

PM2.5 Induces Cardiomyoblast Senescence via AhR-Mediated Oxidative Stress


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Collection of PM2.5 and EOM Purification
2.3. Culture of the H9c2 Rat Cardiomyoblasts
2.4. Cell Viability Determination by MTT Assay
2.5. Apoptosis Analysis by Flow Cytometry via Annexin V/Propidium Iodide Staining
2.6. Western Blotting
2.7. EdU Cell Incorporation Assay
2.8. Determination of Cell Cycle Distribution
2.9. Immunofluorescence Staining
2.10. RNA Extraction, cDNA Synthesis, and qPCR Reactions
2.11. β-Galactosidase Staining
2.12. Mitochondrial ROS (mtROS) and Intracellular ROS Detection
2.13. Statistical Analysis
3. Results
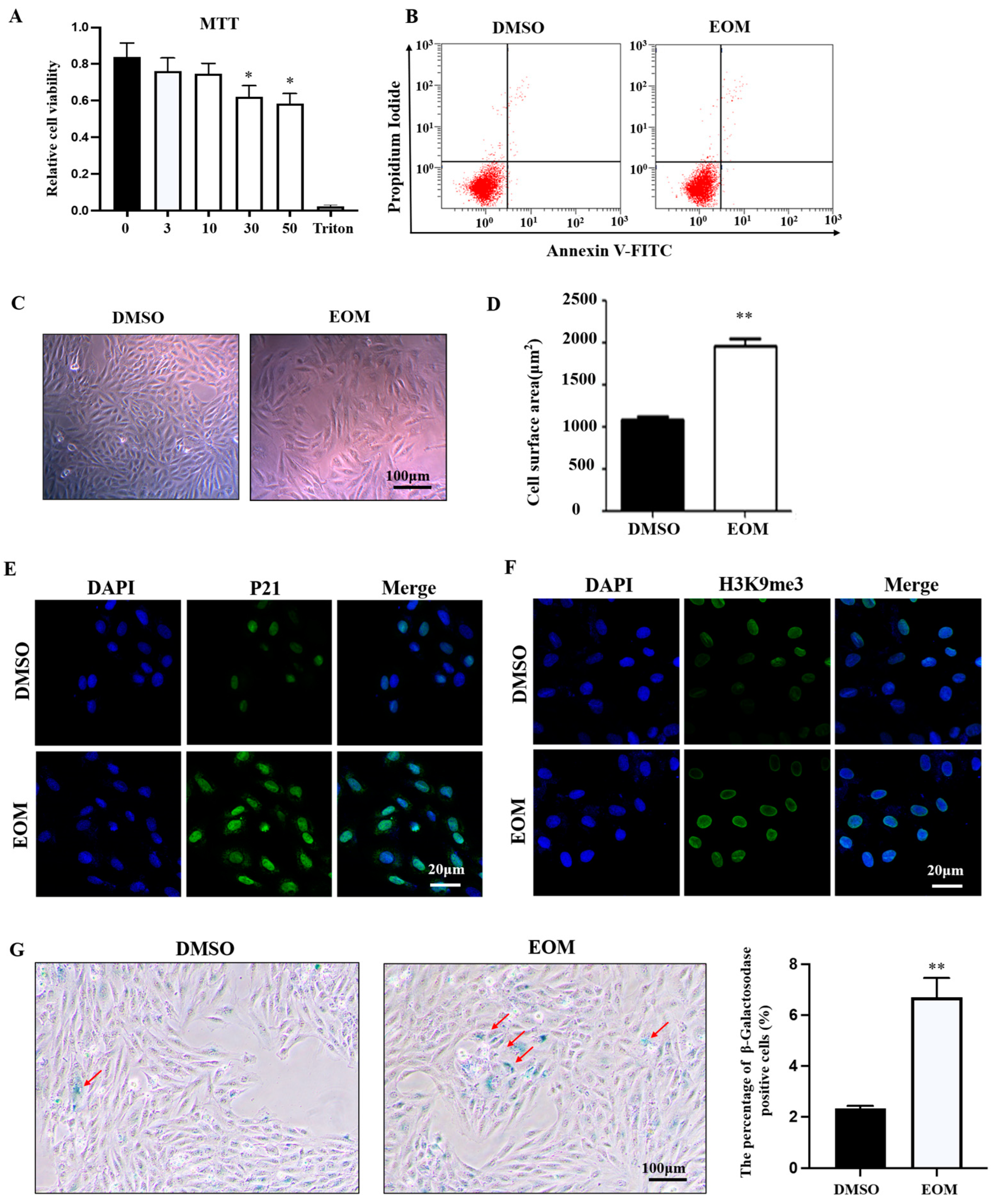
3.1. Effects of EOM on Cell Viability and Morphology
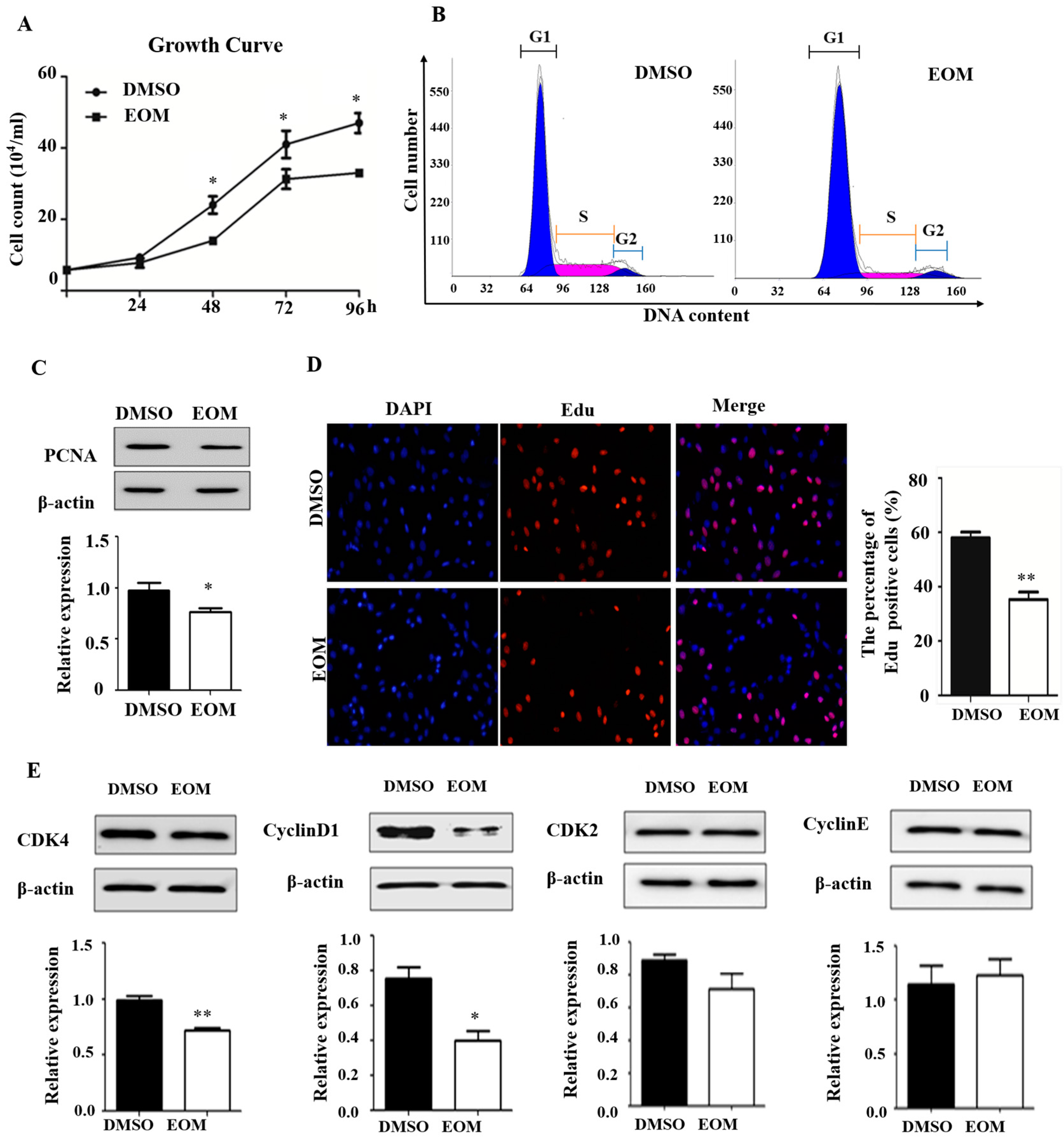
3.2. EOM Exposure Induces Cell Cycle Arrest
3.3. Oxidative Stress Mediates EOM-Induced Cardiac Senescence
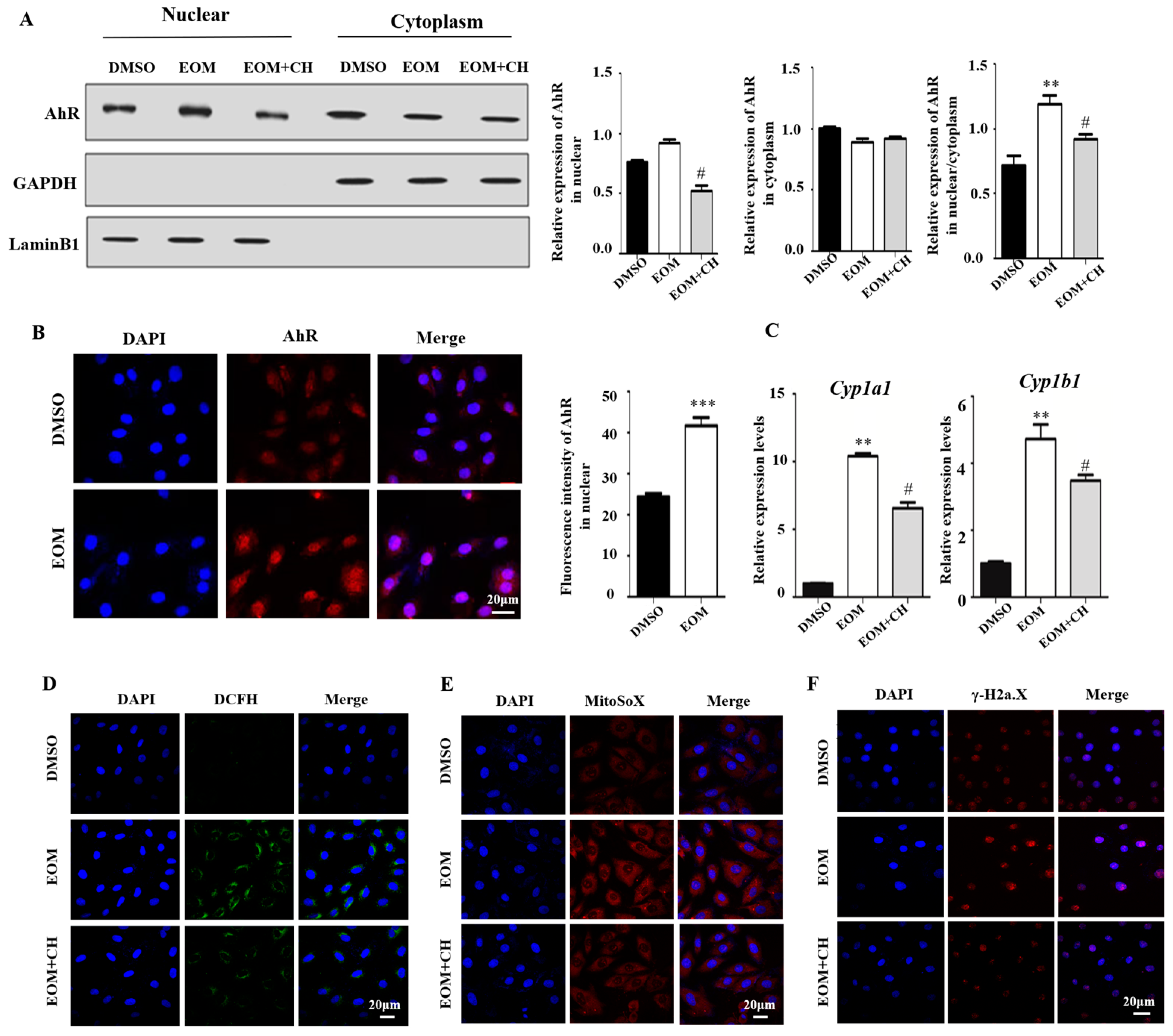
3.4. EOM Exposure Activates the AhR Signaling Pathway and Mediated Oxidative Stress
3.5. AhR Mediates EOM-Induced H9c2 Senescence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feng, S.; Gao, D.; Liao, F.; Zhou, F.; Wang, X. The health effects of ambient PM2.5 and potential mechanisms. Ecotoxicol. Environ. Saf. 2016, 128, 67–74. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Particulate Matter (PM(2.5) and PM(10)), Ozone, Nitrogen Dioxide, Sulfur Dioxide and Carbon Monoxide; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Fushimi, A.; Nakajima, D.; Furuyama, A.; Suzuki, G.; Ito, T.; Sato, K.; Fujitani, Y.; Kondo, Y.; Yoshino, A.; Ramasamy, S.; et al. Source contributions to multiple toxic potentials of atmospheric organic aerosols. Sci. Total Environ. 2021, 773, 145614. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Zhang, Q.; Zhang, J.; Liu, W.; Chen, L.; Wang, M.; Li, R.; Qin, S.; Song, X.; Ji, Y. Diesel exhaust PM2.5 greatly deteriorates fibrosis process in pre-existing pulmonary fibrosis via ferroptosis. Environ. Int. 2023, 171, 107706. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Tao, Y.; Li, X.; Wang, J.; Chen, J.; Aniagu, S.; Jiang, Y.; Chen, T. AHR-mediated m(6)A RNA methylation contributes to PM(2.5)-induced cardiac malformations in zebrafish larvae. J. Hazard. Mater. 2023, 457, 131749. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Cosentino, F. Ageing, metabolism and cardiovascular disease. J. Physiol. 2016, 594, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Mordukhovich, I.; Coull, B.; Kloog, I.; Koutrakis, P.; Vokonas, P.; Schwartz, J. Exposure to sub-chronic and long-term particulate air pollution and heart rate variability in an elderly cohort: The Normative Aging Study. Environ. Health 2015, 14, 87. [Google Scholar] [CrossRef] [PubMed]
- Peralta, A.A.; Schwartz, J.; Gold, D.R.; Coull, B.; Koutrakis, P. Associations between acute and long-term exposure to PM2.5 components and temperature with QT interval length in the VA Normative Aging Study. Eur. J. Prev. Cardiol. 2021, 28, 1610–1617. [Google Scholar] [CrossRef]
- Henneberger, A.; Zareba, W.; Ibald-Mulli, A.; Rückerl, R.; Cyrys, J.; Couderc, J.P.; Mykins, B.; Woelke, G.; Wichmann, H.E.; Peters, A. Repolarization changes induced by air pollution in ischemic heart disease patients. Environ. Health Perspect. 2005, 113, 440–446. [Google Scholar] [CrossRef]
- Schikowski, T.; Hüls, A. Air Pollution and Skin Aging. Curr. Environ. Health Rep. 2020, 7, 58–64. [Google Scholar] [CrossRef]
- Wilker, E.H.; Preis, S.R.; Beiser, A.S.; Wolf, P.A.; Au, R.; Kloog, I.; Li, W.; Schwartz, J.; Koutrakis, P.; DeCarli, C.; et al. Long-term exposure to fine particulate matter, residential proximity to major roads and measures of brain structure. Stroke 2015, 46, 1161–1166. [Google Scholar] [CrossRef]
- Gude, N.A.; Broughton, K.M.; Firouzi, F.; Sussman, M.A. Cardiac ageing: Extrinsic and intrinsic factors in cellular renewal and senescence. Nat. Rev. Cardiol. 2018, 15, 523–542. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Lee, R.T.; Garbern, J.C. Senescence mechanisms and targets in the heart. Cardiovasc. Res. 2022, 118, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Chadda, K.R.; Ajijola, O.A.; Vaseghi, M.; Shivkumar, K.; Huang, C.L.; Jeevaratnam, K. Ageing, the autonomic nervous system and arrhythmia: From brain to heart. Ageing Res. Rev. 2018, 48, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gan, X.; Li, F.; Chen, Y.; Fu, W.; Zhu, X.; Xu, D.; Long, M.; Xu, D. PM(2.5) Exposure Induces More Serious Apoptosis of Cardiomyocytes Mediated by Caspase3 through JNK/P53 Pathway in Hyperlipidemic Rats. Int. J. Biol. Sci. 2019, 15, 24–33. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Ji, C.; Huang, Y.; Aniagu, S.; Jiang, Y.; Chen, T. AHR-mediated ROS production contributes to the cardiac developmental toxicity of PM2.5 in zebrafish embryos. Sci. Total Environ. 2020, 719, 135097. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Peng, V.; Sudan, R.; Ulezko Antonova, A.; Di Luccia, B.; Ohara, T.E.; Fachi, J.L.; Grajales-Reyes, G.E.; Jaeger, N.; Trsan, T.; et al. Repression of the aryl-hydrocarbon receptor prevents oxidative stress and ferroptosis of intestinal intraepithelial lymphocytes. Immunity 2023, 56, 797–812.e4. [Google Scholar] [CrossRef] [PubMed]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef]
- Guo, Y.; Guan, T.; Shafiq, K.; Yu, Q.; Jiao, X.; Na, D.; Li, M.; Zhang, G.; Kong, J. Mitochondrial dysfunction in aging. Ageing Res. Rev. 2023, 88, 101955. [Google Scholar] [CrossRef]
- Bernard, M.; Yang, B.; Migneault, F.; Turgeon, J.; Dieudé, M.; Olivier, M.A.; Cardin, G.B.; El-Diwany, M.; Underwood, K.; Rodier, F.; et al. Autophagy drives fibroblast senescence through MTORC2 regulation. Autophagy 2020, 16, 2004–2016. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, M.; Zou, H.; Aniagu, S.; Jiang, Y.; Chen, T. PM2.5 induces mitochondrial dysfunction via AHR-mediated cyp1a1 overexpression during zebrafish heart development. Toxicology 2023, 487, 153466. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Chen, T.; Jiang, B.; Feng, H.; Zhu, Z.; Li, M.; Zhang, G.; Jiang, Y. 6PPDQ induces cardiomyocyte senescence via AhR/ROS-mediated autophagic flux blockage. Environ. Pollut. 2024, 349, 123872. [Google Scholar] [CrossRef] [PubMed]
- Teng, Z.; Jiang, B.; Wang, J.; Liu, T.; Aniagu, S.; Zhu, Z.; Chen, T.; Jiang, Y. Regulation of Cx43 and its role in trichloroethylene-induced cardiac toxicity in H9C2 rat cardiomyocytes. Chemosphere 2023, 323, 138249. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D.; Rajagopalan, S.; Pope, C.A., 3rd; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Lv, X.; Qiu, C.; Li, J.; Wu, X.; Zhang, H.; Yue, D.; Liu, K.; Eshak, E.S. The effect of China’s Clean Air Act on cognitive function in older adults: A population-based, quasi-experimental study. Lancet Healthy Longev. 2022, 3, e98–e108. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Nepovimova, E.; Heger, Z.; Valko, M.; Wu, Q.; Kuca, K.; Adam, V. Role of hypoxia in cellular senescence. Pharmacol. Res. 2023, 194, 106841. [Google Scholar] [CrossRef] [PubMed]
- Nwanaji-Enwerem, J.C.; Cardenas, A.; Chai, P.R.; Weisskopf, M.G.; Baccarelli, A.A.; Boyer, E.W. Relationships of Long-Term Smoking and Moist Snuff Consumption With a DNA Methylation Age Relevant Smoking Index: An Analysis in Buccal Cells. Nicotine Tob. Res. 2019, 21, 1267–1273. [Google Scholar] [CrossRef]
- Zhou, Z.; Shao, T.; Qin, M.; Miao, X.; Chang, Y.; Sheng, W.; Wu, F.; Yu, Y. The effects of autophagy on vascular endothelial cells induced by airborne PM2.5. J. Environ. Sci. 2018, 66, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wang, Y.; Li, L.; Zhou, W.; Tian, D.; Lu, C.; Yu, S.; Zhao, J.; Peng, S. PM(2.5) induces embryonic growth retardation: Potential involvement of ROS-MAPKs-apoptosis and G0/G1 arrest pathways. Environ. Toxicol. 2016, 31, 2028–2044. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Jia, R.; Wei, M.; Meng, X.; Zhang, X.; Du, R.; Sun, W.; Wang, L.; Song, L. Oxidative stress activates Ryr2-Ca(2+) and apoptosis to promote PM(2.5)-induced heart injury of hyperlipidemia mice. Ecotoxicol. Environ. Saf. 2022, 232, 113228. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, X.; Teng, T.; Ma, Z.G.; Tang, Q.Z. Cellular Senescence in Cardiovascular Diseases: A Systematic Review. Aging. Dis. 2022, 13, 103–128. [Google Scholar] [CrossRef] [PubMed]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shi, Y.; Asweto, C.O.; Feng, L.; Yang, X.; Zhang, Y.; Hu, H.; Duan, J.; Sun, Z. Fine particle matters induce DNA damage and G2/M cell cycle arrest in human bronchial epithelial BEAS-2B cells. Environ. Sci. Pollut. Res. Int. 2017, 24, 25071–25081. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Dong, T.; Yan, K.; Ci, X.; Peng, L. PM2.5 increases susceptibility to acute exacerbation of COPD via NOX4/Nrf2 redox imbalance-mediated mitophagy. Redox Biol. 2023, 59, 102587. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zeng, Z.; Liu, J.; Pi, Z.; Zou, P.; Deng, Q.; Ma, X.; Qiao, F.; Xiong, W.; Zhou, C. Particulate matter promotes hyperpigmentation via AhR/MAPK signaling activation and by increasing α-MSH paracrine levels in keratinocytes. Environ. Pollut. 2021, 278, 116850. [Google Scholar] [CrossRef]
- Vogeley, C.; Esser, C.; Tüting, T.; Krutmann, J.; Haarmann-Stemmann, T. Role of the Aryl Hydrocarbon Receptor in Environmentally Induced Skin Aging and Skin Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 6005. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Jiang, B.; Fu, B.; Shang, C.; Feng, H.; Chen, T.; Jiang, Y. PM2.5 Induces Cardiomyoblast Senescence via AhR-Mediated Oxidative Stress. Antioxidants 2024, 13, 786. https://doi.org/10.3390/antiox13070786
Liu T, Jiang B, Fu B, Shang C, Feng H, Chen T, Jiang Y. PM2.5 Induces Cardiomyoblast Senescence via AhR-Mediated Oxidative Stress. Antioxidants. 2024; 13(7):786. https://doi.org/10.3390/antiox13070786
Chicago/Turabian StyleLiu, Tiantian, Bin Jiang, Baoqiang Fu, Changyi Shang, Haobin Feng, Tao Chen, and Yan Jiang. 2024. "PM2.5 Induces Cardiomyoblast Senescence via AhR-Mediated Oxidative Stress" Antioxidants 13, no. 7: 786. https://doi.org/10.3390/antiox13070786
APA StyleLiu, T., Jiang, B., Fu, B., Shang, C., Feng, H., Chen, T., & Jiang, Y. (2024). PM2.5 Induces Cardiomyoblast Senescence via AhR-Mediated Oxidative Stress. Antioxidants, 13(7), 786. https://doi.org/10.3390/antiox13070786