

The Interplay of Protein Aggregation, Genetics, and Oxidative Stress in Alzheimer’s Disease: Role for Natural Antioxidants and Immunotherapeutics
 ,
,
Abstract
:1. Introduction
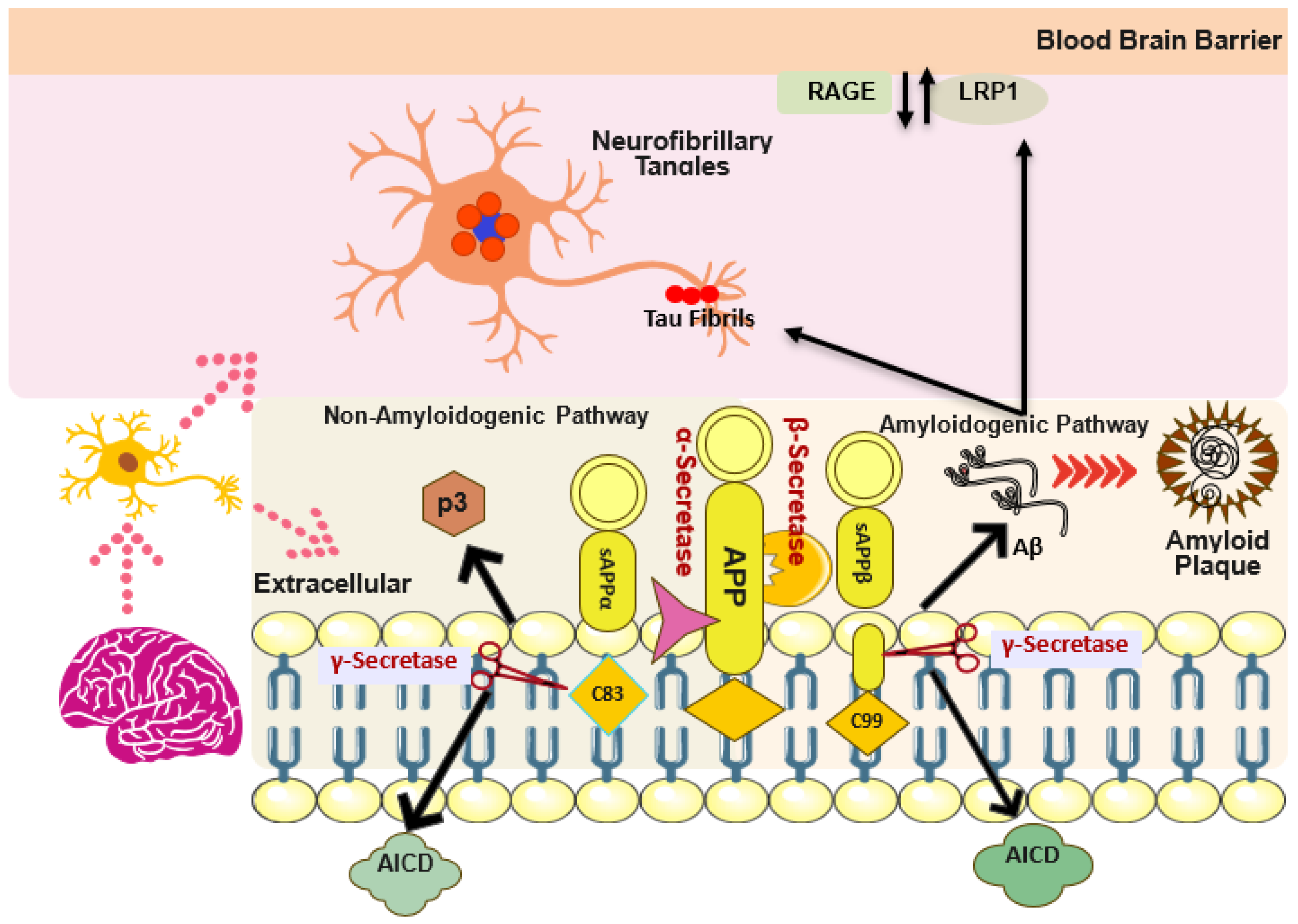
2. Structural and Pathological Aggregation Cascade of Aβ
3. Structural and Pathological Aggregation, Phosphorylation of Tau Protein
4. Aβ and Tau Interplay
5. Early and Late-Onset of AD and Genetics
6. Aβ and Tau Interaction with Synapse
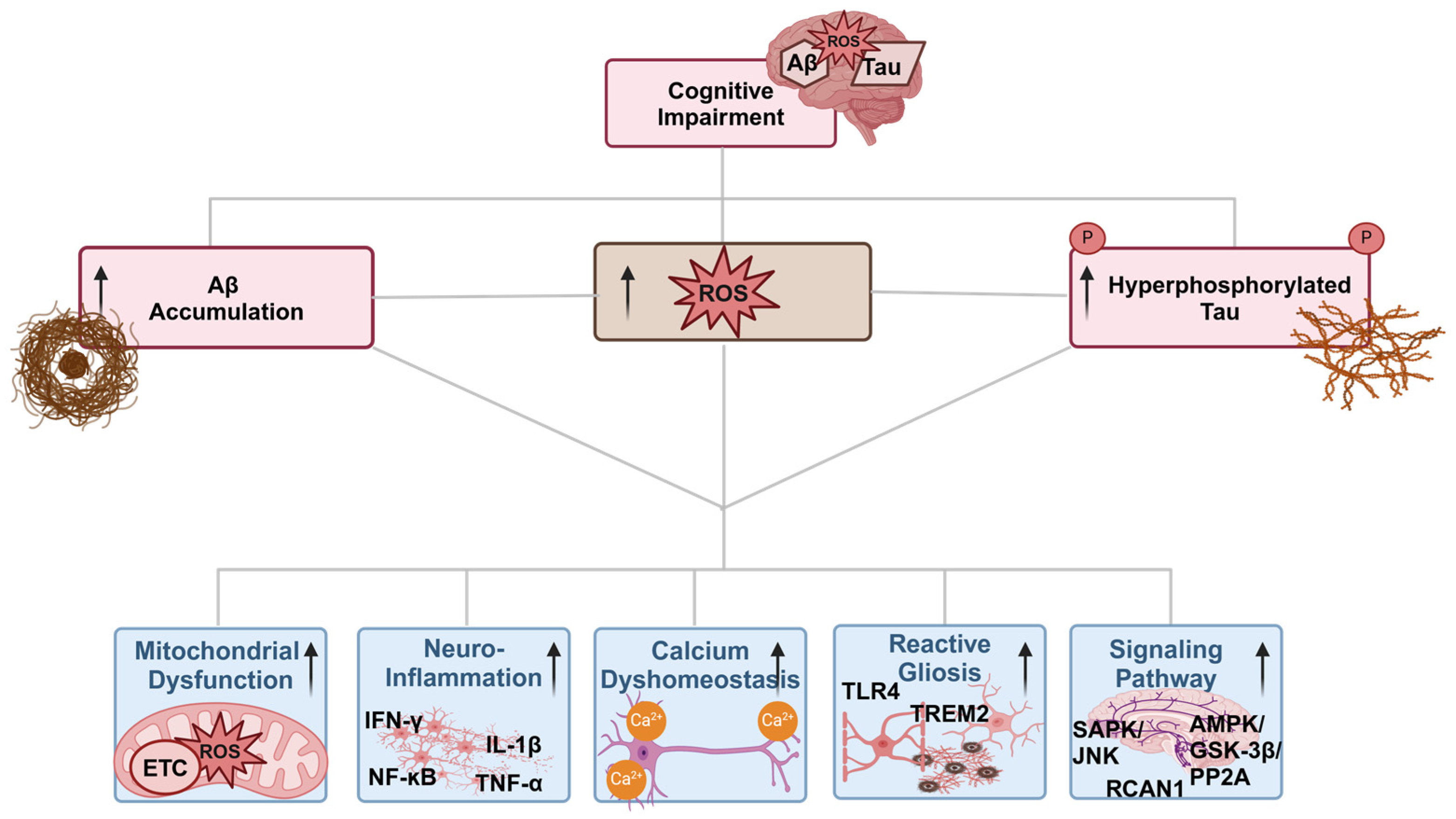
7. Oxidative Stress and Different Pathways in AD
7.1. Oxidative Stress and Different Pathways in AD
7.1.1. Oxidative Stress and Mitochondria Dysfunction in AD
7.1.2. Oxidative Stress and Aβ Interplay
7.1.3. Oxidative Stress and Tau Interplay
7.1.4. Oxidative Stress and Calcium Dyshomeostasis Interplay
7.1.5. Oxidative Stress and Signaling Pathways Interplay
7.1.6. Oxidative Stress and Neuroinflammation Interplay
8. Antioxidant Therapy
9. Targeting Aβ with Various Approaches
9.1. Targeting Aβ at Various Prospectives
9.1.1. Aβ and BACE1 Inhibitors
9.1.2. Aβ and γ-Secretase Inhibitors
10. Limitations, Low Specificity, and Future Directions
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Small, D.H.; McLean, C.A. Alzheimer’s disease and the amyloid β protein: What is the role of amyloid? J. Neurochem. 1999, 73, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Z.; Cai, F.; Zhang, M.; Wu, Y.; Zhang, J.; Song, W. BACE1 cleavage site selection critical for amyloidogenesis and Alzheimer’s pathogenesis. J. Neurosci. 2017, 37, 6915–6925. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.; Quinlan, M.; Wisniewski, H.; Binder, L. Abnormal phosphorylation of the microtubule-associated protein? (tau) in Alzheimer cytoskeletal pathology. Alzheimer Dis. Assoc. Disord. 1987, 1, 202. [Google Scholar] [CrossRef]
- Bird, T.D. Genetic aspects of Alzheimer disease. Genet. Med. 2008, 10, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Rogaeva, E.; Beecham, G.W. Late-onset vs nonmendelian early-onset Alzheimer disease: A distinction without a difference? Neurol. Genet. 2020, 6, e512. [Google Scholar] [CrossRef]
- Xin, S.-H.; Tan, L.; Cao, X.; Yu, J.-T.; Tan, L. Clearance of amyloid beta and tau in Alzheimer’s disease: From mechanisms to therapy. Neurotox. Res. 2018, 34, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.M.; Sharom, F.J.; Reiner, P.B. β-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Ullah, R.; Lee, E.J. Advances in Amyloid-β Clearance in the Brain and Periphery: Implications for Neurodegenerative Diseases. Exp. Neurobiol. 2023, 32, 216. [Google Scholar] [CrossRef] [PubMed]
- Baranello, J.R.; Bharani, L.K.; Padmaraju, V.; Chopra, N.; Lahiri, K.D.; Greig, H.N.; Pappolla, A.M.; Sambamurti, K. Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Malpetti, M.; Kievit, R.A.; Passamonti, L.; Jones, P.S.; Tsvetanov, K.A.; Rittman, T.; Mak, E.; Nicastro, N.; Bevan-Jones, W.R.; Su, L. Microglial activation and tau burden predict cognitive decline in Alzheimer’s disease. Brain 2020, 143, 1588–1602. [Google Scholar] [CrossRef] [PubMed]
- Skender-Gazibara, M.; Slobodan, D. Neuropathological hallmarks of Alzheimer’s disease. Arch Oncol. 2001, 9, 5–7. [Google Scholar]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Noori, T.; Dehpour, A.R.; Sureda, A.; Sobarzo-Sanchez, E.; Shirooie, S. Role of natural products for the treatment of Alzheimer’s disease. Eur. J. Pharmacol. 2021, 898, 173974. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Jiang, Z.-F. Amyloid-β protein precursor family members: A review from homology to biological function. J. Alzheimer’s Dis. 2011, 26, 607–626. [Google Scholar] [CrossRef]
- Bayer, T.A.; Wirths, O.; Majtényi, K.; Hartmann, T.; Multhaup, G.; Beyreuther, K.; Czech, C. Key factors in Alzheimer’s disease: β-amyloid precursor protein processing, metabolism and intraneuronal transport. Brain Pathol. 2001, 11, 1–11. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Chen, G.-f.; Xu, T.-h.; Yan, Y.; Zhou, Y.-r.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Kahle, P.J.; De Strooper, B. Attack on amyloid: International Titisee Conference on Alzheimer’s and Parkinson’s Disease: From Basic Science to Therapeutic Treatment. EMBO Rep. 2003, 4, 747–752. [Google Scholar] [CrossRef]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Caillet-Boudin, M.-L.; Buée, L.; Sergeant, N.; Lefebvre, B. Regulation of human MAPT gene expression. Mol. Neurodegener. 2015, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Ghag, G.; Bhatt, N.; Cantu, D.V.; Guerrero-Munoz, M.J.; Ellsworth, A.; Sengupta, U.; Kayed, R. Soluble tau aggregates, not large fibrils, are the toxic species that display seeding and cross-seeding behavior. Protein Sci. 2018, 27, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- León-Espinosa, G.; Garcia, E.; García-Escudero, V.; Hernandez, F.; DeFelipe, J.; Avila, J. Changes in tau phosphorylation in hibernating rodents. J. Neurosci. Res. 2013, 91, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Roda, A.R.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666–1674. [Google Scholar]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated tau in Alzheimer’s disease and other tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and tau in the pathogenesis of Alzheimer’s disease. Int. J. Biol. Sci. 2021, 17, 2181. [Google Scholar] [CrossRef]
- Ferrer, I.; Blanco, R.; Carmona, M.; Puig, B.; Barrachina, M.; Gomez, C.; Ambrosio, S. Active, phosphorylation-dependent mitogen-activated protein kinase (MAPK/ERK), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), and p38 kinase expression in Parkinson’s disease and Dementia with Lewy bodies. J. Neural Transm. 2001, 108, 1383–1396. [Google Scholar] [CrossRef]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Nilson, A.N.; English, K.C.; Gerson, J.E.; Barton Whittle, T.; Nicolas Crain, C.; Xue, J.; Sengupta, U.; Castillo-Carranza, D.L.; Zhang, W.; Gupta, P. Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. J. Alzheimer’s Dis. 2017, 55, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Klein, A.N.; Vu, A.; Arifin, M.I.; Hannaoui, S.; Gilch, S. Peptide aptamer targeting Aβ–PrP–Fyn axis reduces Alzheimer’s disease pathologies in 5XFAD transgenic mouse model. Cell. Mol. Life Sci. 2023, 80, 139. [Google Scholar] [CrossRef]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Theuns, J.; Van Broeckhoven, C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 2012, 33, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.; Saunders, A.M.; Risch, N.; Strittmatter, W.; Schmechel, D.; Gaskell, P., Jr.; Rimmler, J.; Locke, P.; Conneally, P.; Schmader, K. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Dinamarca, M.C.; Ríos, J.A.; Inestrosa, N.C. Postsynaptic receptors for amyloid-β oligomers as mediators of neuronal damage in Alzheimer’s disease. Front. Physiol. 2012, 3, 34576. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.-Y.; Hyman, B.T.; Bacskai, B.J. Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Fagiani, F.; Lanni, C.; Racchi, M.; Govoni, S. (Dys) regulation of Synaptic Activity and Neurotransmitter Release by β-Amyloid: A Look Beyond Alzheimer’s Disease Pathogenesis. Front. Mol. Neurosci. 2021, 14, 635880. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Davies, C.; Sirisi, S.; Lee, J.-E.; Simzer, E.M.; Tzioras, M.; Querol-Vilaseca, M.; Sanchez-Aced, E.; Chang, Y.Y.; Holt, K. Synaptic oligomeric tau in Alzheimer’s disease—A potential culprit in the spread of tau pathology through the brain. Neuron 2023, 111, 2170–2183.e6. [Google Scholar] [CrossRef]
- Jurcău, M.C.; Andronie-Cioara, F.L.; Jurcău, A.; Marcu, F.; Ţiț, D.M.; Pașcalău, N.; Nistor-Cseppentö, D.C. The link between oxidative stress, mitochondrial dysfunction and neuroinflammation in the pathophysiology of Alzheimer’s disease: Therapeutic implications and future perspectives. Antioxidants 2022, 11, 2167. [Google Scholar] [CrossRef]
- Ali, J.; Khan, A.U.; Shah, F.A.; Ali, H.; Islam, S.U.; Kim, Y.S.; Khan, S. Mucoprotective effects of Saikosaponin-A in 5-fluorouracil-induced intestinal mucositis in mice model. Life Sci. 2019, 239, 116888. [Google Scholar] [CrossRef]
- Sultana, R.; Butterfield, D.A. Protein Oxidation in Aging and Alzheimer’s Disease Brain. Antioxidants 2024, 13, 574. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.; Hirai, K.; Aliev, G.; Drew, K.L.; Nunomura, A.; Takeda, A.; Cash, A.D.; Obrenovich, M.E.; Perry, G.; Smith, M.A. Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Shokouhi, S.; Claassen, D.; Kang, H.; Ding, Z.; Rogers, B.; Mishra, A.; Riddle, W.R.; Initiative, A.s.D.N. Longitudinal progression of cognitive decline correlates with changes in the spatial pattern of brain 18F-FDG PET. J. Nucl. Med. 2013, 54, 1564–1569. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial dysfunction as a driver of cognitive impairment in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; McKee, A.C.; Beal, M.F.; Graham, B.H.; Wallace, D.C. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics 1994, 23, 471–476. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.-g.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, K.; Ohta, Y.; Inufusa, H.; Loon, A.F.N.; Abe, K. Prevention of cognitive decline in Alzheimer’s disease by novel antioxidative supplements. Int. J. Mol. Sci. 2020, 21, 1974. [Google Scholar] [CrossRef]
- Cheignon, C.m.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Hemmelgarn, B.T.; Chuang, C.-C.; Best, T.M. The role of oxidative stress-induced epigenetic alterations in amyloid-β production in Alzheimer’s disease. Oxidative Med. Cell. Longev. 2015, 2015, 604658. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Yu, Q.; Kanaan, N.M.; Yan, S.S. Mitochondrial oxidative stress contributes to the pathological aggregation and accumulation of tau oligomers in Alzheimer’s disease. Hum. Mol. Genet. 2022, 31, 2498–2507. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, F.; Carro, E.; Alquezar, C. Oxidative stress in tauopathies: From cause to therapy. Antioxidants 2022, 11, 1421. [Google Scholar] [CrossRef]
- Yang, J.; Luo, J.; Tian, X.; Zhao, Y.; Li, Y.; Wu, X. Progress in Understanding Oxidative Stress, Aging, and Aging-Related Diseases. Antioxidants 2024, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; von Bernhardi, R.; Godoy, J.A.; Inestrosa, N.C.; Johnson, G.V. Phosphorylated tau potentiates Aβ-induced mitochondrial damage in mature neurons. Neurobiol. Dis. 2014, 71, 260–269. [Google Scholar] [CrossRef]
- Gamblin, T.C.; King, M.E.; Kuret, J.; Berry, R.W.; Binder, L.I. Oxidative regulation of fatty acid-induced tau polymerization. Biochemistry 2000, 39, 14203–14210. [Google Scholar] [CrossRef]
- Fani, G.; La Torre, C.E.; Cascella, R.; Cecchi, C.; Vendruscolo, M.; Chiti, F. Misfolded protein oligomers induce an increase of intracellular Ca2+ causing an escalation of reactive oxidative species. Cell. Mol. Life Sci. 2022, 79, 500. [Google Scholar] [CrossRef] [PubMed]
- Baracaldo-Santamaría, D.; Avendaño-Lopez, S.S.; Ariza-Salamanca, D.F.; Rodriguez-Giraldo, M.; Calderon-Ospina, C.A.; González-Reyes, R.E.; Nava-Mesa, M.O. Role of calcium modulation in the pathophysiology and treatment of Alzheimer’s disease. Int. J. Mol. Sci. 2023, 24, 9067. [Google Scholar] [CrossRef]
- Dhapola, R.; Beura, S.K.; Sharma, P.; Singh, S.K.; HariKrishnaReddy, D. Oxidative stress in Alzheimer’s disease: Current knowledge of signaling pathways and therapeutics. Mol. Biol. Rep. 2024, 51, 48. [Google Scholar] [CrossRef]
- Zhu, X.; Raina, A.K.; Lee, H.-g.; Casadesus, G.; Smith, M.A.; Perry, G. Oxidative stress signalling in Alzheimer’s disease. Brain Res. 2004, 1000, 32–39. [Google Scholar] [CrossRef]
- Zhang, L.; Jope, R.S. Oxidative stress differentially modulates phosphorylation of ERK, p38 and CREB induced by NGF or EGF in PC12 cells☆. Neurobiol. Aging 1999, 20, 271–278. [Google Scholar] [CrossRef]
- Ermak, G.; Davies, K.J. Chronic high levels of the RCAN1-1 protein may promote neurodegeneration and Alzheimer disease. Free Radic. Biol. Med. 2013, 62, 47–51. [Google Scholar] [CrossRef]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2–ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Gameiro, I.; Michalska, P.; Tenti, G.; Cores, Á.; Buendia, I.; Rojo, A.I.; Georgakopoulos, N.D.; Hernández-Guijo, J.M.; Teresa Ramos, M.; Wells, G. Discovery of the first dual GSK3β inhibitor/Nrf2 inducer. A new multitarget therapeutic strategy for Alzheimer’s disease. Sci. Rep. 2017, 7, 45701. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction, oxidative stress, and neuroinflammation: Intertwined roads to neurodegeneration. Antioxidants 2020, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4+ T cells in neurodegenerative diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef]
- Mhatre, M.; Floyd, R.A.; Hensley, K. Oxidative stress and neuroinflammation in Alzheimer’s disease and amyotrophic lateral sclerosis: Common links and potential therapeutic targets. J. Alzheimer’s Dis. 2004, 6, 147–157. [Google Scholar] [CrossRef]
- Qin, Q.; Teng, Z.; Liu, C.; Li, Q.; Yin, Y.; Tang, Y. TREM2, microglia, and Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111438. [Google Scholar] [CrossRef]
- García-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernández, L.; García-Domínguez, I.; Roca-Ceballos, M.A.; Santiago, M. Reformulating pro-oxidant microglia in neurodegeneration. J. Clin. Med. 2019, 8, 1719. [Google Scholar] [CrossRef]
- Simpson, D.S.; Oliver, P.L. ROS generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Wang, L.; Yang, J.-W.; Lin, L.-T.; Huang, J.; Wang, X.-R.; Su, X.-T.; Cao, Y.; Fisher, M.; Liu, C.-Z. Acupuncture attenuates inflammation in microglia of vascular dementia rats by inhibiting miR-93-mediated TLR4/MyD88/NF-κB signaling pathway. Oxidative Med. Cell. Longev. 2020, 2020, 8253904. [Google Scholar] [CrossRef]
- Hanslik, K.L.; Ulland, T.K. The role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimer’s Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Sawda, C.; Moussa, C.; Turner, R.S. Resveratrol for Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2017, 1403, 142–149. [Google Scholar] [CrossRef]
- Valverde-Salazar, V.; Ruiz-Gabarre, D.; García-Escudero, V. Alzheimer’s disease and green tea: Epigallocatechin-3-gallate as a modulator of inflammation and oxidative stress. Antioxidants 2023, 12, 1460. [Google Scholar] [CrossRef]
- Zaplatic, E.; Bule, M.; Shah, S.Z.A.; Uddin, M.S.; Niaz, K. Molecular mechanisms underlying protective role of quercetin in attenuating Alzheimer’s disease. Life Sci. 2019, 224, 109–119. [Google Scholar] [CrossRef]
- Ishige, K.; Schubert, D.; Sagara, Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic. Biol. Med. 2001, 30, 433–446. [Google Scholar] [CrossRef]
- Shah, S.A.; Amin, F.U.; Khan, M.; Abid, M.N.; Rehman, S.U.; Kim, T.H.; Kim, M.W.; Kim, M.O. Anthocyanins abrogate glutamate-induced AMPK activation, oxidative stress, neuroinflammation, and neurodegeneration in postnatal rat brain. J. Neuroinflammation 2016, 13, 286. [Google Scholar] [CrossRef]
- Shah, S.A.; Khan, M.; Jo, M.H.; Jo, M.G.; Amin, F.U.; Kim, M.O. Melatonin stimulates the SIRT 1/Nrf2 signaling pathway counteracting lipopolysaccharide (LPS)-induced oxidative stress to rescue postnatal rat brain. CNS Neurosci. Ther. 2017, 23, 33–44. [Google Scholar] [CrossRef]
- Ratto, F.; Franchini, F.; Musicco, M.; Caruso, G.; Di Santo, S.G. A narrative review on the potential of tomato and lycopene for the prevention of Alzheimer’s disease and other dementias. Crit. Rev. Food Sci. Nutr. 2022, 62, 4970–4981. [Google Scholar] [CrossRef]
- Shah, S.; Yoon, G.; Chung, S.; Abid, M.; Kim, T.; Lee, H.; Kim, M. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol. Psychiatry 2017, 22, 407–416. [Google Scholar] [CrossRef]
- Shah, S.A.; Yoon, G.H.; Kim, H.-O.; Kim, M.O. Vitamin C neuroprotection against dose-dependent glutamate-induced neurodegeneration in the postnatal brain. Neurochem. Res. 2015, 40, 875–884. [Google Scholar] [CrossRef]
- Lee, H.Y.; Naha, N.; Ullah, N.; Jin, G.Z.; Kong, I.K.; Koh, P.O.; Seong, H.H.; Kim, M.O. Effect of the co-administration of vitamin C and vitamin E on tyrosine hydroxylase and Nurr1 expression in the prenatal rat ventral mesencephalon. J. Vet. Med. Sci. 2008, 70, 791–797. [Google Scholar] [CrossRef]
- Fišar, Z.; Hroudová, J. CoQ10 and Mitochondrial Dysfunction in Alzheimer’s Disease. Antioxidants 2024, 13, 191. [Google Scholar] [CrossRef]
- Kagan, V.; Fabisiak, J.; Quinn, P. Coenzyme Q and vitamin E need each other as antioxidants. Protoplasma 2000, 214, 11–18. [Google Scholar] [CrossRef]
- Pritam, P.; Deka, R.; Bhardwaj, A.; Srivastava, R.; Kumar, D.; Jha, A.K.; Jha, N.K.; Villa, C.; Jha, S.K. Antioxidants in Alzheimer’s disease: Current therapeutic significance and future prospects. Biology 2022, 11, 212. [Google Scholar] [CrossRef]
- Wei, P.; Li, X.; Wang, S.; Dong, Y.; Yin, H.; Gu, Z.; Na, X.; Wei, X.; Yuan, J.; Cao, J. Silibinin ameliorates formaldehyde-induced cognitive impairment by inhibiting oxidative stress. Oxidative Med. Cell. Longev. 2022, 2022, 5981353. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Tonk, S.; Kuruva, C.S.; Bhatti, J.S.; Kandimalla, R.; Vijayan, M. Protective effects of Indian spice curcumin against amyloid-β in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 61, 843–866. [Google Scholar] [CrossRef]
- Holmquist, L.; Stuchbury, G.; Berbaum, K.; Muscat, S.; Young, S.; Hager, K.; Engel, J.; Münch, G. Lipoic acid as a novel treatment for Alzheimer’s disease and related dementias. Pharmacol. Ther. 2007, 113, 154–164. [Google Scholar] [CrossRef]
- Diwakar, L.; Ravindranath, V. Protein glutathionylation and glutaredoxin: Role in neurodegenerative diseases. Antioxidants 2022, 11, 2334. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, H.; Luo, Y. Anti-aging implications of Astragalus membranaceus (Huangqi): A well-known Chinese tonic. Aging Dis. 2017, 8, 868. [Google Scholar] [CrossRef]
- Levin, E.D.; Christopher, N.C.; Briggs, S.J.; Rose, J.E. Chronic nicotine reverses working memory deficits caused by lesions of the fimbria or medial basalocortical projection. Cogn. Brain Res. 1993, 1, 137–143. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, J.; Zhu, H.; Qin, C.; Chen, Q.; Zhao, B. Dissecting the signaling pathway of nicotine-mediated neuroprotection in a mouse Alzheimer disease model. FASEB J. 2007, 21, 61–73. [Google Scholar] [CrossRef]
- Chaves, S.K.M.; Afzal, M.I.; Islam, M.T.; Hameed, A.; Da Mata, A.M.O.F.; da Silva Araújo, L.; Ali, S.W.; Rolim, H.M.L.; De Medeiros, M.d.G.F.; Costa, E.V. Palmatine antioxidant and anti-acetylcholinesterase activities: A pre-clinical assessment. Cell. Mol. Biol. 2020, 66, 54–59. [Google Scholar] [CrossRef]
- Han, L.; Chen, W.; Li, J.; Zhao, Y.; Zong, Y.; He, Z.; Du, R. Palmatine improves cognitive dysfunction in Alzheimer’s disease model rats through autophagy pathway and regulation of gut microbiota. Brain Res. 2024, 1835, 148932. [Google Scholar] [CrossRef]
- Ali, J.; Khan, A.; Park, J.S.; Tahir, M.; Ahmad, W.; Choe, K.; Kim, M.O. Neuroprotective Effects of N-methyl-(2S, 4R)-trans-4-hydroxy-L-proline (NMP) against Amyloid-β-Induced Alzheimer’s Disease Mouse Model. Nutrients 2023, 15, 4986. [Google Scholar] [CrossRef]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 625–630. [Google Scholar] [CrossRef]
- Hsieh, H.-M.; Wu, W.-M.; Hu, M.-L. Soy isoflavones attenuate oxidative stress and improve parameters related to aging and Alzheimer’s disease in C57BL/6J mice treated with D-galactose. Food Chem. Toxicol. 2009, 47, 625–632. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, Q.; Zhao, B. P4-330 Genistein ameliorated β-amyloid peptide (25–35)-induced hippocampal neuronal apoptosis. Neurobiol. Aging 2004, 36, 180–188. [Google Scholar]
- Wang, Y.; Huang, Y.; Ma, A.; You, J.; Miao, J.; Li, J. Natural Antioxidants: An Effective Strategy for the Treatment of Alzheimer’s Disease at the Early Stage. J. Agric. Food Chem. 2024, 72, 11854–11870. [Google Scholar] [CrossRef]
- Zou, Y.; Yang, M.; Gong, D.-Z.; Guan, L.-L.; Tian, N. Effects of vagotomy on UCP2 mRNA expression and gastric acid secretion in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi = Zhongguo Yingyong Shenglixue Zazhi = Chin. J. Appl. Physiol. 2005, 21, 290–292. [Google Scholar]
- Min, J.-y.; Min, K.-b. Serum lycopene, lutein and zeaxanthin, and the risk of Alzheimer’s disease mortality in older adults. Dement. Geriatr. Cogn. Disord. 2014, 37, 246–256. [Google Scholar] [CrossRef]
- Yu, L.; Wang, W.; Pang, W.; Xiao, Z.; Jiang, Y.; Hong, Y. Dietary lycopene supplementation improves cognitive performances in tau transgenic mice expressing P301L mutation via inhibiting oxidative stress and tau hyperphosphorylation. J. Alzheimer’s Dis. 2017, 57, 475–482. [Google Scholar] [CrossRef]
- Li, S.; Chen, G.; Zhang, C.; Wu, M.; Wu, S.; Liu, Q. Research progress of natural antioxidants in foods for the treatment of diseases. Food Sci. Hum. Wellness 2014, 3, 110–116. [Google Scholar] [CrossRef]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef]
- Tappel, A.L. Vitamin E as the biological lipid antioxidant. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 1962; Volume 20, pp. 493–510. [Google Scholar]
- Kim, H.; Kim, G.; Jang, W.; Kim, S.Y.; Chang, N. Association between intake of B vitamins and cognitive function in elderly Koreans with cognitive impairment. Nutr. J. 2014, 13, 1–11. [Google Scholar] [CrossRef]
- Duan, S.; Guan, X.; Lin, R.; Liu, X.; Yan, Y.; Lin, R.; Zhang, T.; Chen, X.; Huang, J.; Sun, X. Silibinin inhibits acetylcholinesterase activity and amyloid β peptide aggregation: A dual-target drug for the treatment of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 1792–1807. [Google Scholar] [CrossRef]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Ono, K.; Yamada, M. Curcumin and Alzheimer’s disease. CNS Neurosci. Ther. 2010, 16, 285–297. [Google Scholar] [CrossRef]
- Kaur, D.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Chigurupati, S.; Alhowail, A.; Abdeen, A.; Ibrahim, S.F.; Vargas-De-La-Cruz, C. Decrypting the potential role of α-lipoic acid in Alzheimer’s disease. Life Sci. 2021, 284, 119899. [Google Scholar] [CrossRef]
- Singh, S.; Gupta, G.; Aran, K.R. Emerging role of antioxidants in Alzheimer’s disease: Insight to physiological, pathological mechanisms and management. Pharm. Sci. Adv. 2023, 2, 100021. [Google Scholar]
- Huang, Y.-C.; Tsay, H.-J.; Lu, M.-K.; Lin, C.-H.; Yeh, C.-W.; Liu, H.-K.; Shiao, Y.-J. Astragalus membranaceus-polysaccharides ameliorates obesity, hepatic steatosis, neuroinflammation and cognition impairment without affecting amyloid deposition in metabolically stressed APPswe/PS1dE9 mice. Int. J. Mol. Sci. 2017, 18, 2746. [Google Scholar] [CrossRef]
- Liu, Q.; Tao, Y.; Zhao, B. ESR study on scavenging effect of nicotine on free radicals. Appl. Magn. Reson. 2003, 24, 105–112. [Google Scholar] [CrossRef]
- Jia, W.; Su, Q.; Cheng, Q.; Peng, Q.; Qiao, A.; Luo, X.; Zhang, J.; Wang, Y. Neuroprotective Effects of Palmatine via the Enhancement of Antioxidant Defense and Small Heat Shock Protein Expression in Aβ-Transgenic Caenorhabditis elegans. Oxidative Med. Cell. Longev. 2021, 2021, 9966223. [Google Scholar] [CrossRef]
- Kishi, T.; Matsunaga, S.; Oya, K.; Nomura, I.; Ikuta, T.; Iwata, N. Memantine for Alzheimer’s disease: An updated systematic review and meta-analysis. J. Alzheimer’s Dis. 2017, 60, 401–425. [Google Scholar] [CrossRef]
- Krafft, G.A.; Jerecic, J.; Siemers, E.; Cline, E.N. ACU193: An immunotherapeutic poised to test the amyloid β oligomer hypothesis of Alzheimer’s disease. Front. Neurosci. 2022, 16, 848215. [Google Scholar] [CrossRef]
- Hey, J.A.; Yu, J.Y.; Versavel, M.; Abushakra, S.; Kocis, P.; Power, A.; Kaplan, P.L.; Amedio, J.; Tolar, M. Clinical pharmacokinetics and safety of ALZ-801, a novel prodrug of tramiprosate in development for the treatment of Alzheimer’s disease. Clin. Pharmacokinet. 2018, 57, 315–333. [Google Scholar] [CrossRef]
- de la Torre, R.; Dierssen, M. Therapeutic approaches in the improvement of cognitive performance in Down syndrome: Past, present, and future. Prog. Brain Res. 2012, 197, 1–14. [Google Scholar]
- Lannfelt, L.; Möller, C.; Basun, H.; Osswald, G.; Sehlin, D.; Satlin, A.; Logovinsky, V.; Gellerfors, P. Perspectives on future Alzheimer therapies: Amyloid-β protofibrils-a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 16. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Budd Haeberlein, S.; Aisen, P.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; Von Hehn, C. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J. Prev. Alzheimer’s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef]
- Miles, L.A.; Crespi, G.A.; Doughty, L.; Parker, M.W. Bapineuzumab captures the N-terminus of the Alzheimer’s disease amyloid-beta peptide in a helical conformation. Sci. Rep. 2013, 3, 1302. [Google Scholar] [CrossRef]
- Holdridge, K.C.; Yaari, R.; Hoban, D.B.; Andersen, S.; Sims, J.R. Targeting amyloid β in Alzheimer’s disease: Meta-analysis of low-dose solanezumab in Alzheimer’s disease with mild dementia studies. Alzheimer’s Dement. 2023, 19, 4619–4628. [Google Scholar] [CrossRef]
- Patton, R.L.; Kalback, W.M.; Esh, C.L.; Kokjohn, T.A.; Van Vickle, G.D.; Luehrs, D.C.; Kuo, Y.-M.; Lopez, J.; Brune, D.; Ferrer, I. Amyloid-β peptide remnants in AN-1792-immunized Alzheimer’s disease patients: A biochemical analysis. Am. J. Pathol. 2006, 169, 1048–1063. [Google Scholar] [CrossRef]
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, aducanumab, and gantenerumab—Binding profiles to different forms of amyloid-beta might explain efficacy and side effects in clinical trials for Alzheimer’s disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- May, P.C.; Willis, B.A.; Lowe, S.L.; Dean, R.A.; Monk, S.A.; Cocke, P.J.; Audia, J.E.; Boggs, L.N.; Borders, A.R.; Brier, R.A. The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 2015, 35, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Matsuki, S.; Matsuguma, K.; Yoshihara, T.; Uchida, N.; Azuma, F.; Russell, M.; Hughes, G.; Haeberlein, S.B.; Alexander, R.C. BACE1 inhibitor lanabecestat (AZD3293) in a phase 1 study of healthy Japanese subjects: Pharmacokinetics and effects on plasma and cerebrospinal fluid Aβ peptides. J. Clin. Pharmacol. 2017, 57, 1460–1471. [Google Scholar] [CrossRef]
- Al-Tel, T.H.; Semreen, M.H.; Al-Qawasmeh, R.A.; Schmidt, M.F.; El-Awadi, R.; Ardah, M.; Zaarour, R.; Rao, S.N.; El-Agnaf, O. Design, synthesis, and qualitative structure–activity evaluations of novel β-Secretase inhibitors as potential Alzheimer’s drug leads. J. Med. Chem. 2011, 54, 8373–8385. [Google Scholar] [CrossRef]
- Nordvall, G.; Lundkvist, J.; Sandin, J. Gamma-secretase modulators: A promising route for the treatment of Alzheimer’s disease. Front. Mol. Neurosci. 2023, 16, 1279740. [Google Scholar] [CrossRef]
- Ross, J.; Sharma, S.; Winston, J.; Nunez, M.; Bottini, G.; Franceschi, M.; Scarpini, E.; Frigerio, E.; Fiorentini, F.; Fernandez, M. CHF5074 reduces biomarkers of neuroinflammation in patients with mild cognitive impairment: A 12-week, double-blind, placebo-controlled study. Curr. Alzheimer Res. 2013, 10, 742–753. [Google Scholar] [CrossRef]
- Nakano-Ito, K.; Fujikawa, Y.; Hihara, T.; Shinjo, H.; Kotani, S.; Suganuma, A.; Aoki, T.; Tsukidate, K. E2012-induced cataract and its predictive biomarkers. Toxicol. Sci. 2014, 137, 249–258. [Google Scholar] [CrossRef]
- Fields, M.; Marcuzzi, A.; Gonelli, A.; Celeghini, C.; Maximova, N.; Rimondi, E. Mitochondria-targeted antioxidants, an innovative class of antioxidant compounds for neurodegenerative diseases: Perspectives and limitations. Int. J. Mol. Sci. 2023, 24, 3739. [Google Scholar] [CrossRef]
- Varesi, A.; Campagnoli, L.I.M.; Carrara, A.; Pola, I.; Floris, E.; Ricevuti, G.; Chirumbolo, S.; Pascale, A. Non-enzymatic antioxidants against Alzheimer’s disease: Prevention, diagnosis and therapy. Antioxidants 2023, 12, 180. [Google Scholar] [CrossRef]
- Sun, X.; He, G.; Song, W. BACE2, as a novel APP θ-secretase, is not responsible for the pathogenesis of Alzheimer’s disease in Down syndrome. FASEB J. 2006, 20, 1369–1376. [Google Scholar] [CrossRef]
- Daviglus, M.L.; Bell, C.C.; Berrettini, W.; Bowen, P.E.; Connolly, E.S., Jr.; Cox, N.J.; Dunbar-Jacob, J.M.; Granieri, E.C.; Hunt, G.; McGarry, K. National Institutes of Health State-of-the-Science Conference statement: Preventing alzheimer disease and cognitive decline. Ann. Intern. Med. 2010, 153, 176–181. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Benzinger, T.L.; Blazey, T.; Jack, C.R., Jr.; Koeppe, R.A.; Su, Y.; Xiong, C.; Raichle, M.E.; Snyder, A.Z.; Ances, B.M.; Bateman, R.J. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, E4502–E4509. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Barbosa, D.J.; Remião, F.; Silva, R. Alzheimer’s disease: Insights and new prospects in disease pathophysiology, biomarkers and disease-modifying drugs. Biochem. Pharmacol. 2023, 211, 115522. [Google Scholar] [CrossRef]
- Pires, P.C.; Paiva-Santos, A.C.; Veiga, F. Liposome-Derived Nanosystems for the Treatment of Behavioral and Neurodegenerative Diseases: The Promise of Niosomes, Transfersomes, and Ethosomes for Increased Brain Drug Bioavailability. Pharmaceuticals 2023, 16, 1424. [Google Scholar] [CrossRef]
- Tiwari, V.; Tiwari, A.; Sharma, A.; Kumar, M.; Kaushik, D.; Sagadevan, S. An optimistic approach to nanotechnology in Alzheimer’s disease management: An overview. J. Drug Deliv. Sci. Technol. 2023, 86, 104722. [Google Scholar] [CrossRef]
- Nguyen, N.K.; Poduska, B.; Franks, M.; Bera, M.; MacCormack, I.; Lin, G.; Petroff, A.P.; Das, S.; Nag, A. A Copper-Selective Sensor and Its Inhibition of Copper-Amyloid Beta Aggregation. Biosensors 2024, 14, 247. [Google Scholar] [CrossRef]
- Yang, M.; Chen, Y.; Sun, H.; Li, D.; Li, Y. A Simple Sandwich Electrochemical Immunosensor for Rapid Detection of the Alzheimer’s Disease Biomarker Tau Protein. Biosensors 2024, 14, 279. [Google Scholar] [CrossRef]
- Ivanov, A.; Shamagsumova, R.; Larina, M.; Evtugyn, G. Electrochemical Acetylcholinesterase Sensors for Anti-Alzheimer’s Disease Drug Determination. Biosensors 2024, 14, 93. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, L.; Fair, S.R.; McElroy, C.A.; Hester, M.E.; Fu, H. Modeling neurodegenerative diseases with cerebral organoids and other three-dimensional culture systems: Focus on Alzheimer’s disease. Stem Cell Rev. Rep. 2020, 18, 696–717. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Ji, C.; Tetlow, A.M.; Jiang, Y.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease: Current status and future directions. Nat. Rev. Neurol. 2023, 19, 715–736. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Active Ingredient | Mechanism of Action (MOA) | Outcome Goals | Reference |
|---|---|---|---|
| Resveratrol | PI3K/Akt/Nrf2 pathway | Reduced ROS, elevated antioxidant enzymes GST and SOD, and activated PI3K, Akt, HO-1, and Nrf2 pathways. | [81] |
| Epicatechin gallate Green tea | Iron-chelating, activating ERK, Akt/PKB, PI3K, and PKC pathways | Reduces both amyloid plaques and ROS, promotes the production of SAPPα | [82] |
| Quercetin | Nrf2/ARE pathway reduces the level of MDA in an animal model | Increase antioxidant enzymes SOD, HO-1, Catalase, and scavenging ROS | [83] |
| Apigenin | C2–C3 double bond on the C ring reduces hydrogen peroxide and ROS ERK1/2/CREB/BDNF pathways | Reduce oxidative stress, Synaptic improvement | [84] |
| Anthocyanins | Reduce glutamate-induced AMPK activation, ros, and pro-inflammatory cytokines | Glutamate-induced neurotoxicity, neuroinflammation | [85] |
| Melatonin | SIRT1/Nrf2 pathway, anti-amyloidogenic properties | oxidative stress damage, apoptotic neurodegeneration | [86] |
| Lycopene | Nrf2/ARE pathway, reducing the phosphorylation of the tau protein | Increased antioxidant enzymes as well as anti-AD benefits | [87] |
| Osmotin | TLR4/NFκB signaling and SREBP2 via the AdipoR1/AMPK/SIRT1 pathway | Neuroinflammation, improved pre- and post-synaptic dysfunction | [88] |
| Vitamin C | Preserves the mitochondria’s cellular membrane integrity, scavenging ROS | Antioxidant potential in AD, change in cognitive function | [89] |
| Vitamin E | Tyrosine hydroxylase and Nurr1 expression, scavenging ROS | Antioxidant, neuroprotective, anti-inflammatory, and cholesterol-lowering properties | [90] |
| Coenzyme Q10 | As a cofactor for electron carriers in ETC, inhibits lipid peroxidation, recycles vitamin E phenoxyl radical | Bioenergetic modulator, antioxidant, boost synaptic connection and neurite outgrowth | [91,92] |
| Silibinin | Aβ and AChE inhibitors, lowering the production of H2O2 | Could increase the number of neuronal precursor cells in the brain, reduce oxidative stress | [93,94] |
| Curcumin | Increased GSH level, reduced insoluble Aβ, soluble Aβ, tau hyperphosphorylation, and intracellular calcium levels | Anti-protein aggregates, antioxidant, epigenetic potential in AD | [95] |
| Alpha-lipoic acid | ChAT enzyme activation, stimulation of phosphokinase C, metal chelation | As a dietary supplement for AD, reduces age-linked cognitive decline | [96] |
| Glutathione | substrate for thiol oxidoreductase and oxidized glutathione disulfide, reduces hydrogen peroxide, peroxynitrite, and lipid hydroperoxides | Maintain the redox state of the brain | [97] |
| Astragalus membranaceus | Increase the number of M-cholinergic receptors in the brain, inhibit reactive gliosis | Alleviating AD-related metabolic pathologies | [98] |
| Nicotine | Cholinergic secretagogue, down-regulation of inducible NOS, inhibition of NF-kB and c-Myc pathways, scavenging of hydroxyl and superoxide radicals | Enhance retention, and learning abilities in AD patients, antioxidant | [99,100] |
| Palmatine | Anti-AChE, AMPK/mTOR autophagy signaling system | Anti-depressant, anti-inflammatory, antioxidant | [101,102] |
| N-methyl-(2S, 4R)-Trans-4-hydroxy-L-proline | Elevate expression levels of NRF2/HO-1 and pre-synaptic and post-synaptic proteins | Anti-inflammatory, antioxidant, improving cognitive improvement | [103] |
| N-acetyl-L-cysteine | Increased GSH level, cysteine donor, cross BBB | Cease apoptosis in CNS, improved dementia rating scale | [104] |
| Active Ingredient | Mechanism of Action (MOA) | Outcome Goals | Reference |
|---|---|---|---|
| Bapineuzumab | Aβ in a monomeric helical conformation at the N-terminus | An early biomarker of AD | [130] |
| Solanezumab | A humanized monoclonal IgG1 antibody that decreases Aβ-induced synaptic toxicity by targeting the Aβ peptide’s mid-domain | elevated levels of plasma Aβ and lowered levels of CSF Aβ40, synaptic re-modelling in phase 2/3 clinical trials | [131] |
| AN1792 | Amyloid-beta (anti-Aβ) vaccine | Senile plaque disruption | [132] |
| Gantenerumab | Human IgG1 antibody that binds to a conformational epitope on Aβ fibrils to stimulate phagocytosis by attracting microglia | Reduced plaque load | [133] |
| BAN2401 | A monoclonal antibody with a humanized version of mAb158, selectivity for protofibrils | Aβ immunotherapy in early AD | [127] |
| Lecanemab | A humanized IgG1 binds to large soluble Aβ protofibrils (Approved by the FDA) | Reduced brain amyloid and improved cognitive decline in phase 3 clinical trials | [134] |
| Aducanumab | Fully humanized IgG1 mAb target conformational epitope present on the N-terminus of Aβ, dissolving β-amyloid clumps into smaller oligopeptides (Approved by FDA in June 2021) | Significantly improved cognitive deficits with high dose of intravenous infusion in randomized phase 1b and phase 3 clinical trials | [128,129] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, J.; Choe, K.; Park, J.S.; Park, H.Y.; Kang, H.; Park, T.J.; Kim, M.O. The Interplay of Protein Aggregation, Genetics, and Oxidative Stress in Alzheimer’s Disease: Role for Natural Antioxidants and Immunotherapeutics. Antioxidants 2024, 13, 862. https://doi.org/10.3390/antiox13070862
Ali J, Choe K, Park JS, Park HY, Kang H, Park TJ, Kim MO. The Interplay of Protein Aggregation, Genetics, and Oxidative Stress in Alzheimer’s Disease: Role for Natural Antioxidants and Immunotherapeutics. Antioxidants. 2024; 13(7):862. https://doi.org/10.3390/antiox13070862
Chicago/Turabian StyleAli, Jawad, Kyonghwan Choe, Jun Sung Park, Hyun Young Park, Heeyoung Kang, Tae Ju Park, and Myeong Ok Kim. 2024. "The Interplay of Protein Aggregation, Genetics, and Oxidative Stress in Alzheimer’s Disease: Role for Natural Antioxidants and Immunotherapeutics" Antioxidants 13, no. 7: 862. https://doi.org/10.3390/antiox13070862