophages in mouse type 2 diabetic nephropathy: Correlation with diabetic state and progressive renal injury. Kidney Int. 2004, 65, 116–128. [Google Scholar] [CrossRef]
- Yu, J.; Wu, H.; Liu, Z.Y.; Zhu, Q.; Shan, C.; Zhang, K.Q. Advanced glycation end products induce the apoptosis of and inflammation in mouse podocytes through CXCL9-mediated JAK2/STAT3 pathway activation. Int. J. Mol. Med. 2017, 40, 1185–1193. [Google Scholar] [CrossRef]
- Wu, L.; Liu, C.; Chang, D.Y.; Zhan, R.; Zhao, M.; Man Lam, S.; Shui, G.; Zhao, M.H.; Zheng, L.; Chen, M. The Attenuation of Diabetic Nephropathy by Annexin A1 via Regulation of Lipid Metabolism through the AMPK/PPARalpha/CPT1b Pathway. Diabetes 2021, 70, 2192–2203. [Google Scholar] [CrossRef]
- Li, F.; Chen, Y.; Li, Y.; Huang, M.; Zhao, W. Geniposide alleviates diabetic nephropathy of mice through AMPK/SIRT1/NF-kappaB pathway. Eur. J. Pharmacol. 2020, 886, 173449. [Google Scholar] [CrossRef]
- Dugan, L.L.; You, Y.H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.E.; Andreyev, A.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Investig. 2013, 123, 4888–4899. [Google Scholar] [CrossRef]
- Entezari, M.; Hashemi, D.; Taheriazam, A.; Zabolian, A.; Mohammadi, S.; Fakhri, F.; Hashemi, M.; Hushmandi, K.; Ashrafizadeh, M.; Zarrabi, A.; et al. AMPK signaling in diabetes mellitus, insulin resistance and diabetic complications: A pre-clinical and clinical investigation. BioMed. Pharmacother. 2022, 146, 112563. [Google Scholar] [CrossRef]
- Han, Y.C.; Tang, S.Q.; Liu, Y.T.; Li, A.M.; Zhan, M.; Yang, M.; Song, N.; Zhang, W.; Wu, X.Q.; Peng, C.H.; et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 2021, 12, 925. [Google Scholar] [CrossRef]
- Natarajan, R. Epigenetic Mechanisms in Diabetic Vascular Complications and Metabolic Memory: The 2020 Edwin Bierman Award Lecture. Diabetes 2021, 70, 328–337. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, J.; Wang, W.; An, X.; Luo, L.; Yu, D.; Sun, W. Epigenetic modification in diabetic kidney disease. Front. Endocrinol. 2023, 14, 1133970. [Google Scholar] [CrossRef]
- Kuo, F.C.; Chao, C.T.; Lin, S.H. The Dynamics and Plasticity of Epigenetics in Diabetic Kidney Disease: Therapeutic Applications Vis-a-Vis. Int. J. Mol. Sci. 2022, 23, 843. [Google Scholar] [CrossRef]
- Oba, S.; Ayuzawa, N.; Nishimoto, M.; Kawarazaki, W.; Ueda, K.; Hirohama, D.; Kawakami-Mori, F.; Shimosawa, T.; Marumo, T.; Fujita, T. Aberrant DNA methylation of Tgfb1 in diabetic kidney mesangial cells. Sci. Rep. 2018, 8, 16338. [Google Scholar] [CrossRef]
- Thiruvengadam, R.; Venkidasamy, B.; Samynathan, R.; Govindasamy, R.; Thiruvengadam, M.; Kim, J.H. Association of nanoparticles and Nrf2 with various oxidative stress-mediated diseases. Chem. Biol. Interact. 2023, 380, 110535. [Google Scholar] [CrossRef]
- Majumder, S.; Advani, A. The epigenetic regulation of podocyte function in diabetes. J. Diabetes Complicat. 2015, 29, 1337–1344. [Google Scholar] [CrossRef]
- Hayashi, K.; Sasamura, H.; Nakamura, M.; Sakamaki, Y.; Azegami, T.; Oguchi, H.; Tokuyama, H.; Wakino, S.; Hayashi, K.; Itoh, H. Renin-angiotensin blockade resets podocyte epigenome through Kruppel-like Factor 4 and attenuates proteinuria. Kidney Int. 2015, 88, 745–753. [Google Scholar] [CrossRef]
- Kim, H.; Bae, J.H.; Park, K.S.; Sung, J.; Kwak, S.H. DNA Methylation Changes Associated with Type 2 Diabetes and Diabetic Kidney Disease in an East Asian Population. J. Clin. Endocrinol. Metab. 2021, 106, e3837–e3851. [Google Scholar] [CrossRef]
- Larkin, B.P.; Glastras, S.J.; Chen, H.; Pollock, C.A.; Saad, S. DNA methylation and the potential role of demethylating agents in prevention of progressive chronic kidney disease. FASEB J. 2018, 32, 5215–5226. [Google Scholar] [CrossRef]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef]
- Siddiqi, F.S.; Majumder, S.; Thai, K.; Abdalla, M.; Hu, P.; Advani, S.L.; White, K.E.; Bowskill, B.B.; Guarna, G.; Dos Santos, C.C.; et al. The Histone Methyltransferase Enzyme Enhancer of Zeste Homolog 2 Protects against Podocyte Oxidative Stress and Renal Injury in Diabetes. J. Am. Soc. Nephrol. 2016, 27, 2021–2034. [Google Scholar] [CrossRef]
- Zhu, Y.; Yu, C.; Zhuang, S. Protein arginine methyltransferase 1 mediates renal fibroblast activation and fibrogenesis through activation of Smad3 signaling. Am. J. Physiol. Ren. Physiol. 2020, 318, F375–F387. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Sun, G. Histone Acetylation and Its Modifiers in the Pathogenesis of Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 4065382. [Google Scholar] [CrossRef]
- Kourtidou, C.; Tziomalos, K. The Role of Histone Modifications in the Pathogenesis of Diabetic Kidney Disease. Int. J. Mol. Sci. 2023, 24, 6007. [Google Scholar] [CrossRef]
- Lazar, A.G.; Vlad, M.L.; Manea, A.; Simionescu, M.; Manea, S.A. Activated Histone Acetyltransferase p300/CBP-Related Signalling Pathways Mediate Up-Regulation of NADPH Oxidase, Inflammation, and Fibrosis in Diabetic Kidney. Antioxidants 2021, 10, 1356. [Google Scholar] [CrossRef]
- Seok, S.; Kim, Y.C.; Byun, S.; Choi, S.; Xiao, Z.; Iwamori, N.; Zhang, Y.; Wang, C.; Ma, J.; Ge, K.; et al. Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid beta-oxidation. J. Clin. Investig. 2018, 128, 3144–3159. [Google Scholar] [CrossRef]
- Zhang, T.; Chi, Y.; Kang, Y.; Lu, H.; Niu, H.; Liu, W.; Li, Y. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1alpha mediated attenuation of mitochondrial oxidative stress. J. Cell Physiol. 2019, 234, 5033–5043. [Google Scholar] [CrossRef]
- Khan, S.; Jena, G.; Tikoo, K.; Kumar, V. Valproate attenuates the proteinuria, podocyte and renal injury by facilitating autophagy and inactivation of NF-kappaB/iNOS signaling in diabetic rat. Biochimie 2015, 110, 1–16. [Google Scholar] [CrossRef]
- Alghamdi, T.A.; Batchu, S.N.; Hadden, M.J.; Yerra, V.G.; Liu, Y.; Bowskill, B.B.; Advani, S.L.; Geldenhuys, L.; Siddiqi, F.S.; Majumder, S.; et al. Histone H3 Serine 10 Phosphorylation Facilitates Endothelial Activation in Diabetic Kidney Disease. Diabetes 2018, 67, 2668–2681. [Google Scholar] [CrossRef]
- Yuan, H.; Reddy, M.A.; Deshpande, S.; Jia, Y.; Park, J.T.; Lanting, L.L.; Jin, W.; Kato, M.; Xu, Z.G.; Das, S.; et al. Epigenetic Histone Modifications Involved in Profibrotic Gene Regulation by 12/15-Lipoxygenase and Its Oxidized Lipid Products in Diabetic Nephropathy. Antioxid. Redox Signal. 2016, 24, 361–375. [Google Scholar] [CrossRef]
- Cao, Q.; Chen, X.M.; Huang, C.; Pollock, C.A. MicroRNA as novel biomarkers and therapeutic targets in diabetic kidney disease: An update. FASEB Bioadv. 2019, 1, 375–388. [Google Scholar] [CrossRef]
- Hagiwara, S.; McClelland, A.; Kantharidis, P. MicroRNA in diabetic nephropathy: Renin angiotensin, aGE/RAGE, and oxidative stress pathway. J. Diabetes Res. 2013, 2013, 173783. [Google Scholar] [CrossRef]
- Gao, C.; Wang, B.; Chen, Q.; Wang, M.; Fei, X.; Zhao, N. Serum exosomes from diabetic kidney disease patients promote pyroptosis and oxidative stress through the miR-4449/HIC1 pathway. Nutr. Diabetes 2021, 11, 33. [Google Scholar] [CrossRef]
- Wu, J.; Lu, K.; Zhu, M.; Xie, X.; Ding, Y.; Shao, X.; Chen, Y.; Liu, J.; Xu, M.; Xu, Y.; et al. miR-485 suppresses inflammation and proliferation of mesangial cells in an in vitro model of diabetic nephropathy by targeting NOX5. BioChem. Biophys. Res. Commun. 2020, 521, 984–990. [Google Scholar] [CrossRef]
- Hu, M.; Ma, Q.; Liu, B.; Wang, Q.; Zhang, T.; Huang, T.; Lv, Z. Long Non-Coding RNAs in the Pathogenesis of Diabetic Kidney Disease. Front. Cell Dev. Biol. 2022, 10, 845371. [Google Scholar] [CrossRef]
- Feng, X.; Zhao, J.; Ding, J.; Shen, X.; Zhou, J.; Xu, Z. LncRNA Blnc1 expression and its effect on renal fibrosis in diabetic nephropathy. Am. J. Transl. Res. 2019, 11, 5664–5672. [Google Scholar]
- Li, N.; Jia, T.; Li, Y.R. LncRNA NEAT1 accelerates the occurrence and development of diabetic nephropathy by sponging miR-23c. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1325–1337. [Google Scholar] [CrossRef]
- Wu, X.; Fan, D.; Chen, B. LncRNA NEAT1 Accelerates the Proliferation, Oxidative Stress, Inflammation, and Fibrosis and Suppresses the Apoptosis through the miR-423-5p/GLIPR2 Axis in Diabetic Nephropathy. J. Cardiovasc. Pharmacol. 2022, 79, 342–354. [Google Scholar] [CrossRef]
- Wang, G.; Wu, B.; Zhang, B.; Wang, K.; Wang, H. LncRNA CTBP1-AS2 alleviates high glucose-induced oxidative stress, ECM accumulation, and inflammation in diabetic nephropathy via miR-155-5p/FOXO1 axis. Biochem. Biophys. Res. Commun. 2020, 532, 308–314. [Google Scholar] [CrossRef]
- Zhang, X.L.; Zhu, H.Q.; Zhang, Y.; Zhang, C.Y.; Jiao, J.S.; Xing, X.Y. LncRNA CASC2 regulates high glucose-induced proliferation, extracellular matrix accumulation and oxidative stress of human mesangial cells via miR-133b/FOXP1 axis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 802–812. [Google Scholar] [CrossRef]
- Shu, S.; Xu, Z.; Lu, H.; Li, Z.; Zhang, Y. CircHOMER1 aggravates oxidative stress, inflammation and extracellular matrix deposition in high glucose-induced human mesangial cells. Nephrology 2022, 27, 983–993. [Google Scholar] [CrossRef]
- Wang, J.; Yang, S.; Li, W.; Zhao, M.; Li, K. Circ_0000491 Promotes Apoptosis, Inflammation, Oxidative Stress, and Fibrosis in High Glucose-Induced Mesangial Cells by Regulating miR-455-3p/Hmgb1 Axis. Nephron 2022, 146, 72–83. [Google Scholar] [CrossRef]
- Zhuang, L.; Jin, G.; Qiong, W.; Ge, X.; Pei, X. Circular RNA COL1A2 Mediates High Glucose-Induced Oxidative Stress and Pyroptosis by Regulating MiR-424-5p/SGK1 in Diabetic Nephropathy. Appl. Biochem. Biotechnol. 2023, 195, 7652–7667. [Google Scholar] [CrossRef]
- Sun, L.; Han, Y.; Shen, C.; Luo, H.; Wang, Z. Emodin alleviates high glucose-induced oxidative stress, inflammation and extracellular matrix accumulation of mesangial cells by the circ_0000064/miR-30c-5p/Lmp7 axis. J. Recept. Signal. Transduct Res. 2022, 42, 302–312. [Google Scholar] [CrossRef]
- Ren, H.; Shao, Y.; Wu, C.; Ma, X.; Lv, C.; Wang, Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol. Cell Endocrinol. 2020, 500, 110628. [Google Scholar] [CrossRef]
- Alshahrani, M.Y.; Ebrahim, H.A.; Alqahtani, S.M.; Bayoumy, N.M.; Kamar, S.S.; ShamsEldeen, A.M.; Haidara, M.A.; Al-Ani, B.; Albawardi, A. Metformin Suppresses Thioacetamide-Induced Chronic Kidney Disease in Association with the Upregulation of AMPK and Downregulation of Oxidative Stress and Inflammation as Well as Dyslipidemia and Hypertension. Molecules 2023, 28, 2756. [Google Scholar] [CrossRef]
- Mima, A. Mitochondria-targeted drugs for diabetic kidney disease. Heliyon 2022, 8, e08878. [Google Scholar] [CrossRef]
- Nam, B.Y.; Jhee, J.H.; Park, J.; Kim, S.; Kim, G.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Yu, J.W.; Han, S.H. PGC-1alpha inhibits the NLRP3 inflammasome via preserving mitochondrial viability to protect kidney fibrosis. Cell Death Dis. 2022, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ma, X.Y.; Han, J.Y.; Yang, M.; Lv, C.; Shao, Y.; Wang, Y.L.; Kang, J.Y.; Wang, Q.Y. Metformin regulates inflammation and fibrosis in diabetic kidney disease through TNC/TLR4/NF-kappaB/miR-155-5p inflammatory loop. World J. Diabetes 2021, 12, 19–46. [Google Scholar] [CrossRef]
- Lee, Y.J.; Han, H.J. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3beta, Snail1, and beta-catenin in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2010, 298, F1263–F1275. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.D.; Yu, S.L.; Wang, R.; Liu, J.N.; Jin, Y.S.; Li, Y.F.; An, R.H. Rosiglitazone Suppresses Calcium Oxalate Crystal Binding and Oxalate-Induced Oxidative Stress in Renal Epithelial Cells by Promoting PPAR-gamma Activation and Subsequent Regulation of TGF-beta1 and HGF Expression. Oxid. Med. Cell Longev. 2019, 2019, 4826525. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, A.; Mozes, M.M.; Calvier, L.; Hansmann, G.; Kokeny, G. The PPARgamma agonist pioglitazone prevents TGF-beta induced renal fibrosis by repressing EGR-1 and STAT3. BMC Nephrol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Sonneveld, R.; Hoenderop, J.G.; Isidori, A.M.; Henique, C.; Dijkman, H.B.; Berden, J.H.; Tharaux, P.L.; van der Vlag, J.; Nijenhuis, T. Sildenafil Prevents Podocyte Injury via PPAR-gamma-Mediated TRPC6 Inhibition. J. Am. Soc. Nephrol. 2017, 28, 1491–1505. [Google Scholar] [CrossRef]
- Ho, C.C.; Yang, Y.S.; Huang, C.N.; Lo, S.C.; Wang, Y.H.; Kornelius, E. The efficacy of pioglitazone for renal protection in diabetic kidney disease. PLoS ONE 2022, 17, e0264129. [Google Scholar] [CrossRef]
- Zhu, C.; Huang, S.; Yuan, Y.; Ding, G.; Chen, R.; Liu, B.; Yang, T.; Zhang, A. Mitochondrial dysfunction mediates aldosterone-induced podocyte damage: A therapeutic target of PPARgamma. Am. J. Pathol. 2011, 178, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Cersosimo, E.; Triplitt, C.; DeFronzo, R.A. Rosiglitazone decreases albuminuria in type 2 diabetic patients. Kidney Int. 2007, 72, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Pistrosch, F.; Passauer, J.; Herbrig, K.; Schwanebeck, U.; Gross, P.; Bornstein, S.R. Effect of thiazolidinedione treatment on proteinuria and renal hemodynamic in type 2 diabetic patients with overt nephropathy. Horm. Metab. Res. 2012, 44, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- The, E.-K.C.G.; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar] [CrossRef]
- Muskiet, M.H.A.; Tonneijck, L.; Huang, Y.; Liu, M.; Saremi, A.; Heerspink, H.J.L.; van Raalte, D.H. Lixisenatide and renal outcomes in patients with type 2 diabetes and acute coronary syndrome: An exploratory analysis of the ELIXA randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2018, 6, 859–869. [Google Scholar] [CrossRef]
- Abdel-Latif, R.G.; Ahmed, A.F.; Heeba, G.H. Low-dose lixisenatide protects against early-onset nephropathy induced in diabetic rats. Life Sci. 2020, 263, 118592. [Google Scholar] [CrossRef] [PubMed]
- Clegg, L.E.; Penland, R.C.; Bachina, S.; Boulton, D.W.; Thuresson, M.; Heerspink, H.J.L.; Gustavson, S.; Sjostrom, C.D.; Ruggles, J.A.; Hernandez, A.F.; et al. Effects of exenatide and open-label SGLT2 inhibitor treatment, given in parallel or sequentially, on mortality and cardiovascular and renal outcomes in type 2 diabetes: Insights from the EXSCEL trial. Cardiovasc. Diabetol. 2019, 18, 138. [Google Scholar] [CrossRef]
- Mann, J.F.E.; Orsted, D.D.; Brown-Frandsen, K.; Marso, S.P.; Poulter, N.R.; Rasmussen, S.; Tornoe, K.; Zinman, B.; Buse, J.B.; Committee, L.S.; et al. Liraglutide and Renal Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 839–848. [Google Scholar] [CrossRef]
- Liu, J.; Guo, S.; Li, H.; Liu, X.Y. Effects of glucagon-like peptide-1 receptor agonists (GLP-1RAs) on podocytes, inflammation, and oxidative stress in patients with diabetic nephropathy (DN). Pak. J. Med. Sci. 2022, 38, 1170–1174. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Bosch-Traberg, H.; Cherney, D.Z.I.; Hadjadj, S.; Lawson, J.; Mosenzon, O.; Rasmussen, S.; Bain, S.C. Post hoc analysis of SUSTAIN 6 and PIONEER 6 trials suggests that people with type 2 diabetes at high cardiovascular risk treated with semaglutide experience more stable kidney function compared with placebo. Kidney Int. 2023, 103, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Rossing, P.; Baeres, F.M.M.; Bakris, G.; Bosch-Traberg, H.; Gislum, M.; Gough, S.C.L.; Idorn, T.; Lawson, J.; Mahaffey, K.W.; Mann, J.F.E.; et al. The rationale, design and baseline data of FLOW, a kidney outcomes trial with once-weekly semaglutide in people with type 2 diabetes and chronic kidney disease. Nephrol. Dial. Transpl. 2023, 38, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; D’Agostino, R.B., Sr.; Granger, C.B.; Jones, N.P.; Leiter, L.A.; Rosenberg, A.E.; Sigmon, K.N.; Somerville, M.C.; et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): A double-blind, randomised placebo-controlled trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef]
- Hoffmann-Petersen, I.T.; Holt, C.B.; Jensen, L.; Hage, C.; Mellbin, L.G.; Thiel, S.; Hansen, T.K.; Ostergaard, J.A. Effect of dipeptidyl peptidase-4 inhibitors on complement activation. Diabetes Metab. Res. Rev. 2021, 37, e3385. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Kanda, H.; Takama, H.; Ichikawa, T.; Hase, H.; Akizawa, T. Randomized Clinical Trial on the Effect of Bardoxolone Methyl on GFR in Diabetic Kidney Disease Patients (TSUBAKI Study). Kidney Int. Rep. 2020, 5, 879–890. [Google Scholar] [CrossRef]
- De Zeeuw, D.; Anzalone, D.A.; Cain, V.A.; Cressman, M.D.; Heerspink, H.J.; Molitoris, B.A.; Monyak, J.T.; Parving, H.H.; Remuzzi, G.; Sowers, J.R.; et al. Renal effects of atorvastatin and rosuvastatin in patients with diabetes who have progressive renal disease (PLANET I): A randomised clinical trial. Lancet Diabetes Endocrinol. 2015, 3, 181–190. [Google Scholar] [CrossRef]
- Yang, J.; Ma, X.; Niu, D.; Sun, Y.; Chai, X.; Deng, Y.; Wang, J.; Dong, J. PCSK9 inhibitors suppress oxidative stress and inflammation in atherosclerotic development by promoting macrophage autophagy. Am. J. Transl. Res. 2023, 15, 5129–5144. [Google Scholar]
- Lee, J.S.; Mukhopadhyay, P.; Matyas, C.; Trojnar, E.; Paloczi, J.; Yang, Y.R.; Blank, B.A.; Savage, C.; Sorokin, A.V.; Mehta, N.N.; et al. PCSK9 inhibition as a novel therapeutic target for alcoholic liver disease. Sci. Rep. 2019, 9, 17167. [Google Scholar] [CrossRef]
- Safaeian, L.; Mirian, M.; Bahrizadeh, S. Evolocumab, a PCSK9 inhibitor, protects human endothelial cells against H2O2-induced oxidative stress. Arch. Physiol. Biochem. 2022, 128, 1681–1686. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Michels, A.J.; Frei, B. Vitamin C. Adv. Nutr. 2014, 5, 16–18. [Google Scholar] [CrossRef]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef]
- Farhangi, M.A.; Mesgari-Abbasi, M.; Hajiluian, G.; Nameni, G.; Shahabi, P. Adipose Tissue Inflammation and Oxidative Stress: The Ameliorative Effects of Vitamin D. Inflammation 2017, 40, 1688–1697. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta 2014, 1840, 2709–2729. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
- Sun, F.; Jiang, D.; Cai, J. Effects of valsartan combined with alpha-lipoic acid on renal function in patients with diabetic nephropathy: A systematic review and meta-analysis. BMC Endocr. Disord. 2021, 21, 178. [Google Scholar] [CrossRef]
- Jiang, Z.; Tan, Z.; Meng, F.; Li, X. Curative Effects of Valsartan Alone or Combined with Alpha-lipoic Acid on Inflammatory Cytokines and Renal Function in Early-stage Diabetic Kidney Disease. J. Coll. Physicians Surg. Pak. 2019, 29, 1009–1011. [Google Scholar] [CrossRef]
- Arambasic, J.; Mihailovic, M.; Uskokovic, A.; Dinic, S.; Grdovic, N.; Markovic, J.; Poznanovic, G.; Bajec, D.; Vidakovic, M. Alpha-lipoic acid upregulates antioxidant enzyme gene expression and enzymatic activity in diabetic rat kidneys through an O-GlcNAc-dependent mechanism. Eur. J. Nutr. 2013, 52, 1461–1473. [Google Scholar] [CrossRef]
- Lewis, E.J.; Greene, T.; Spitalewiz, S.; Blumenthal, S.; Berl, T.; Hunsicker, L.G.; Pohl, M.A.; Rohde, R.D.; Raz, I.; Yerushalmy, Y.; et al. Pyridorin in type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 131–136. [Google Scholar] [CrossRef]
- Beddhu, S.; Filipowicz, R.; Wang, B.; Wei, G.; Chen, X.; Roy, A.C.; DuVall, S.L.; Farrukh, H.; Habib, A.N.; Bjordahl, T.; et al. A Randomized Controlled Trial of the Effects of Febuxostat Therapy on Adipokines and Markers of Kidney Fibrosis in Asymptomatic Hyperuricemic Patients With Diabetic Nephropathy. Can. J. Kidney Health Dis. 2016, 3, 2054358116675343. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Koenig, W.; Libby, P.; Everett, B.M.; Lefkowitz, M.; Thuren, T.; Cornel, J.H. Inhibition of Interleukin-1beta by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. J. Am. Coll. Cardiol. 2018, 71, 2405–2414. [Google Scholar] [CrossRef]
- Ray, K.K.; Nicholls, S.J.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Effect of Apabetalone Added to Standard Therapy on Major Adverse Cardiovascular Events in Patients with Recent Acute Coronary Syndrome and Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2020, 323, 1565–1573. [Google Scholar] [CrossRef]
- Martinez-Moreno, J.M.; Fontecha-Barriuso, M.; Martin-Sanchez, D.; Guerrero-Mauvecin, J.; Goma-Garces, E.; Fernandez-Fernandez, B.; Carriazo, S.; Sanchez-Nino, M.D.; Ramos, A.M.; Ruiz-Ortega, M.; et al. Epigenetic Modifiers as Potential Therapeutic Targets in Diabetic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 4113. [Google Scholar] [CrossRef]
- Jordan, S.C.; Choi, J.; Aubert, O.; Haas, M.; Loupy, A.; Huang, E.; Peng, A.; Kim, I.; Louie, S.; Ammerman, N.; et al. A phase I/II, double-blind, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceased donor kidney transplant recipients. Am. J. Transpl. 2018, 18, 2955–2964. [Google Scholar] [CrossRef]
- Yue, Y.; Yeh, J.N.; Chiang, J.Y.; Sung, P.H.; Chen, Y.L.; Liu, F.; Yip, H.K. Intrarenal arterial administration of human umbilical cord-derived mesenchymal stem cells effectively preserved the residual renal function of diabetic kidney disease in rat. Stem Cell Res. Ther. 2022, 13, 186. [Google Scholar] [CrossRef]
- He, J.; Liu, B.; Du, X.; Wei, Y.; Kong, D.; Feng, B.; Guo, R.; Asiamah, E.A.; Griffin, M.D.; Hynes, S.O.; et al. Amelioration of diabetic nephropathy in mice by a single intravenous injection of human mesenchymal stromal cells at early and later disease stages is associated with restoration of autophagy. Stem. Cell Res. Ther. 2024, 15, 66. [Google Scholar] [CrossRef]
- Han, X.; Wang, J.; Li, R.; Huang, M.; Yue, G.; Guan, L.; Deng, Y.; Cai, W.; Xu, J. Placental Mesenchymal Stem Cells Alleviate Podocyte Injury in Diabetic Kidney Disease by Modulating Mitophagy via the SIRT1-PGC-1alpha-TFAM Pathway. Int. J. Mol. Sci. 2023, 24, 4696. [Google Scholar] [CrossRef]
- Yu, S.; Cheng, Y.; Zhang, L.; Yin, Y.; Xue, J.; Li, B.; Gong, Z.; Gao, J.; Mu, Y. Treatment with adipose tissue-derived mesenchymal stem cells exerts anti-diabetic effects, improves long-term complications, and attenuates inflammation in type 2 diabetic rats. Stem Cell Res. Ther. 2019, 10, 333. [Google Scholar] [CrossRef]
- Hickson, L.J.; Eirin, A.; Conley, S.M.; Taner, T.; Bian, X.; Saad, A.; Herrmann, S.M.; Mehta, R.A.; McKenzie, T.J.; Kellogg, T.A.; et al. Diabetic Kidney Disease Alters the Transcriptome and Function of Human Adipose-Derived Mesenchymal Stromal Cells but Maintains Immunomodulatory and Paracrine Activities Important for Renal Repair. Diabetes 2021, 70, 1561–1574. [Google Scholar] [CrossRef]
- Khamis, T.; Abdelkhalek, A.; Abdellatif, H.; Dwidar, N.; Said, A.; Ahmed, R.; Wagdy, K.; Elgarhy, R.; Eltahan, R.; Mohamed, H.; et al. BM-MSCs alleviate diabetic nephropathy in male rats by regulating ER stress, oxidative stress, inflammation, and apoptotic pathways. Front. Pharmacol. 2023, 14, 1265230. [Google Scholar] [CrossRef]
- Packham, D.K.; Fraser, I.R.; Kerr, P.G.; Segal, K.R. Allogeneic Mesenchymal Precursor Cells (MPC) in Diabetic Nephropathy: A Randomized, Placebo-controlled, Dose Escalation Study. EBioMedicine 2016, 12, 263–269. [Google Scholar] [CrossRef]
- Scheele, W.; Diamond, S.; Gale, J.; Clerin, V.; Tamimi, N.; Le, V.; Walley, R.; Grover-Paez, F.; Perros-Huguet, C.; Rolph, T.; et al. Phosphodiesterase Type 5 Inhibition Reduces Albuminuria in Subjects with Overt Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 3459–3468. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, J.; Zhou, F.; Wang, W.; Chen, N. PGC-1alpha ameliorates kidney fibrosis in mice with diabetic kidney disease through an antioxidative mechanism. Mol. Med. Rep. 2018, 17, 4490–4498. [Google Scholar] [CrossRef]
- Koniari, I.; Velissaris, D.; Kounis, N.G.; Koufou, E.; Artopoulou, E.; de Gregorio, C.; Mplani, V.; Paraskevas, T.; Tsigkas, G.; Hung, M.Y.; et al. Anti-Diabetic Therapy, Heart Failure and Oxidative Stress: An Update. J. Clin. Med. 2022, 11, 4660. [Google Scholar] [CrossRef]
- Maeda, S.; Matsui, T.; Takeuchi, M.; Yamagishi, S. Sodium-glucose cotransporter 2-mediated oxidative stress augments advanced glycation end products-induced tubular cell apoptosis. Diabetes Metab. Res. Rev. 2013, 29, 406–412. [Google Scholar] [CrossRef]
- Song, Y.; Guo, F.; Liu, Y.; Huang, F.; Fan, X.; Zhao, L.; Qin, G. Identification of circular RNAs and functional competing endogenous RNA networks in human proximal tubular epithelial cells treated with sodium-glucose cotransporter 2 inhibitor dapagliflozin in diabetic kidney disease. Bioengineered 2022, 13, 3911–3929. [Google Scholar] [CrossRef]
- Jaikumkao, K.; Thongnak, L.; Htun, K.T.; Pengrattanachot, N.; Phengpol, N.; Sutthasupha, P.; Promsan, S.; Montha, N.; Sriburee, S.; Kothan, S.; et al. Dapagliflozin and metformin in combination ameliorates diabetic nephropathy by suppressing oxidative stress, inflammation, and apoptosis and activating autophagy in diabetic rats. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 166912. [Google Scholar] [CrossRef]
- Leng, W.; Ouyang, X.; Lei, X.; Wu, M.; Chen, L.; Wu, Q.; Deng, W.; Liang, Z. The SGLT-2 Inhibitor Dapagliflozin Has a Therapeutic Effect on Atherosclerosis in Diabetic ApoE−/− Mice. Mediat. Inflamm. 2016, 2016, 6305735. [Google Scholar] [CrossRef]
- Maki, T.; Maeno, S.; Maeda, Y.; Yamato, M.; Sonoda, N.; Ogawa, Y.; Wakisaka, M.; Inoguchi, T. Amelioration of diabetic nephropathy by SGLT2 inhibitors independent of its glucose-lowering effect: A possible role of SGLT2 in mesangial cells. Sci. Rep. 2019, 9, 4703. [Google Scholar] [CrossRef]
- Corremans, R.; Vervaet, B.A.; Dams, G.; D’Haese, P.C.; Verhulst, A. Metformin and Canagliflozin Are Equally Renoprotective in Diabetic Kidney Disease but Have No Synergistic Effect. Int. J. Mol. Sci. 2023, 24, 9043. [Google Scholar] [CrossRef]
- van der Hoek, S.; Jongs, N.; Oshima, M.; Neuen, B.L.; Stevens, J.; Perkovic, V.; Levin, A.; Mahaffey, K.W.; Pollock, C.; Greene, T.; et al. Glycemic Control and Effects of Canagliflozin in Reducing Albuminuria and eGFR: A Post Hoc Analysis of the CREDENCE Trial. Clin. J. Am. Soc. Nephrol. 2023, 18, 748–758. [Google Scholar] [CrossRef]
- Yi, T.W.; Wong, M.M.Y.; Neuen, B.L.; Arnott, C.; Poirier, P.; Seufert, J.; Slee, A.; Rapattoni, W.; Ang, F.G.; Wheeler, D.C.; et al. Effects of canagliflozin on cardiovascular and kidney events in patients with chronic kidney disease with and without peripheral arterial disease: Integrated analysis from the CANVAS Program and CREDENCE trial. Diabetes Obes. Metab. 2023, 25, 2043–2047. [Google Scholar] [CrossRef]
- Alharbi, S.H. Anti-inflammatory role of glucagon-like peptide 1 receptor agonists and its clinical implications. Ther. Adv. Endocrinol. Metab. 2024, 15, 20420188231222367. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Ryden, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Yang, S.; Lin, C.; Zhuo, X.; Wang, J.; Rao, S.; Xu, W.; Cheng, Y.; Yang, L. Glucagon-like peptide-1 alleviates diabetic kidney disease through activation of autophagy by regulating AMP-activated protein kinase-mammalian target of rapamycin pathway. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E1019–E1030. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Ruggenenti, P. The CARDINAL Trial of Bardoxolone Methyl in Alport Syndrome: When Marketing Interests Prevail over Patients Clinical Needs. Nephron 2023, 147, 465–469. [Google Scholar] [CrossRef]
- Golbidi, S.; Ebadi, S.A.; Laher, I. Antioxidants in the treatment of diabetes. Curr. Diabetes Rev. 2011, 7, 106–125. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Lambris, J.D. Complement-targeted therapeutics. Nat. Biotechnol. 2007, 25, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Avacopan for the Treatment of ANCA-Associated Vasculitis. N. Engl. J. Med. 2024, 390, 388. [CrossRef]
- Hellmich, B.; Sanchez-Alamo, B.; Schirmer, J.H.; Berti, A.; Blockmans, D.; Cid, M.C.; Holle, J.U.; Hollinger, N.; Karadag, O.; Kronbichler, A.; et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Ann. Rheum. Dis. 2024, 83, 30–47. [Google Scholar] [CrossRef]
- Henry Li, H.; Riedl, M.; Kashkin, J. Update on the Use of C1-Esterase Inhibitor Replacement Therapy in the Acute and Prophylactic Treatment of Hereditary Angioedema. Clin. Rev. Allergy Immunol. 2019, 56, 207–218. [Google Scholar] [CrossRef]
- Ogden, C.A.; deCathelineau, A.; Hoffmann, P.R.; Bratton, D.; Ghebrehiwet, B.; Fadok, V.A.; Henson, P.M. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 2001, 194, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Sikora, J.; Wroblewska-Czech, A.; Smycz-Kubanska, M.; Mielczarek-Palacz, A.; Cygal, A.; Witek, A.; Kondera-Anasz, Z. The role of complement components C1q, MBL and C1 inhibitor in pathogenesis of endometriosis. Arch. Gynecol. Obs. 2018, 297, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.K.; Claes, K.; Goh, Y.T.; Kwong, Y.L.; Leung, N.; Mendrek, W.; Nakamura, R.; Sathar, J.; Ng, E.; Nangia, N.; et al. Narsoplimab, a Mannan-Binding Lectin-Associated Serine Protease-2 Inhibitor, for the Treatment of Adult Hematopoietic Stem-Cell Transplantation-Associated Thrombotic Microangiopathy. J. Clin. Oncol. 2022, 40, 2447–2457. [Google Scholar] [CrossRef]
- Russo, G.; Piscitelli, P.; Giandalia, A.; Viazzi, F.; Pontremoli, R.; Fioretto, P.; De Cosmo, S. Atherogenic dyslipidemia and diabetic nephropathy. J. Nephrol. 2020, 33, 1001–1008. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [CrossRef]
- Tunnicliffe, D.J.; Palmer, S.C.; Cashmore, B.A.; Saglimbene, V.M.; Krishnasamy, R.; Lambert, K.; Johnson, D.W.; Craig, J.C.; Strippoli, G.F. HMG CoA reductase inhibitors (statins) for people with chronic kidney disease not requiring dialysis. Cochrane Database Syst. Rev. 2023, 11, CD007784. [Google Scholar] [CrossRef]
- Cammisotto, V.; Baratta, F.; Simeone, P.G.; Barale, C.; Lupia, E.; Galardo, G.; Santilli, F.; Russo, I.; Pignatelli, P. Proprotein Convertase Subtilisin Kexin Type 9 (PCSK9) Beyond Lipids: The Role in Oxidative Stress and Thrombosis. Antioxidants 2022, 11, 569. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, S.; Wang, X.; Theus, S.; Deng, X.; Fan, Y.; Zhou, S.; Mehta, J.L. PCSK9 regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc. Res. 2018, 114, 1145–1153. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Prattichizzo, F.; Marfella, R.; Sardu, C.; Martino, E.; Scisciola, L.; Marfella, L.; Grotta, R.; Frige, C.; Paolisso, G.; et al. SIRT3 mediates the effects of PCSK9 inhibitors on inflammation, autophagy, and oxidative stress in endothelial cells. Theranostics 2023, 13, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Cammisotto, V.; Pastori, D.; Nocella, C.; Bartimoccia, S.; Castellani, V.; Marchese, C.; Scavalli, A.S.; Ettorre, E.; Viceconte, N.; Violi, F.; et al. PCSK9 Regulates Nox2-Mediated Platelet Activation via CD36 Receptor in Patients with Atrial Fibrillation. Antioxidants 2020, 9, 296. [Google Scholar] [CrossRef] [PubMed]
- Cammisotto, V.; Baratta, F.; Castellani, V.; Bartimoccia, S.; Nocella, C.; D’Erasmo, L.; Cocomello, N.; Barale, C.; Scicali, R.; Di Pino, A.; et al. Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors Reduce Platelet Activation Modulating ox-LDL Pathways. Int. J. Mol. Sci. 2021, 22, 7193. [Google Scholar] [CrossRef]
- Elewa, U.; Fernandez-Fernandez, B.; Mahillo-Fernandez, I.; Martin-Cleary, C.; Sanz, A.B.; Sanchez-Nino, M.D.; Ortiz, A. PCSK9 in diabetic kidney disease. Eur. J. Clin. Investig. 2016, 46, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Liao, X.; Peng, J.; Quan, J.; Zhang, H.; Huang, Z.; Yi, B. PCSK9 causes inflammation and cGAS/STING pathway activation in diabetic nephropathy. FASEB J. 2023, 37, e23127. [Google Scholar] [CrossRef] [PubMed]
- Habiba, U.E.; Khan, N.; Greene, D.L.; Shamim, S.; Umer, A. The therapeutic effect of mesenchymal stem cells in diabetic kidney disease. J. Mol. Med. 2024, 102, 537–570. [Google Scholar] [CrossRef]
- Xiang, E.; Han, B.; Zhang, Q.; Rao, W.; Wang, Z.; Chang, C.; Zhang, Y.; Tu, C.; Li, C.; Wu, D. Human umbilical cord-derived mesenchymal stem cells prevent the progression of early diabetic nephropathy through inhibiting inflammation and fibrosis. Stem Cell Res. Ther. 2020, 11, 336. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Li, Q.; Liu, K.; Hou, J.; Shao, C.; Wang, Y. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat. Rev. Nephrol. 2018, 14, 493–507. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, F.; Zhang, W.; Lv, J.; Lv, J.; Yin, A. BMSCs and miR-124a ameliorated diabetic nephropathy via inhibiting notch signalling pathway. J. Cell Mol. Med. 2018, 22, 4840–4855. [Google Scholar] [CrossRef] [PubMed]
- Li, F.X.; Lin, X.; Xu, F.; Shan, S.K.; Guo, B.; Lei, L.M.; Zheng, M.H.; Wang, Y.; Xu, Q.S.; Yuan, L.Q. The Role of Mesenchymal Stromal Cells-Derived Small Extracellular Vesicles in Diabetes and Its Chronic Complications. Front. Endocrinol. 2021, 12, 780974. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Hu, H.; Guo, R.; Wang, H.; Jiang, H. Mesenchymal Stem Cell Exosomes as a New Strategy for the Treatment of Diabetes Complications. Front. Endocrinol. 2021, 12, 646233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Wang, H.; Lv, S.; Liu, Q.; Li, S.; Yang, X.; Liu, G. Mesenchymal Stem Cell-Derived Exosomes Ameliorate Diabetic Kidney Disease Through the NLRP3 Signaling Pathway. Stem Cells 2023, 41, 368–383. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, S.; Wang, C.; Wang, Y.; Wan, M.; Liu, F.; Gong, M.; Yuan, Y.; Chen, Y.; Cheng, J.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Attenuate Mitochondrial Damage and Inflammation by Stabilizing Mitochondrial DNA. ACS Nano 2021, 15, 1519–1538. [Google Scholar] [CrossRef]
- Savio-Silva, C.; Soinski-Sousa, P.E.; Simplicio-Filho, A.; Bastos, R.M.C.; Beyerstedt, S.; Rangel, E.B. Therapeutic Potential of Mesenchymal Stem Cells in a Pre-Clinical Model of Diabetic Kidney Disease and Obesity. Int. J. Mol. Sci. 2021, 22, 1546. [Google Scholar] [CrossRef]
- Perico, N.; Remuzzi, G.; Griffin, M.D.; Cockwell, P.; Maxwell, A.P.; Casiraghi, F.; Rubis, N.; Peracchi, T.; Villa, A.; Todeschini, M.; et al. Safety and Preliminary Efficacy of Mesenchymal Stromal Cell (ORBCEL-M) Therapy in Diabetic Kidney Disease: A Randomized Clinical Trial (NEPHSTROM). J. Am. Soc. Nephrol. 2023, 34, 1733–1751. [Google Scholar] [CrossRef] [PubMed]
- Harloff, M.; Pruschenk, S.; Seifert, R.; Schlossmann, J. Activation of soluble guanylyl cyclase signalling with cinaciguat improves impaired kidney function in diabetic mice. Br. J. Pharmacol. 2022, 179, 2460–2475. [Google Scholar] [CrossRef]
- Balzer, M.S.; Pavkovic, M.; Frederick, J.; Abedini, A.; Freyberger, A.; Vienenkotter, J.; Mathar, I.; Siudak, K.; Eitner, F.; Sandner, P.; et al. Treatment effects of soluble guanylate cyclase modulation on diabetic kidney disease at single-cell resolution. Cell Rep. Med. 2023, 4, 100992. [Google Scholar] [CrossRef]
- Hu, L.; Chen, Y.; Zhou, X.; Hoek, M.; Cox, J.; Lin, K.; Liu, Y.; Blumenschein, W.; Grein, J.; Swaminath, G. Effects of soluble guanylate cyclase stimulator on renal function in ZSF-1 model of diabetic nephropathy. PLoS ONE 2022, 17, e0261000. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, G.A.; Harrison, P.C.; Lincoln, K.; Chen, H.; Sun, P.; Hill, J.; Qian, H.S.; McHugh, M.C.; Clifford, H.; Ng, K.J.; et al. The Novel, Clinical-Stage Soluble Guanylate Cyclase Activator BI 685509 Protects from Disease Progression in Models of Renal Injury and Disease. J. Pharmacol. Exp. Ther. 2023, 384, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Ott, I.M.; Alter, M.L.; von Websky, K.; Kretschmer, A.; Tsuprykov, O.; Sharkovska, Y.; Krause-Relle, K.; Raila, J.; Henze, A.; Stasch, J.P.; et al. Effects of stimulation of soluble guanylate cyclase on diabetic nephropathy in diabetic eNOS knockout mice on top of angiotensin II receptor blockade. PLoS ONE 2012, 7, e42623. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Shea, C.M.; Jones, J.E.; Price, G.M.; Warren, W.; Lonie, E.; Yan, S.; Currie, M.G.; Profy, A.T.; Masferrer, J.L.; et al. Praliciguat inhibits progression of diabetic nephropathy in ZSF1 rats and suppresses inflammation and apoptosis in human renal proximal tubular cells. Am. J. Physiol. Ren. Physiol. 2020, 319, F697–F711. [Google Scholar] [CrossRef] [PubMed]
- Guoguo, S.; Akaike, T.; Tao, J.; Qi, C.; Nong, Z.; Hui, L. HGF-mediated inhibition of oxidative stress by 8-nitro-cGMP in high glucose-treated rat mesangial cells. Free Radic. Res. 2012, 46, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Gevezova, M.; Sarafian, V.; Maes, M. Redox regulation of the immune response. Cell Mol. Immunol. 2022, 19, 1079–1101. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, K.; Garg, S.S.; Gupta, J. Targeting epigenetic regulators for treating diabetic nephropathy. Biochimie 2022, 202, 146–158. [Google Scholar] [CrossRef]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef]
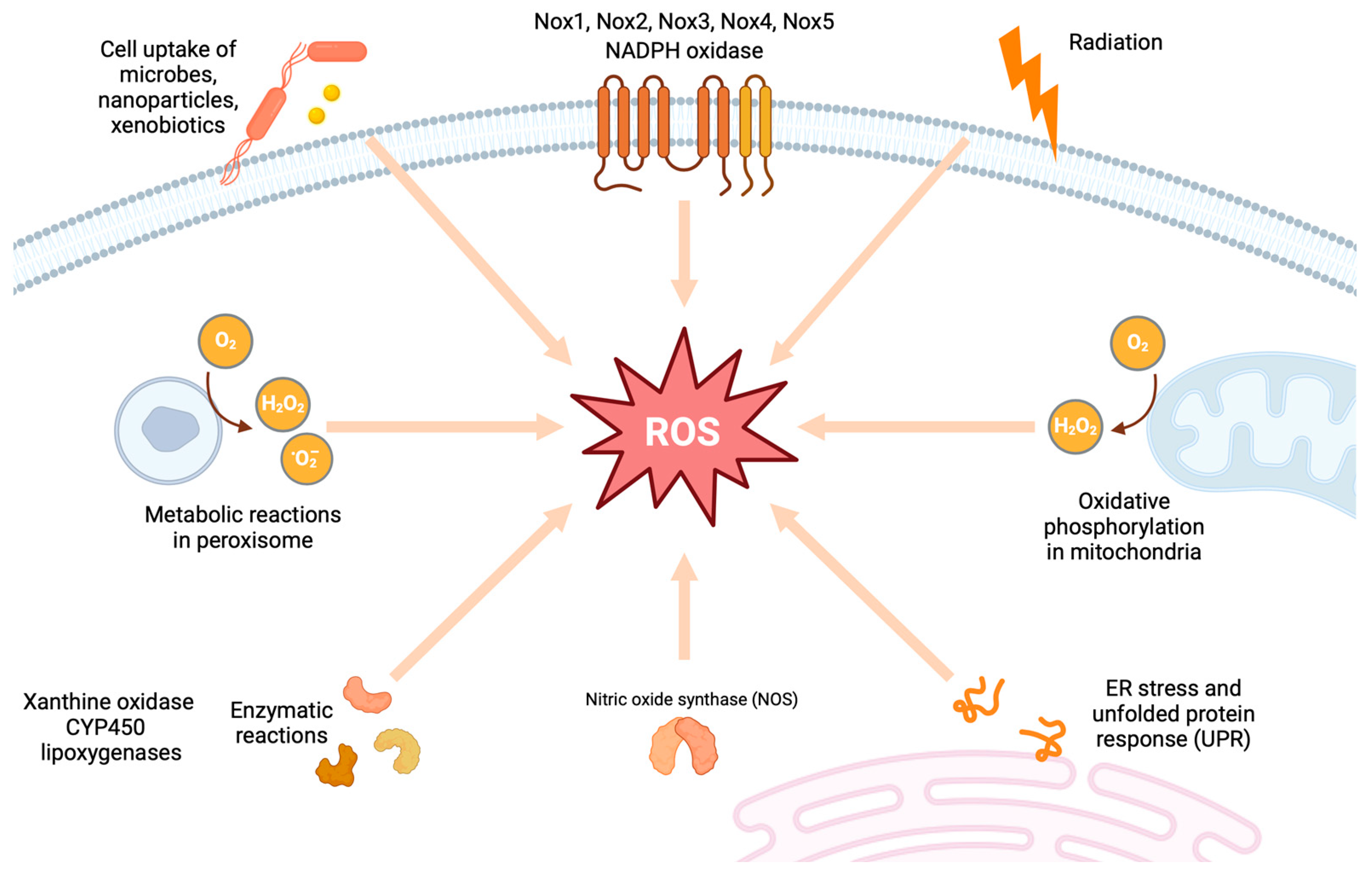

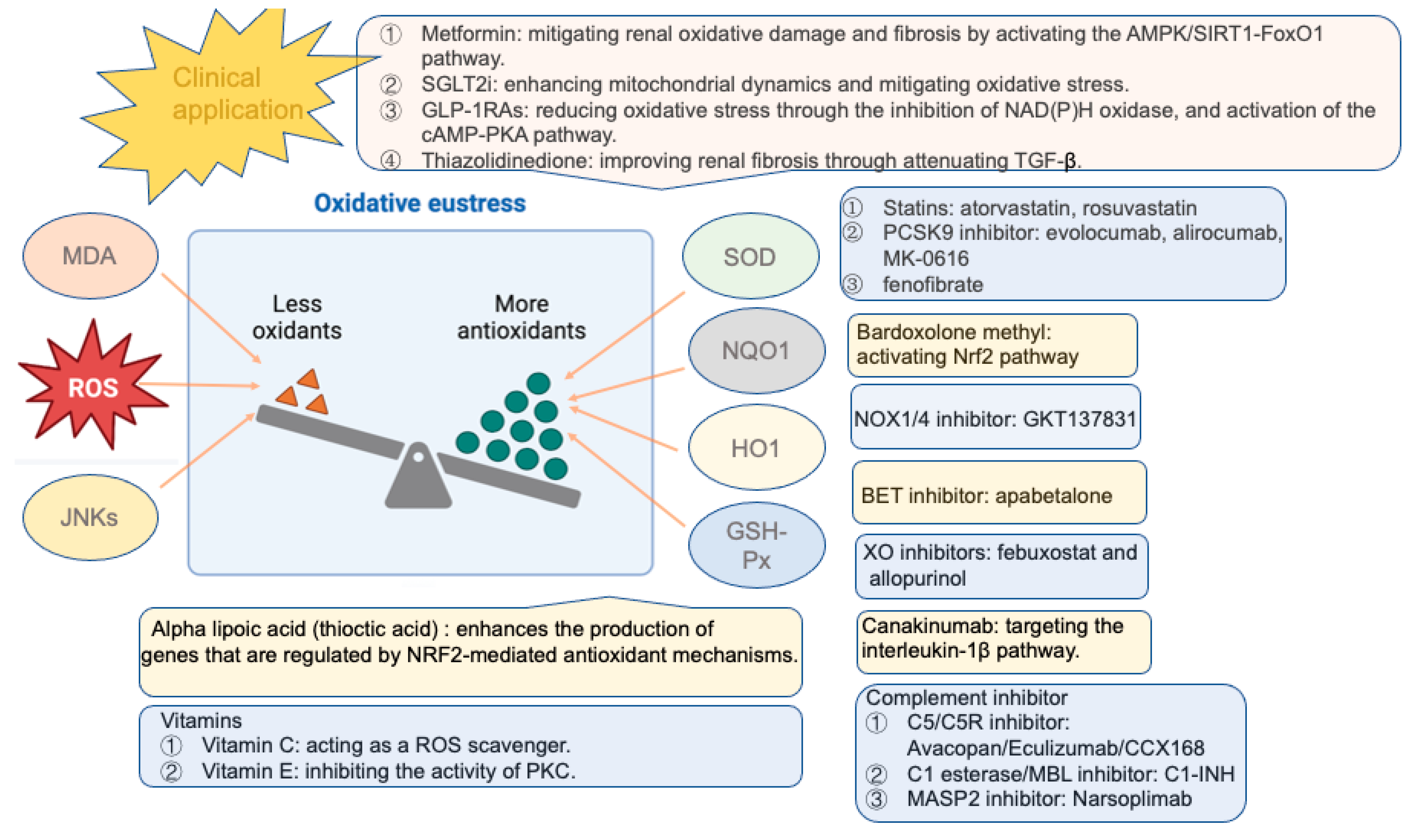

{kind=link}
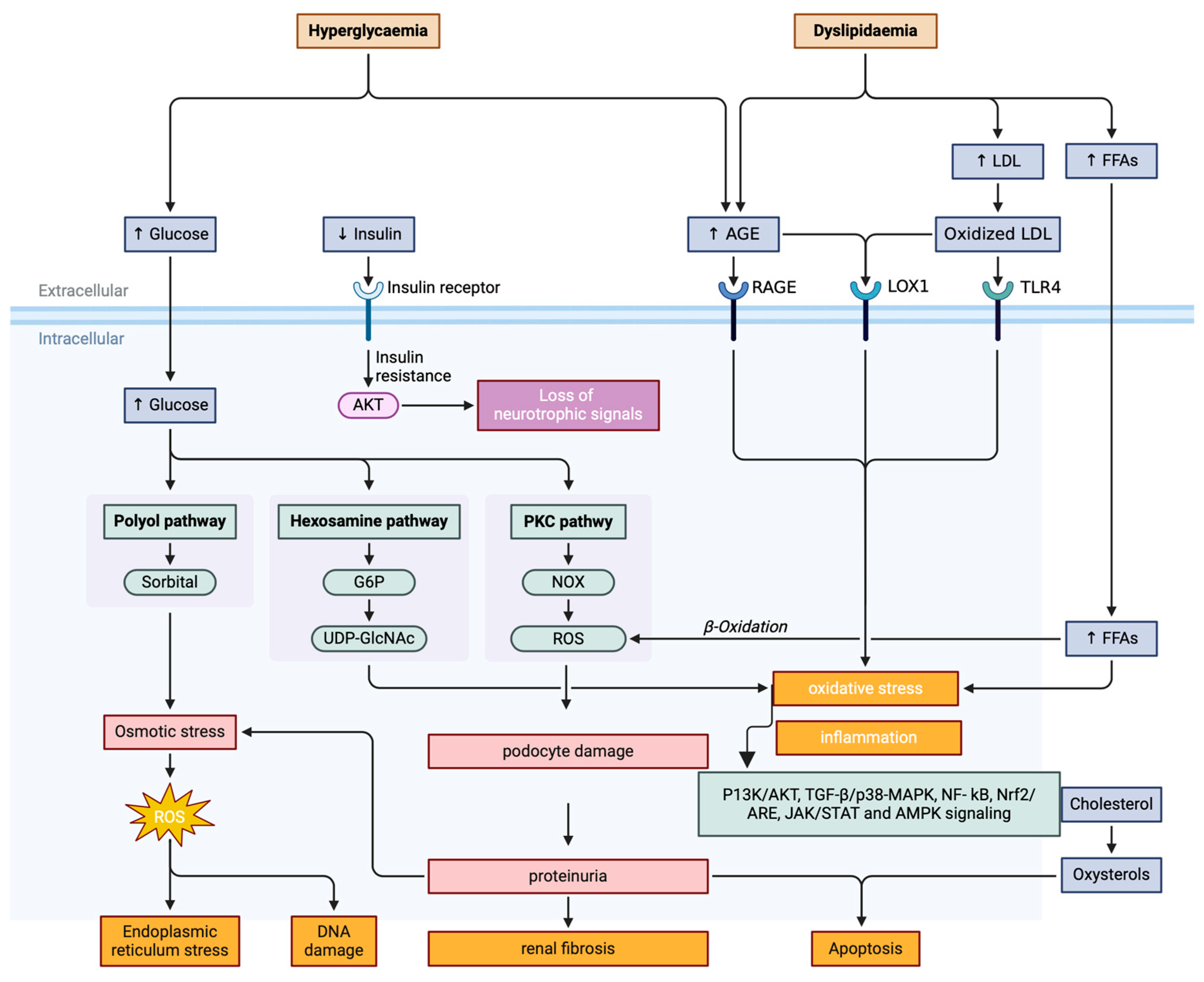{kind=link}
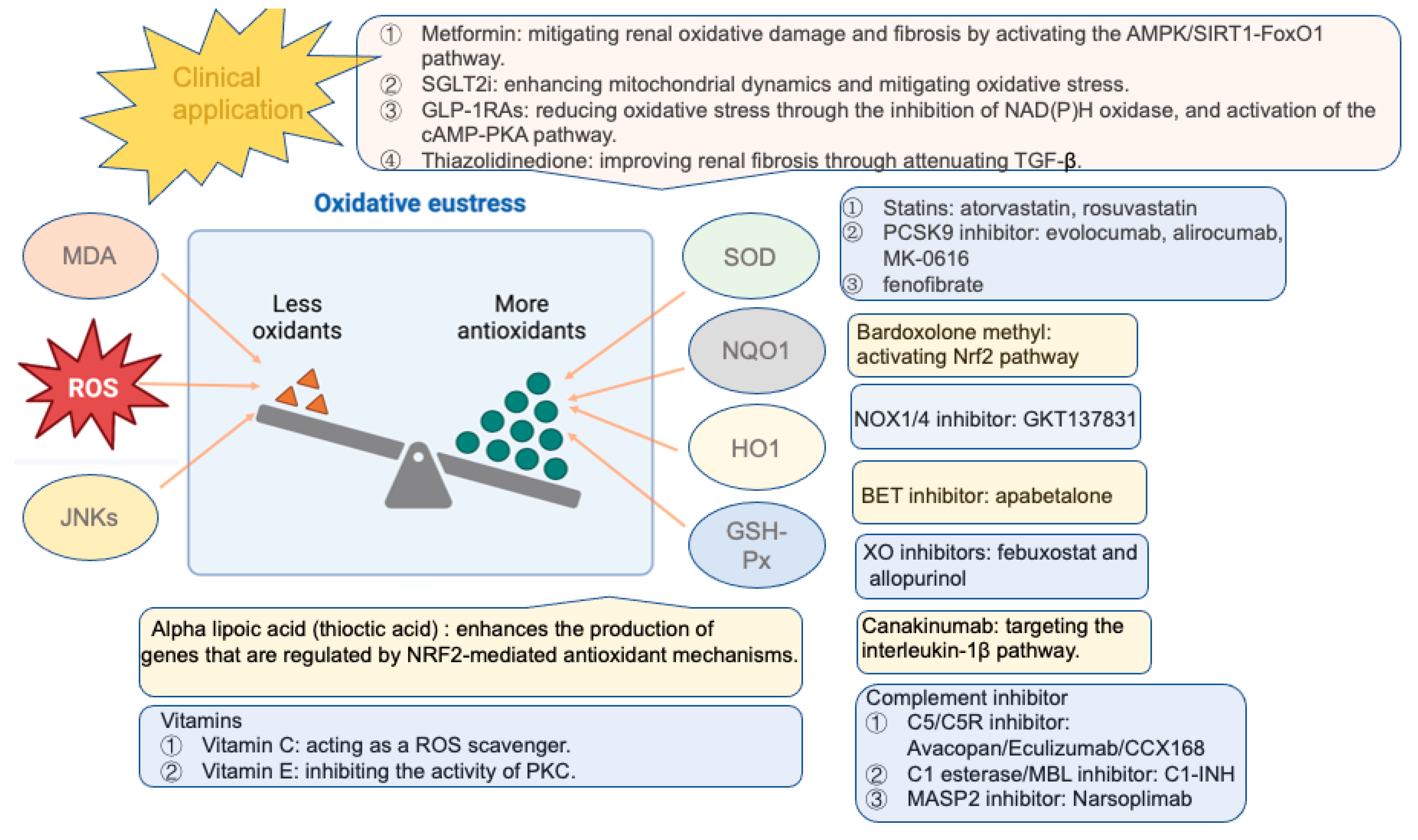{kind=link}
| Target Drugs | Type of Study | Promising Effects | Reference |
|---|---|---|---|
| Glucose-lowering drugs | |||
| Metformin | Animal studies | Ameliorating renal oxidative damage and fibrosis | [186,221,222,223,224,225] |
| Troglitazone | In vitro | Reducing EMT in renal tubule cells | [226] |
| Rosiglitazone | In vitro | Suppressing oxidative stress in renal epithelial cells | [227] |
| Animal studies | Protecting against oxidative stress and renal fibrosis | [227,228] | |
| Animal studies | Mitigating glomerular injury and proteinuria in podocytes | [229] | |
| Clinical study | Exhibiting no influence on albuminuria | [230] | |
| Rosiglitazone | In vitro/vivo | Protecting against podocyte injury | [231] |
| Clinical trials | Reducing albuminuria | [232,233] | |
| SGLT2 inhibitors | |||
| Tofogliflozin | In vitro | Attenuating oxidative stress | [17] |
| Dapagliflozin | Clinical trial | Improving renal events | [234,235] |
| Canagliflozin | Clinical trial | Reduction in death due to renal events | [234] |
| Empagliflozin | Clinical trial | Lower incidence of DKD | [236] |
| GLP-1 RAs | |||
| GLP-1 receptor | Animal study | Protecting against oxidative stress | [14] |
| Lixisenatide | Clinical trial | Reducing albuminuria (post hoc analysis) | [237] |
| Animal study | Enhancing antioxidant effect | [238] | |
| Exenatide | Clinical trial | Improving decline in eGFR (post hoc analysis) | [239] |
| Animal study | Protecting against the progression of DKD | [15] | |
| Liraglutide | Clinical trial | Lower rates of progression of DKD (secondary analysis) | [240] |
| Clinical study | Reducing inflammation and oxidative stress | [241] | |
| Animal study | Reducing albuminuria and attenuating oxidative stress | [14] | |
| Semaglutide | Clinical trial | Reducing the rate of eGFR decline (post hoc analysis) | [242] |
| Clinical trial | Ongoing (primary renal endpoints) | [243] | |
| Albuglutide | Clinical trial | Presenting no significant secondary renal events | [244] |
| DDP-4 inhibitors | Clinical trial | Modulating complement activation | [245] |
| Nrf2 activator | |||
| Sulforaphane/cinnamic | Animal study | Reducing renal damage | [169] |
| Bardoxolone | Clinical trial | Improving eGFR | [246] |
| Clinical trial | Being terminated due to cardiovascular events | [247] | |
| Clinical trial | Improving eGFR without heart failure | [248] | |
| Clinical trial | Being terminated due to high incidence of heart failure | NCT03550443 | |
| Lipid-lowering drugs | |||
| Atorvastatin | Clinical trial | Improving proteinuria | [249] |
| Rosuvastatin | Clinical trial | Exhibiting renoprotective effects | [249] |
| Evolocumab (PCSK9i) | Animal study | Suppressing oxidative stress and inflammation | [250] |
| Alirocumab (PCSK9i) | Animal study | Reducing oxidative stress | [251] |
| Evolocumab (PCSK9i) | In vitro | Protecting against oxidative damage | [252] |
| MK-0616 (PCSK9i) | Clinical trial | Having been completed | NCT05934292 |
| Fenofibrate | Clinical trial | Unknown status | NCT03869931 |
| Vitamins | Animal study | Regulating the levels of ROS | [253,254,255,256,257] |
| Thioctic acid | Clinical study | Reducing urinary albuminuria | [258] |
| Clinical trial | Improving renal functions in early DKD | [259] | |
| Clinical trial | Recruiting patients | NCT06253429 | |
| Animal study | Exerting antioxidative properties | [260] | |
| Pyridorin | Clinical trial | Showing no influence on serum creatinine | [261] |
| Clinical trial | Being terminated with no results | NCT02156843 | |
| Nox inhibitor | |||
| GKT137831(Nox1/4) | Clinical trial | Exhibiting no influence on albuminuria | NCT02010242 |
| Animal studies | Mitigating glomerular injury and proteinuria in podocytes | [229] | |
| XO inhibitor | |||
| Febuxostat | Animal study | Slowing the progression of albuminuria | [57] |
| Febuxostat | Clinical trial | Having no influence in albuminuria or eGFR | [262] |
| Clinical trial | Having been completed without results | NCT01350388 | |
| Allopurinol | Animal study | Reducing albuminuria and attenuating renal injury | [56] |
| Clinical trial | Having been completed without results | NCT02017171; NCT02829177 | |
| AGE/RAGE inhibitor | |||
| Aminoguanidine (AGE) | Animal study | Reducing albuminuria and renal damage | [113] |
| Pyridoxamine (AGE) | Animal study | Slowing the progression of DKD | [114] |
| Alagebrium (AGE) | Clinical trial | Having been terminated | NCT00557518 |
| Animal study | Ameliorating renal damage and inflammation | [115] | |
| RAGE antibody | Animal study | Attenuating albuminuria and renal injury | [116] |
| Green tea (RAGE) | Clinical trial | Without status of the study | NCT03622762 |
| Canakinumab (IL-1β) | Clinical trial | With no adverse renal events (post hoc analysis) | [263] |
| Apabetalone (BET) | Clinical trial | Improving kidney function (post hoc analysis) | [264,265] |
| Clinical trial | Not yet recruiting | NCT03160430 | |
| C3a/C5aR antagonist | In vitro/vivo | Ameliorating EMT | [104] |
| In vitro/vivo | Modulating inflammation and metabolic function | [106] | |
| C3aR antagonism | Animal study | Improving proteinuria and kidney function | [97] |
| C1INH | Clinical trial | Improving renal function | [266] |
| Mesenchymal stem cells | |||
| HUCDMSCs | Animal study | Ameliorating renal fibrosis | [267,268] |
| p-MSCs | Animal study | Attenuating renal damage | [168,269] |
| ADMSCs | Animal study | Enhancing renal repair | [270,271] |
| BM-MSCs | Animal study | Alleviating renal damage | [272] |
| MSCs | Clinical trial | Being safe and tolerated for clinical use | [273] |
| Guanylate cyclase activator | |||
| Cinaciguat | Animal study | Improving proteinuria | [48] |
| PF-00489791(PDE5i) | Clinical trial | Improving albuminuria | [274] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Zhang, C. Oxidative Stress: A Culprit in the Progression of Diabetic Kidney Disease. Antioxidants 2024, 13, 455. https://doi.org/10.3390/antiox13040455
Wang N, Zhang C. Oxidative Stress: A Culprit in the Progression of Diabetic Kidney Disease. Antioxidants. 2024; 13(4):455. https://doi.org/10.3390/antiox13040455
Chicago/Turabian StyleWang, Na, and Chun Zhang. 2024. "Oxidative Stress: A Culprit in the Progression of Diabetic Kidney Disease" Antioxidants 13, no. 4: 455. https://doi.org/10.3390/antiox13040455
APA StyleWang, N., & Zhang, C. (2024). Oxidative Stress: A Culprit in the Progression of Diabetic Kidney Disease. Antioxidants, 13(4), 455. https://doi.org/10.3390/antiox13040455