

TNFα Induces DNA and Histone Hypomethylation and Pulmonary Artery Smooth Muscle Cell Proliferation Partly via Excessive Superoxide Formation
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
3. Results
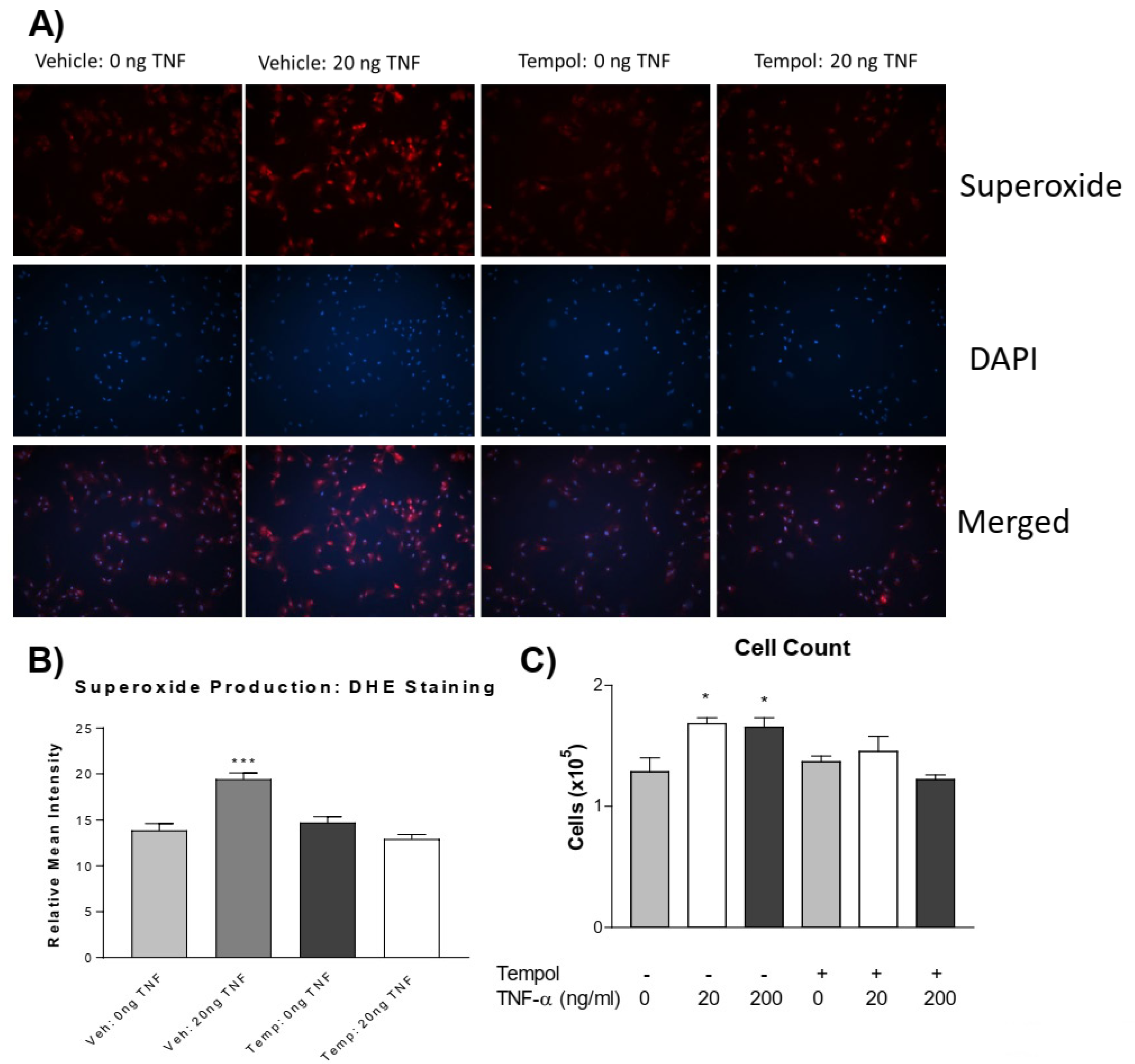
3.1. TNF-α Treatment Increases Superoxide Production in PASMCs
3.2. TNF-α Treatment Increases PASMC Proliferation
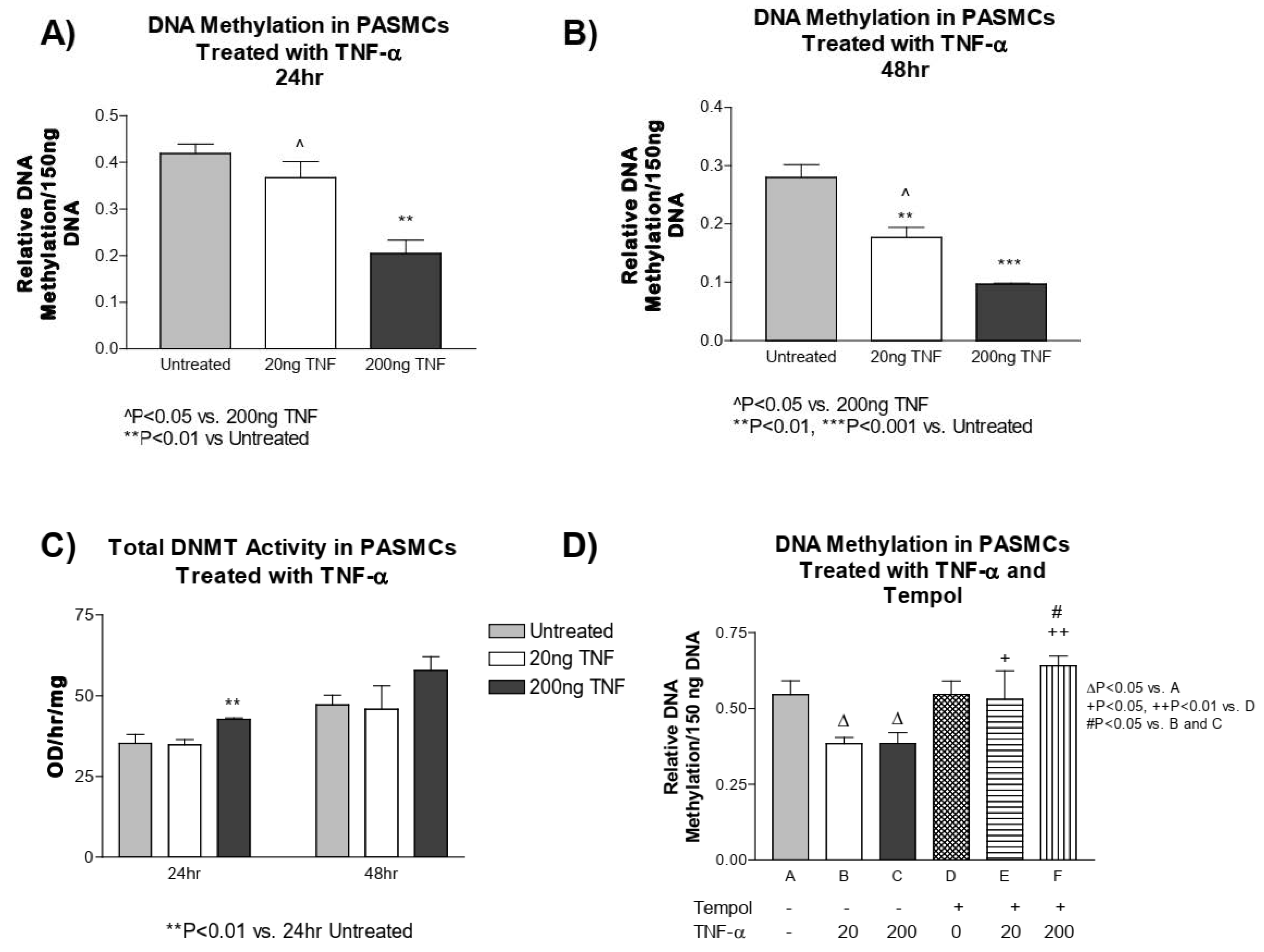
3.3. Tempol Prevents TNF-α-Induced Superoxide Increase and PASMC Proliferation
3.4. TNF-α Treatment Does Not Alter Histone Acetylation in PASMCs
3.5. TNF-α Treatment Decreases DNA Methylation in PASMCs
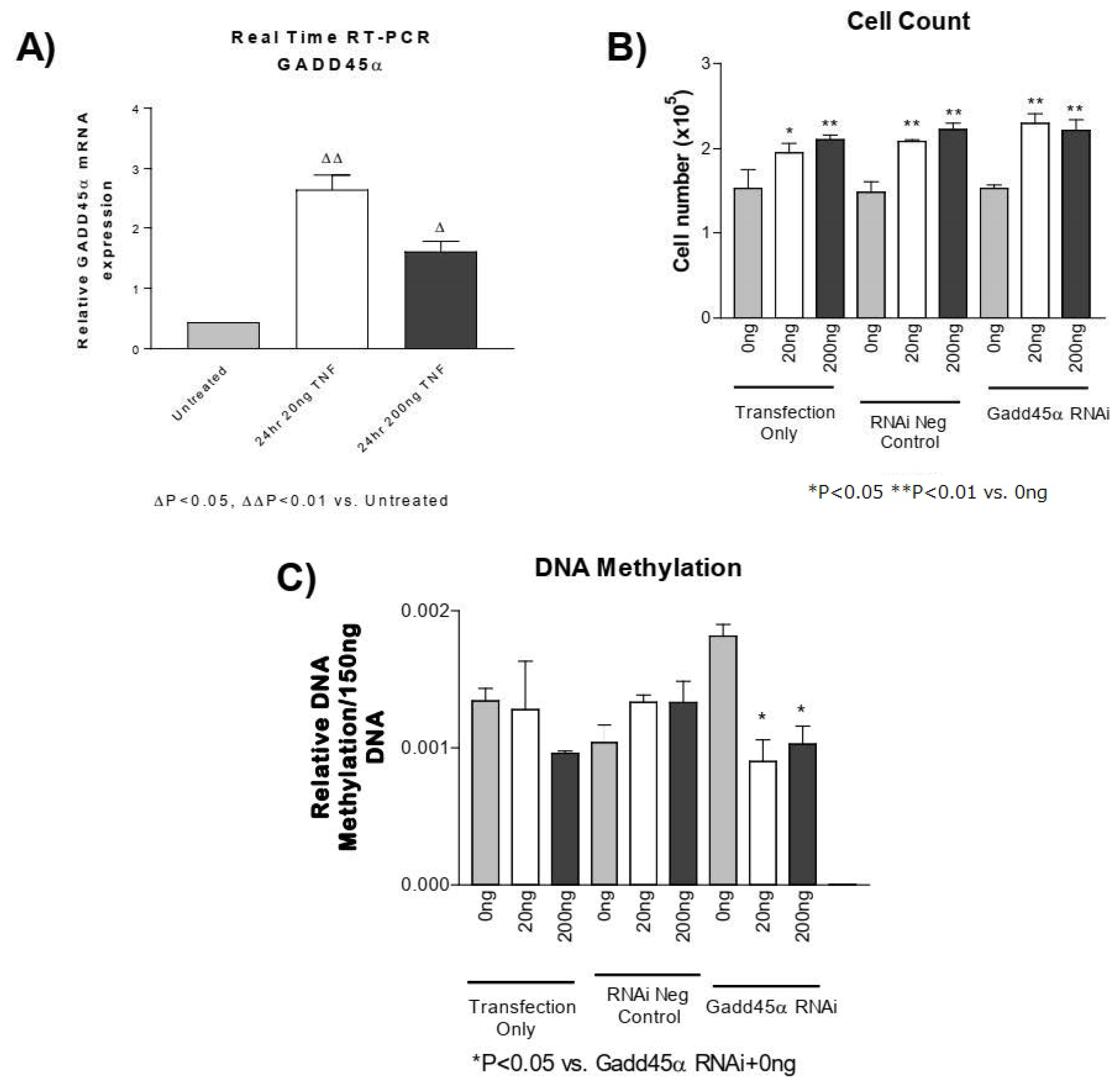
3.6. GADD45-α siRNA Does Not Prevent the TNF-α-Induced Decrease in DNA Methylation
3.7. TNF-α Treatment Decreases H3-K4 Methylation in PASMCs
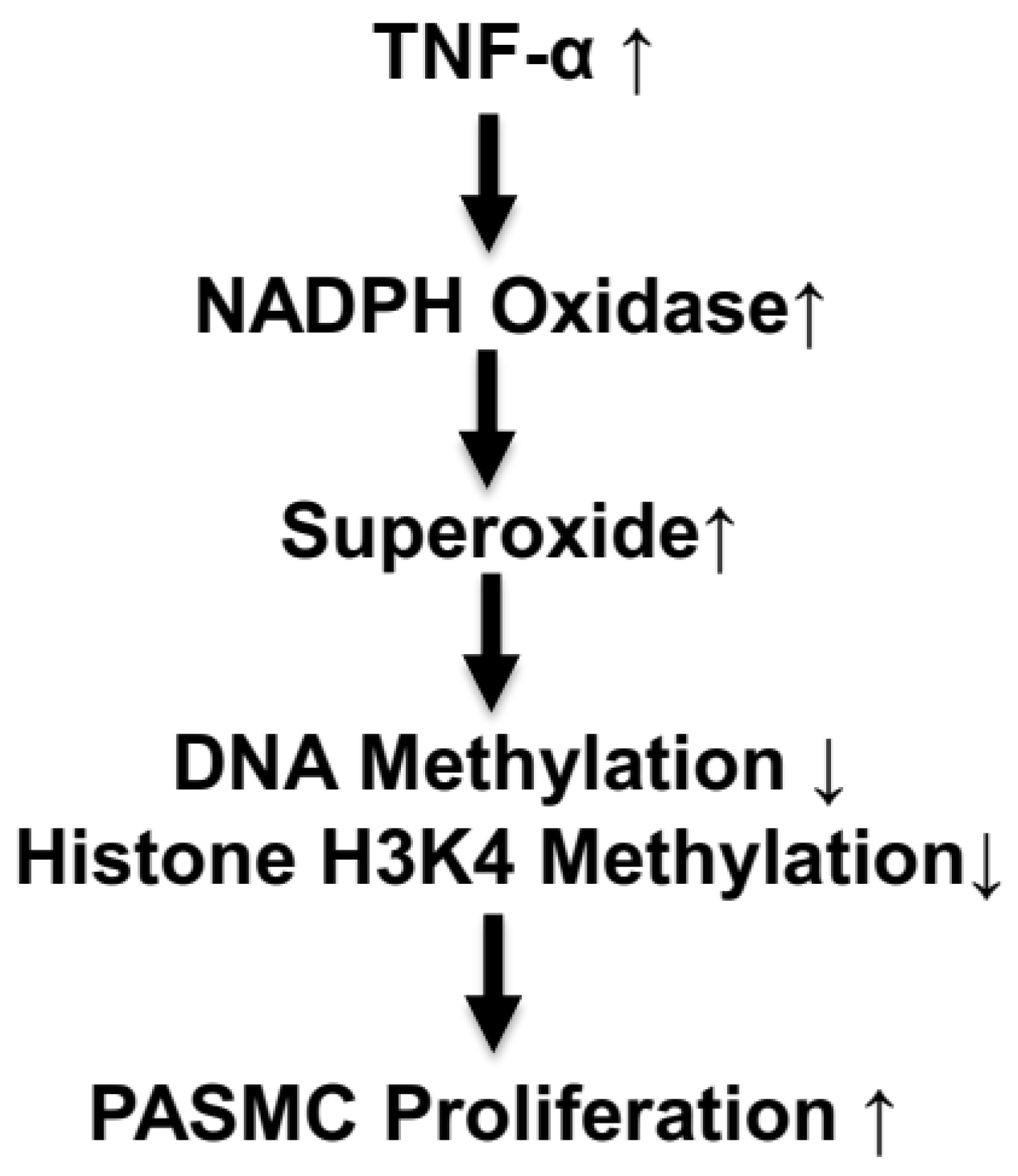
4. Discussion
5. Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Crosswhite, P.; Sun, Z. Molecular mechanisms of pulmonary arterial remodeling. Mol. Med. 2014, 20, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.W.; Sun, Z. Stem cell therapy for pulmonary arterial hypertension: An update. J. Heart Lung Transplant. 2022, 41, 692–703. [Google Scholar] [CrossRef]
- Hjortnaes, J.; Butcher, J.; Figueiredo, J.L.; Riccio, M.; Kohler, R.H.; Kozloff, K.M.; Weissleder, R.; Aikawa, E. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: A role for inflammation. Eur. Heart J. 2010, 31, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Crosswhite, P.; Sun, Z. Inhibition of phosphodiesterase-1 attenuates cold-induced pulmonary hypertension. Hypertension 2013, 61, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Crosswhite, P.; Chen, K.; Sun, Z. AAV delivery of tumor necrosis factor-alpha short hairpin RNA attenuates cold-induced pulmonary hypertension and pulmonary arterial remodeling. Hypertension 2014, 64, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Ryan, J.J.; Marsboom, G.; Archer, S.L. Epigenetic mechanisms of pulmonary hypertension. Pulm. Circ. 2011, 1, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.F.; Cheng, F.; Du, L.Z. Epigenetic regulation of pulmonary arterial hypertension. Hypertens. Res. 2011, 34, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.F.; Ma, X.L.; Shen, Z.; Wu, X.L.; Cheng, F.; Du, L.Z. Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J. Hypertens. 2011, 28, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.F.; Du, L.Z. Epigenetics in neonatal diseases. Chin. Med. J. 2010, 123, 2948–2954. [Google Scholar]
- Sims, R.J., 3rd; Nishioka, K.; Reinberg, D. Histone lysine methylation: A signature for chromatin function. Trends Genet. TIG 2003, 19, 629–639. [Google Scholar] [CrossRef]
- Spencer, V.A.; Davie, J.R. Role of covalent modifications of histones in regulating gene expression. Gene 1999, 240, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J.A. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed]
- David, R. All wrapped up in histones. Nat. Rev. Mol. Cell Biol. 2010, 11, 680–681. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.B.; Klinger, J.R.; Rounds, S.I. Pulmonary arterial hypertension: New insights and new hope. Respirology 2006, 11, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. TIG 2000, 16, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Wild, L.; Flanagan, J.M. Genome-wide hypomethylation in cancer may be a passive consequence of transformation. Biochim. Et Biophys. Acta 2010, 1806, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Symonds, M.E.; Sebert, S.; Budge, H. The obesity epidemic: From the environment to epigenetics—Not simply a response to dietary manipulation in a thermoneutral environment. Front. Genet. 2011, 2, 24. [Google Scholar] [CrossRef]
- Junien, C. Early determinants of health and disease: Epigenetics and environment. Bull. l‘Acad. Natl. Med. 2011, 195, 511–526; discussion 526–527. [Google Scholar]
- Toyokawa, S.; Uddin, M.; Koenen, K.C.; Galea, S. How does the social environment ‘get into the mind’? Epigenetics at the intersection of social and psychiatric epidemiology. Soc. Sci. Med. 2012, 74, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Binder, E.B. Current research trends in early life stress and depression: Review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp. Neurol. 2012, 233, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Furrow, R.E.; Christiansen, F.B.; Feldman, M.W. Environment-sensitive epigenetics and the heritability of complex diseases. Genetics 2011, 189, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Woroniecki, R.; Gaikwad, A.B.; Susztak, K. Fetal environment, epigenetics, and pediatric renal disease. Pediatr. Nephrol. 2011, 26, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.C.; Mill, J.; Uher, R.; Schmidt, U. Eating disorders, gene-environment interactions and epigenetics. Neurosci. Biobehav. Rev. 2011, 35, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Matura, L.A.; Ventetuolo, C.E.; Palevsky, H.I.; Lederer, D.J.; Horn, E.M.; Mathai, S.C.; Pinder, D.; Archer-Chicko, C.; Bagiella, E.; Roberts, K.E.; et al. Interleukin-6 and tumor necrosis factor-alpha are associated with quality of life-related symptoms in pulmonary arterial hypertension. Ann. Am. Thorac. Soc. 2015, 12, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Deng, C.; Wu, D.; Zhong, Z.; Lv, X.; Huang, Z.; Lian, N.; Liu, K.; Zhang, Q. The role of mononuclear cell tissue factor and inflammatory cytokines in patients with chronic thromboembolic pulmonary hypertension. J. Thromb. Thrombolysis 2015, 42, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, Z. Thyroid hormone induces artery smooth muscle cell proliferation: Discovery of a new TRalpha1-Nox1 pathway. J. Cell Mol. Med. 2010, 14, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Dammanahalli, J.K.; Sun, Z. Endothelin (ET)-1 inhibits nicotinamide adenine dinucleotide phosphate oxidase activity in human abdominal aortic endothelial cells: A novel function of ETB1 receptors. Endocrinology 2008, 149, 4979–4987. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Q.; Sun, Z. Normal IgG downregulates the intracellular superoxide level and attenuates migration and permeability in human aortic endothelial cells isolated from a hypertensive patient. Hypertension 2012, 60, 818–826. [Google Scholar] [CrossRef]
- Wang, Y.; Kuro-o, M.; Sun, Z. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP-PKA pathway. Aging Cell 2012, 11, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Crosswhite, P.; Sun, Z. Nitric oxide, oxidative stress and inflammation in pulmonary arterial hypertension. J. Hypertens. 2010, 28, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Sun, Z. The Anti-aging Gene Klotho Regulates Proliferation and Differentiation of Adipose-derived Stem Cells. Stem Cells 2016, 34, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sun, Z. Antiaging Gene Klotho Attenuates Pancreatic beta-Cell Apoptosis in Type 1 Diabetes. Diabetes 2015, 64, 4298–4311. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lin, Y.; Sun, Z. Inhibition of miR-101-3p prevents human aortic valve interstitial cell calcification through regulation of CDH11/SOX9 expression. Mol. Med. 2023, 29, 24. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Sun, Z. Activation of DNA demethylases attenuates aging-associated arterial stiffening and hypertension. Aging Cell 2018, 17, e12762. [Google Scholar] [CrossRef]
- Chen, K.; Sun, Z. Autophagy plays a critical role in Klotho gene deficiency-induced arterial stiffening and hypertension. J. Mol. Med. 2019, 97, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Sun, Z. Estrogen inhibits renal Na-Pi Co-transporters and improves klotho deficiency-induced acute heart failure. Redox Biol. 2021, 47, 102173. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, S.; Sun, Q.W.; Zhang, B.; Ullah, M.; Sun, Z. Klotho Deficiency Causes Heart Aging via Impairing the Nrf2-GR Pathway. Circ. Res. 2021, 128, 492–507. [Google Scholar] [CrossRef]
- Chen, K.; Wang, S.; Sun, Z. In Vivo Cardiac-specific Expression of Adenylyl Cyclase 4 Gene Protects against Klotho Deficiency-induced Heart Failure. Transl. Res. 2022, 244, 101–113. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, B.; Sun, Z. MicroRNA 379 Regulates Klotho Deficiency-Induced Cardiomyocyte Apoptosis Via Repression of Smurf1. Hypertension 2021, 78, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Skelley, L.; Wang, B.; Mejia, A.; Sapozhnikov, V.; Sun, Z. AAV-Based RNAi Silencing of NADPH Oxidase gp91(phox) Attenuates Cold-Induced Cardiovascular Dysfunction. Hum. Gene Ther. 2012, 23, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension 2009, 54, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; Rousset, F.; Banchereau, J. Evolving principles in immunopathology: Interleukin 10 and its relationship to Epstein-Barr virus protein BCRF1. Springer Semin. Immunopathol. 1991, 13, 157–166. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, Z. In vivo pancreatic beta-cell-specific expression of antiaging gene Klotho: A novel approach for preserving beta-cells in type 2 diabetes. Diabetes 2015, 64, 1444–1458. [Google Scholar] [CrossRef]
- Han, X.; Sun, Z. Epigenetic Regulation of KL (Klotho) via H3K27me3 (Histone 3 Lysine [K] 27 Trimethylation) in Renal Tubule Cells. Hypertension 2020, 75, 1233–1241. [Google Scholar] [CrossRef]
- Han, X.; Sun, Z. Adult Mouse Kidney Stem Cells Orchestrate the De Novo Assembly of a Nephron via Sirt2-Modulated Canonical Wnt/β-Catenin Signaling. Adv. Sci. 2022, 9, e2104034. [Google Scholar] [CrossRef]
- Han, X.; Akinseye, L.; Sun, Z. KDM6A Demethylase Regulates Renal Sodium Excretion and Blood Pressure. Hypertension 2024, 81, 541–551. [Google Scholar] [CrossRef]
- Fan, J.; Wang, S.; Chen, K.; Sun, Z. Aging impairs arterial compliance via Klotho-mediated downregulation of B-cell population and IgG levels. Cell. Mol. Life Sci. 2022, 79, 494. [Google Scholar] [CrossRef]
- Fan, J.; Wang, S.; Lu, X.; Sun, Z. Transplantation of bone marrow cells from miR150 knockout mice improves senescence-associated humoral immune dysfunction and arterial stiffness. Metabolism. 2022, 134, 155249. [Google Scholar] [CrossRef]
- Niehrs, C.; Schafer, A. Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol. 2012, 22, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Vairapandi, M.; Azam, N.; Balliet, A.G.; Hoffman, B.; Liebermann, D.A. Characterization of MyD118, Gadd45, and proliferating cell nuclear antigen (PCNA) interacting domains. PCNA impedes MyD118 AND Gadd45-mediated negative growth control. J. Biol. Chem. 2000, 275, 16810–16819. [Google Scholar] [CrossRef] [PubMed]
- Azam, N.; Vairapandi, M.; Zhang, W.; Hoffman, B.; Liebermann, D.A. Interaction of CR6 (GADD45gamma) with proliferating cell nuclear antigen impedes negative growth control. J. Biol. Chem. 2001, 276, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Chen, I.C.; Hsieh, C.M.; Nepali, K.; Lai, R.W.; Hsu, K.C.; Lin, T.E.; Pan, S.L.; Chen, M.C.; Liou, J.P. Installation of Pargyline, a LSD1 Inhibitor, in the HDAC Inhibitory Template Culminated in the Identification of a Tractable Antiprostate Cancer Agent. J. Med. Chem. 2021, 64, 17824–17845. [Google Scholar] [CrossRef] [PubMed]
- Bonow, R.O.; Carabello, B.A.; Chatterjee, K.; de Leon, A.C., Jr.; Faxon, D.P.; Freed, M.D.; Gaasch, W.H.; Lytle, B.W.; Nishimura, R.A.; O‘Gara, P.T.; et al. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): Endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 2008, 118, e523–e661. [Google Scholar] [PubMed]
- Sutendra, G.; Dromparis, P.; Bonnet, S.; Haromy, A.; McMurtry, M.S.; Bleackley, R.C.; Michelakis, E.D. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFα contributes to the pathogenesis of pulmonary arterial hypertension. J. Mol. Med. 2011, 89, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Dammanahalli, K.J.; Sun, Z. Endothelins and NADPH oxidases in the cardiovascular system. Clin. Exp. Pharmacol. Physiol. 2008, 35, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Park, W.H. Tempol differently affects cellular redox changes and antioxidant enzymes in various lung-related cells. Sci. Rep. 2021, 11, 14869. [Google Scholar] [CrossRef]
- Hajkova, P.; Jeffries, S.J.; Lee, C.; Miller, N.; Jackson, S.P.; Surani, M.A. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science 2010, 329, 78–82. [Google Scholar] [CrossRef]
- Popp, C.; Dean, W.; Feng, S.; Cokus, S.J.; Andrews, S.; Pellegrini, M.; Jacobsen, S.E.; Reik, W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 2010, 463, 1101–1105. [Google Scholar] [CrossRef]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Liebermann, D.A.; Tront, J.S.; Sha, X.; Mukherjee, K.; Mohamed-Hadley, A.; Hoffman, B. Gadd45 stress sensors in malignancy and leukemia. Crit. Rev. Oncog. 2011, 16, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Vallbohmer, D.; Warnecke-Eberz, U.; Hokita, S.; Xi, H.; Brabender, J.; Metzger, R.; Baldus, S.E.; Natsugoe, S.; Aikou, T.; et al. Down-regulation of Gadd45 expression is associated with tumor differentiation in non-small cell lung cancer. Anticancer. Res. 2006, 26, 2143–2147. [Google Scholar] [PubMed]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Rigal, M.; Kevei, Z.; Pelissier, T.; Mathieu, O. DNA methylation in an intron of the IBM1 histone demethylase gene stabilizes chromatin modification patterns. EMBO J. 2012, 31, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Z.; Huang, Z.; Cui, Q.Y.; Song, X.H.; Du, L.; Jeltsch, A.; Chen, P.; Li, G.; Li, E.; Xu, G.L. Histone tails regulate DNA methylation by allosterically activating de novo methyltransferase. Cell Res. 2011, 21, 1172–1181. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Alzayady, K.; Stewart, R.; Ye, P.; Yang, S.; Li, W.; Shi, Y. Histone demethylase LSD1 regulates neural stem cell proliferation. Mol. Cell. Biol. 2010, 30, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, R.; Park, G.; Park, J.W.; Kim, J.E. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J. Biol. Chem. 2012, 287, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Janzer, A.; Lim, S.; Fronhoffs, F.; Niazy, N.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) and histone deacetylase 1 (HDAC1) synergistically repress proinflammatory cytokines and classical complement pathway components. Biochem. Biophys. Res. Commun. 2012, 421, 665–670. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crosswhite, P.; Sun, Z. TNFα Induces DNA and Histone Hypomethylation and Pulmonary Artery Smooth Muscle Cell Proliferation Partly via Excessive Superoxide Formation. Antioxidants 2024, 13, 677. https://doi.org/10.3390/antiox13060677
Crosswhite P, Sun Z. TNFα Induces DNA and Histone Hypomethylation and Pulmonary Artery Smooth Muscle Cell Proliferation Partly via Excessive Superoxide Formation. Antioxidants. 2024; 13(6):677. https://doi.org/10.3390/antiox13060677
Chicago/Turabian StyleCrosswhite, Patrick, and Zhongjie Sun. 2024. "TNFα Induces DNA and Histone Hypomethylation and Pulmonary Artery Smooth Muscle Cell Proliferation Partly via Excessive Superoxide Formation" Antioxidants 13, no. 6: 677. https://doi.org/10.3390/antiox13060677