
Pinitol Improves Diabetic Foot Ulcers in Streptozotocin-Induced Diabetes Rats Through Upregulation of Nrf2/HO-1 Signaling



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. Cell Proliferation Assay
2.4. Wound Healing Assay
2.5. Measurement of ROS
2.6. Measurement of Mitochondrial Membrane Potential (JC-1)
2.7. Measurement of Mitochondrial ATP Content
2.8. Nuclear and Cytoplasmic Protein Extraction
2.9. Western Blot Analysis
2.10. Immunofluorescence Analysis
2.11. Real-Time Quantitative Reverse Transcriptase-Polymerase Chain Reaction
2.12. Animal Models of Diabetes
2.13. Wound-Healing Model
2.14. Measurement of MDA, and GSH Levels
2.15. Histological Analysis
2.16. Statistical Analysis
3. Results
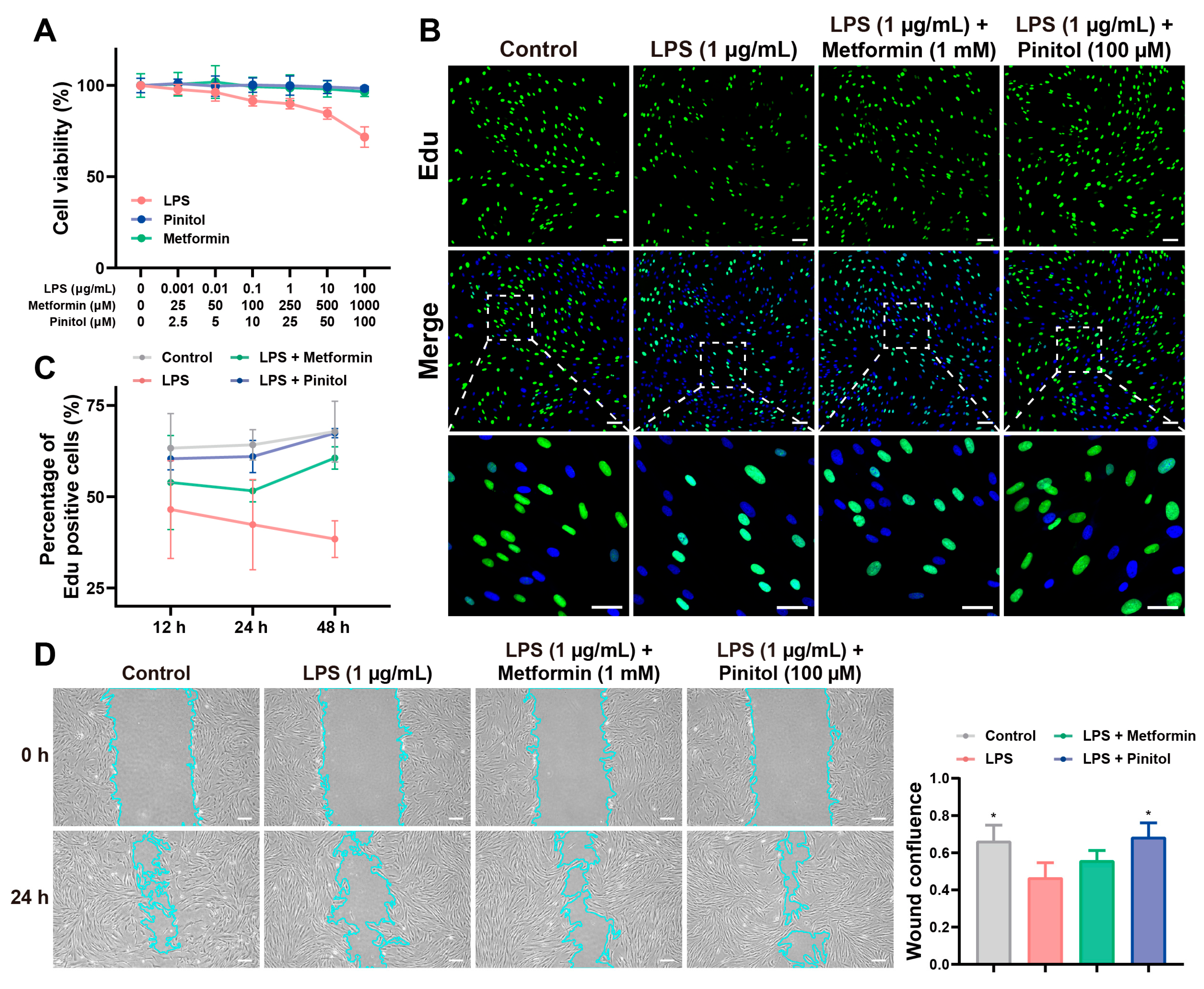
3.1. Pinitol Promoted Proliferation and Wound Healing
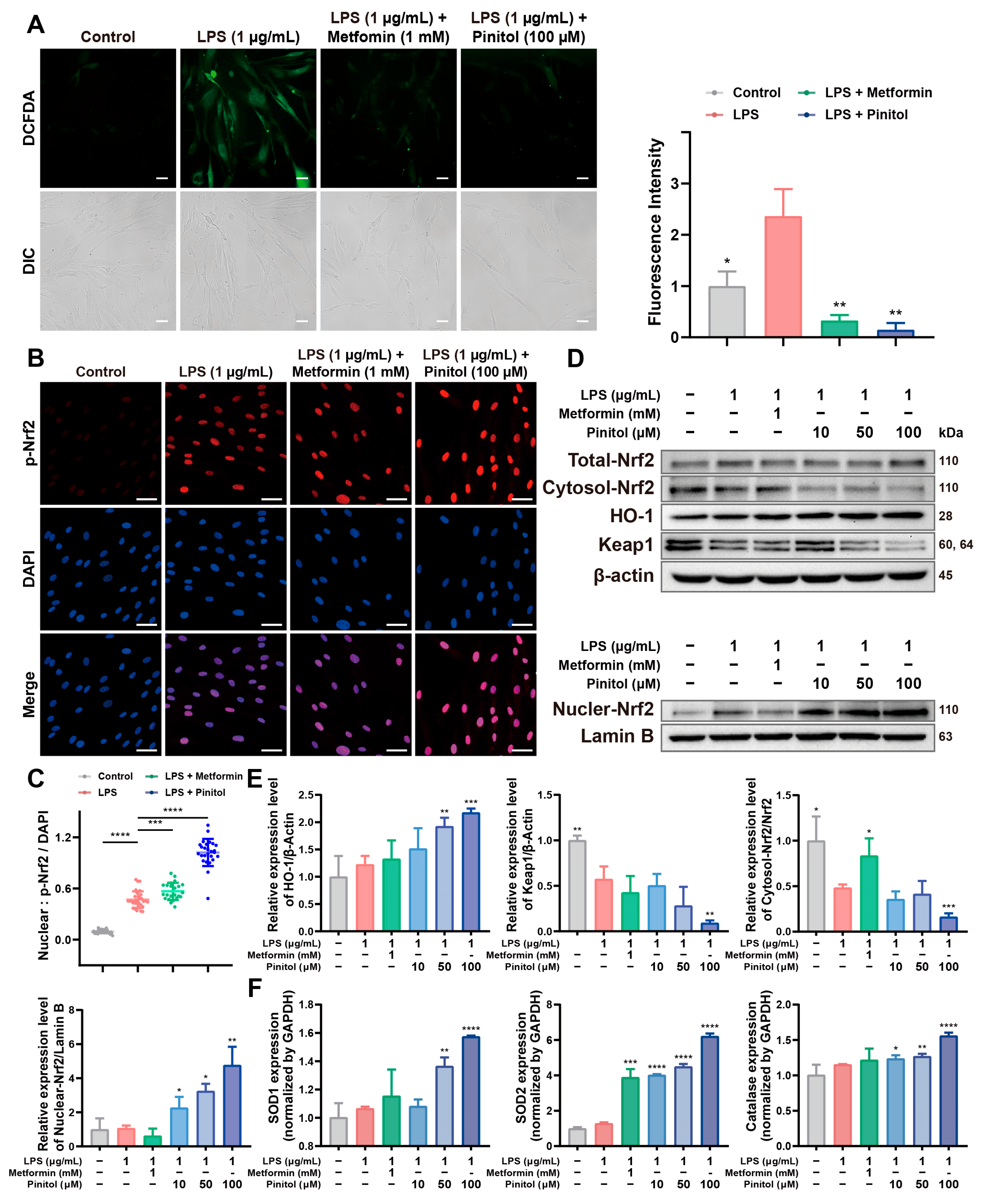
3.2. Pinitol Enhanced the Upregulation of Nrf2 to Mediate the Antioxidant Response
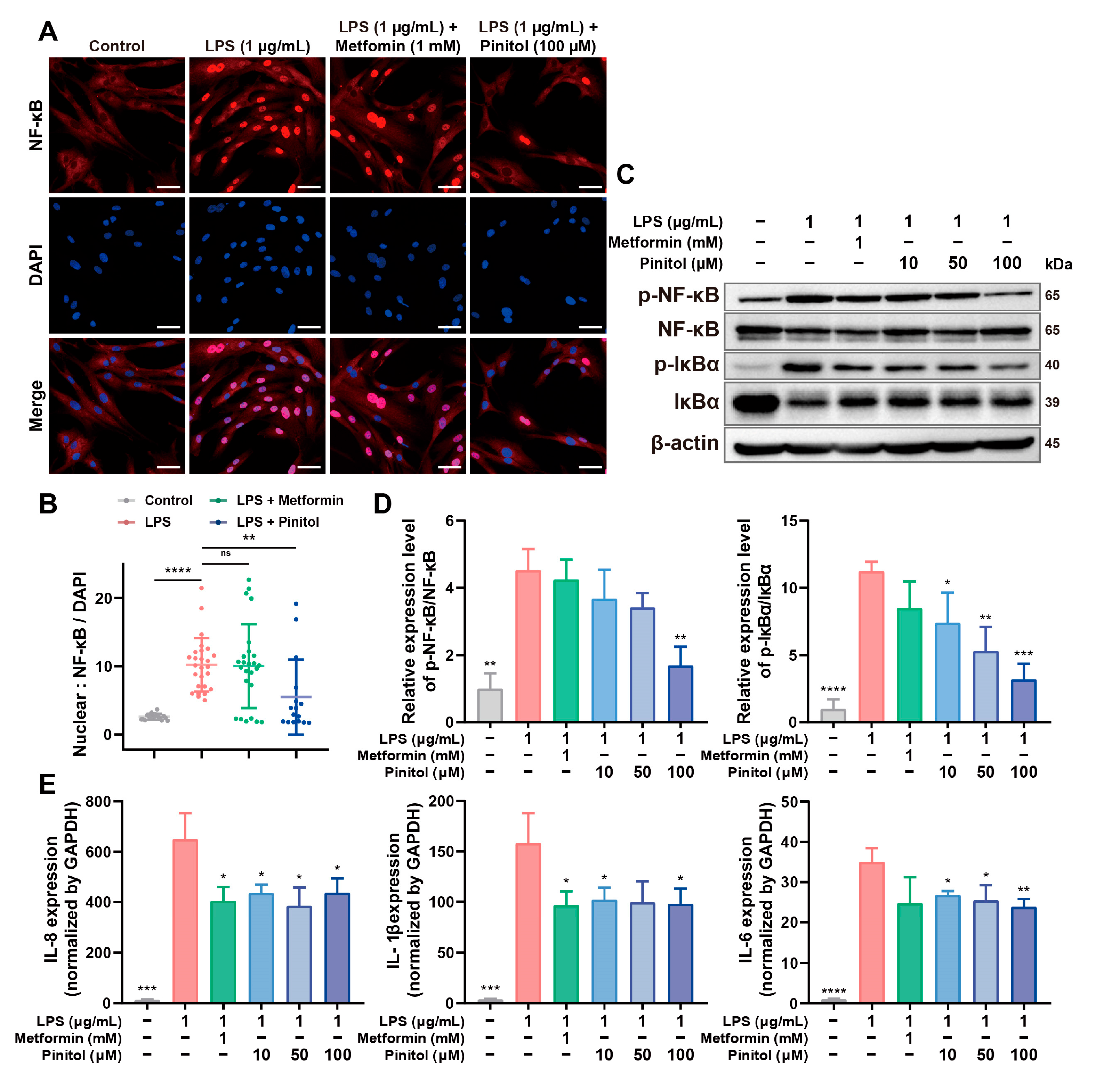
3.3. Pinitol Downregulated the IκBα/NF-κB Signaling to Reduce LPS-Induced Inflammation
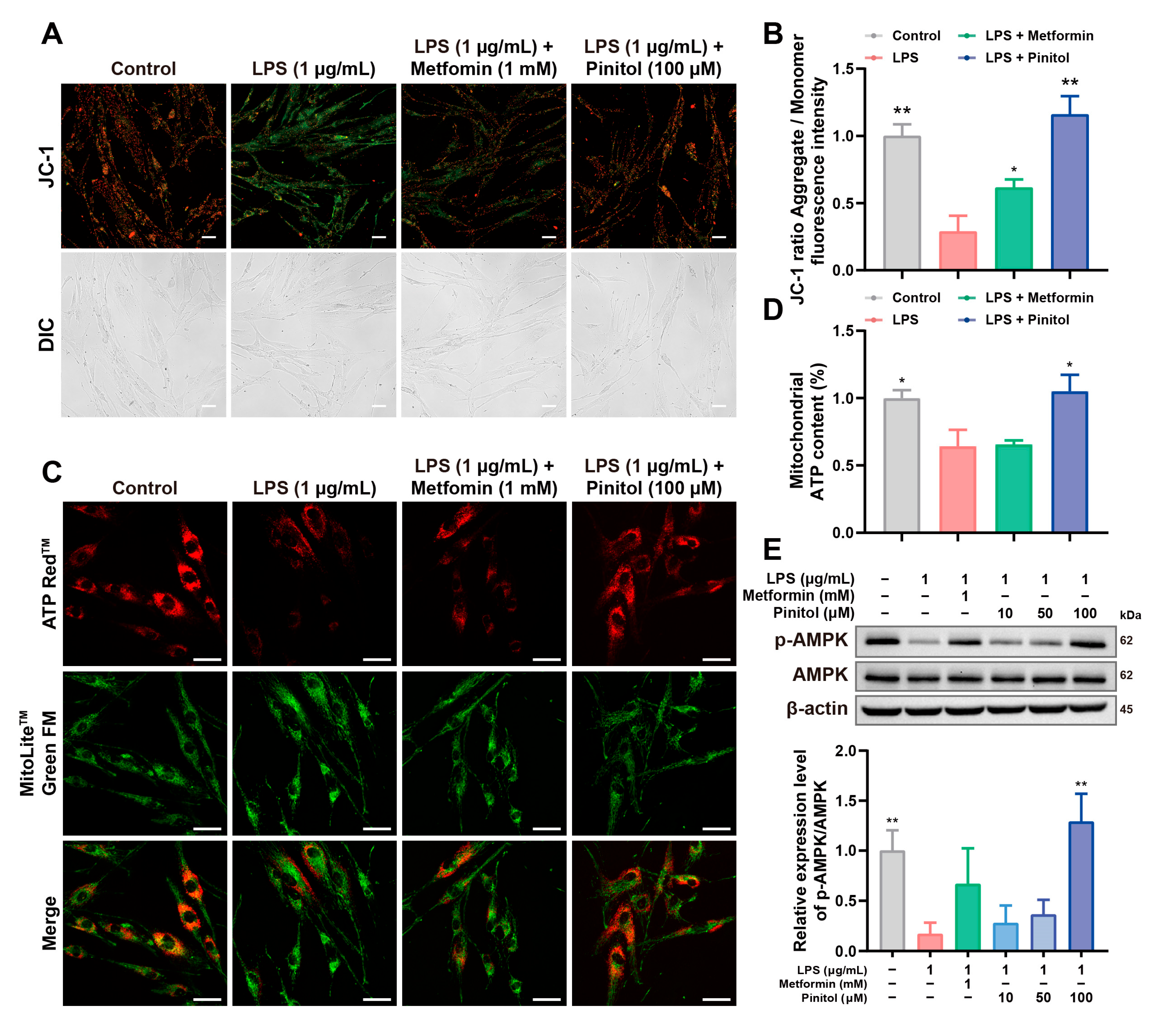
3.4. Pinitol Improved LPS-Induced Mitochondrial Dysfunction
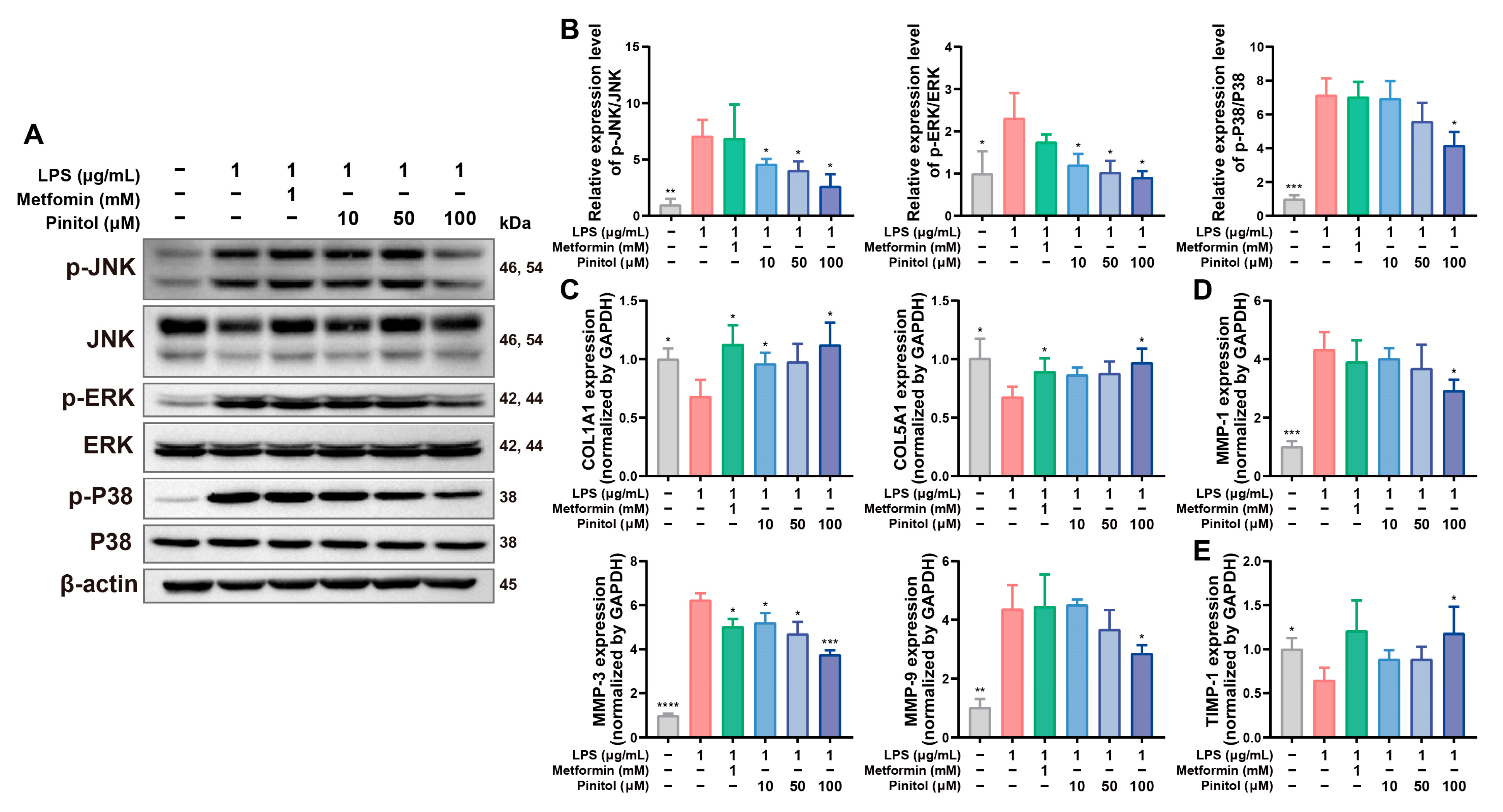
3.5. Pinitol Downregulated the MAPK Pathway, Reduced MMP Levels, and Protected Collagen
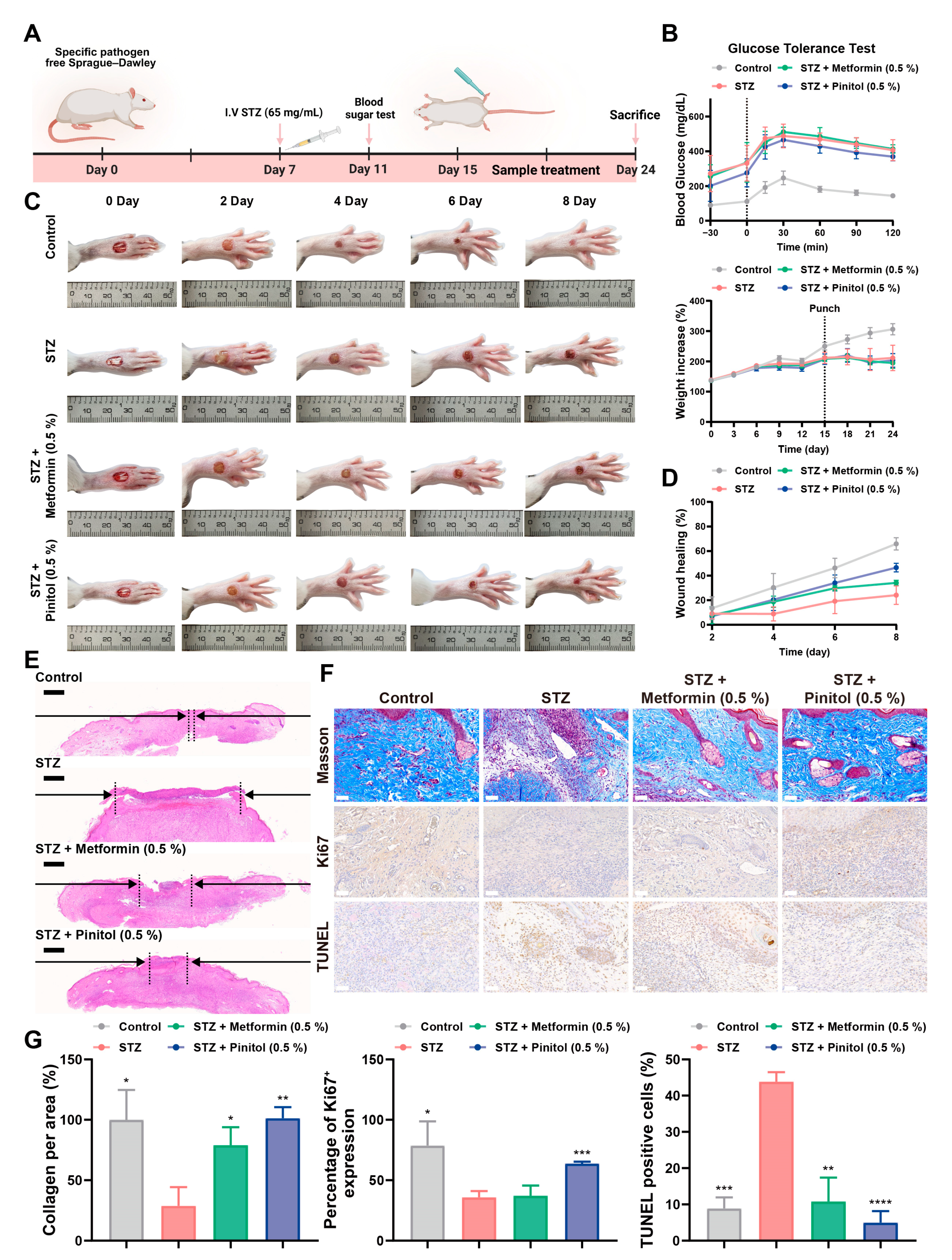
3.6. Pinitol Accelerated Wound Healing in Diabetic Foot Ulcers
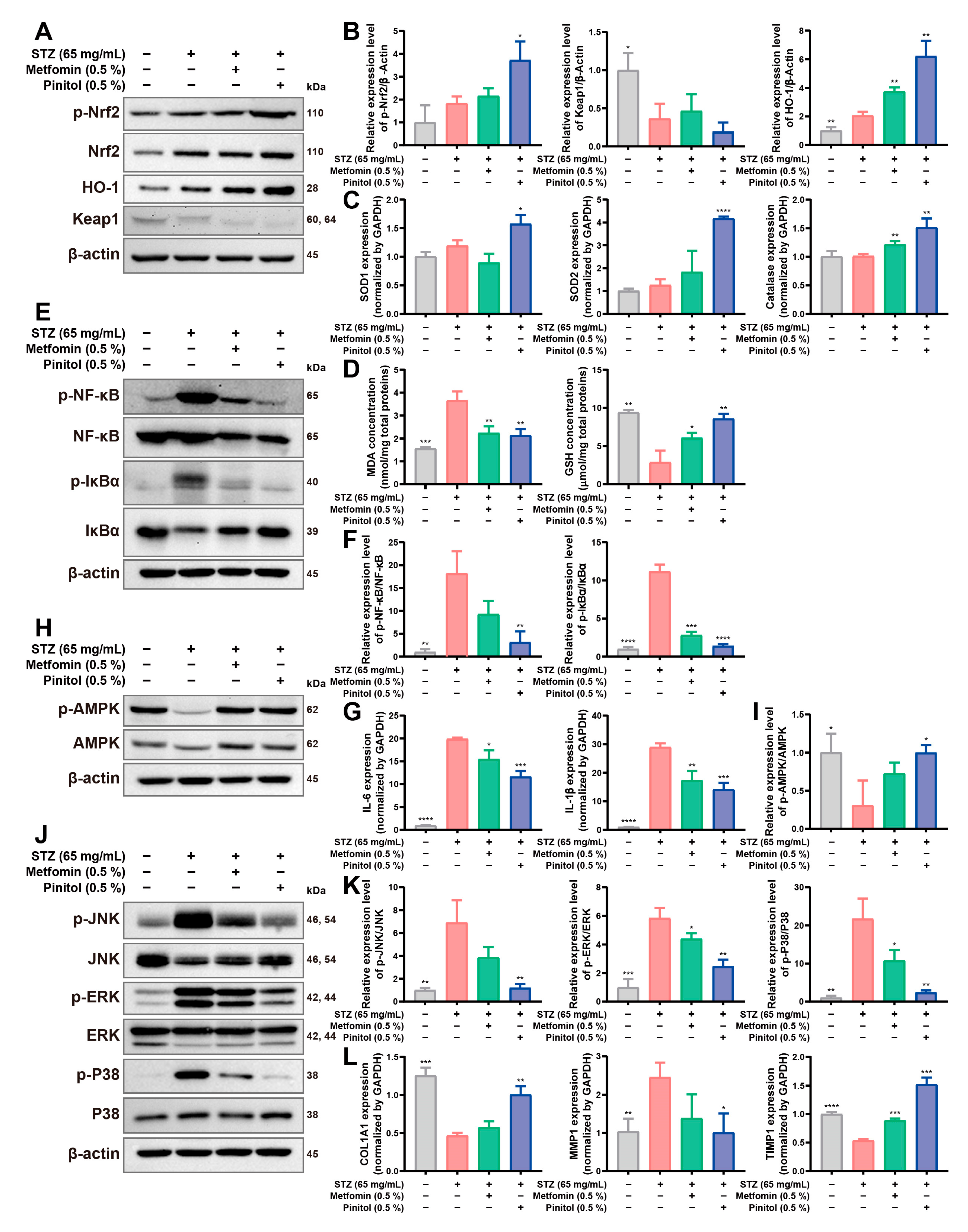
3.7. Pinitol Upregulated Nrf2 to Reduce Oxidative Stress and Inflammation, Enhancing Diabetic Foot Ulcers
3.8. Pinitol Restored Impaired Angiogenesis in Diabetic Foot Ulcers
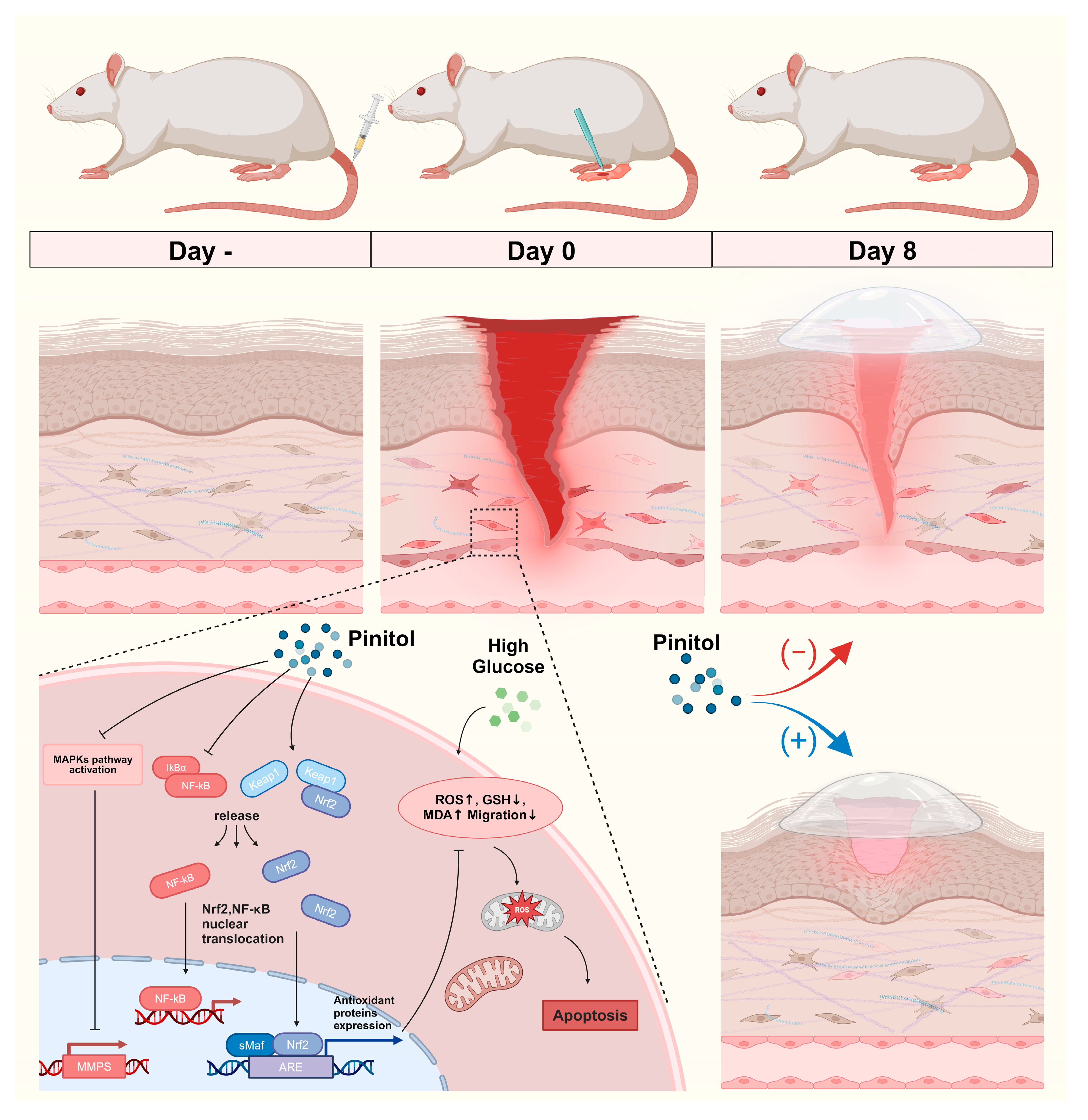
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zimmet, P.Z.; Magliano, D.J.; Herman, W.H.; Shaw, J.E. Diabetes: A 21st century challenge. Lancet Diabetes Endocrinol. 2014, 2, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, A.; Singh, S.K.; Kumar, B.; Gulati, M.; Kumar, R.; Wadhwa, S.; Khursheed, R.; Corrie, L.; Kr, A.; Kumar, R.; et al. Treatment Strategies Against Diabetic Foot Ulcer: Success so Far and the Road Ahead. Curr. Diabetes Rev. 2021, 17, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Soares, M.; Boyko, E.J.; Jeffcoate, W.; Mills, J.L.; Russell, D.; Morbach, S.; Game, F. Diabetic foot ulcer classifications: A critical review. Diabetes Metab. Res. Rev. 2020, 36 (Suppl. S1), e3272. [Google Scholar] [CrossRef]
- Boulton, A.J.M.; Vileikyte, L.; Ragnarson-Tennvall, G.; Apelqvist, J. The global burden of diabetic foot disease. Lancet 2005, 366, 1719–1724. [Google Scholar] [CrossRef]
- Malone, M.; Schultz, G. Challenges in the diagnosis and management of wound infection. Br. J. Dermatol. 2022, 187, 159–166. [Google Scholar] [CrossRef]
- Mohsin, F.; Javaid, S.; Tariq, M.; Mustafa, M. Molecular immunological mechanisms of impaired wound healing in diabetic foot ulcers (DFU), current therapeutic strategies and future directions. Int. Immunopharmacol. 2024, 139, 112713. [Google Scholar] [CrossRef]
- Acosta, J.B.; del Barco, D.G.; Vera, D.C.; Savigne, W.; Lopez-Saura, P.; Guillen Nieto, G.; Schultz, G.S. The pro-inflammatory environment in recalcitrant diabetic foot wounds. Int. Wound J. 2008, 5, 530–539. [Google Scholar] [CrossRef]
- Mendoza-Mari, Y.; Garcia-Ojalvo, A.; Fernandez-Mayola, M.; Rodriguez-Rodriguez, N.; Martinez-Jimenez, I.; Berlanga-Acosta, J. Epidermal growth factor effect on lipopolysaccharide-induced inflammation in fibroblasts derived from diabetic foot ulcer. Scars Burn. Heal. 2022, 8, 20595131211067380. [Google Scholar] [CrossRef]
- Hiebert, P.; Werner, S. Regulation of Wound Healing by the NRF2 Transcription Factor-More Than Cytoprotection. Int. J. Mol. Sci. 2019, 20, 3856. [Google Scholar] [CrossRef]
- Ambrozova, N.; Ulrichova, J.; Galandakova, A. Models for the study of skin wound healing. The role of Nrf2 and NF-kappaB. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 2017, 161, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Yagishita, Y.; Yamamoto, M. The Keap1-Nrf2 system and diabetes mellitus. Arch. Biochem. Biophys. 2015, 566, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Azab, A. D-Pinitol-Active Natural Product from Carob with Notable Insulin Regulation. Nutrients 2022, 14, 1453. [Google Scholar] [CrossRef]
- Pandi, A.; Sattu, K.; Kalappan, V.M.; Lal, V.; Varikasuvu, S.R.; Ganguly, A.; Prasad, J. Pharmacological effects of D-Pinitol—A comprehensive review. J. Food Biochem. 2022, 46, e14282. [Google Scholar] [CrossRef]
- Antonowski, T.; Osowski, A.; Lahuta, L.; Gorecki, R.; Rynkiewicz, A.; Wojtkiewicz, J. Health-Promoting Properties of Selected Cyclitols for Metabolic Syndrome and Diabetes. Nutrients 2019, 11, 2314. [Google Scholar] [CrossRef]
- Jung, J.; Shim, J.H.; Cho, S.H.; Bae, I.; Yang, S.H.; Kim, J.; Lim, H.W.; Shin, D.W. The Anti-Diabetic Pinitol Improves Damaged Fibroblasts. Biomol. Ther. 2024, 32, 224–230. [Google Scholar] [CrossRef]
- Ghasemi, A.; Jeddi, S. Streptozotocin as a tool for induction of rat models of diabetes: A practical guide. EXCLI J. 2023, 22, 274–294. [Google Scholar] [CrossRef]
- Hostalek, U.; Gwilt, M.; Hildemann, S. Therapeutic Use of Metformin in Prediabetes and Diabetes Prevention. Drugs 2015, 75, 1071–1094. [Google Scholar] [CrossRef]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Roohi, T.F.; Faizan, S.; Parray, Z.A.; Baig, M.D.A.I.; Mehdi, S.; Kinattingal, N.; Krishna, K.L. Beyond Glucose: The Dual Assault of Oxidative and ER Stress in Diabetic Disorders. High Blood Press. Cardiovasc. Prev. 2023, 30, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Xiao, C.; Feng, S.; Bai, J. Artemisitene Alters LPS-Induced Oxidative stress, inflammation and Ferroptosis in Liver Through Nrf2/HO-1 and NF-kB Pathway. Front. Pharmacol. 2023, 14, 1177542. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.L.; Murphy, J.T. Reactive oxygen species mediate endotoxin-induced human dermal endothelial NF-kappaB activation. J. Surg. Res. 2003, 111, 120–126. [Google Scholar] [CrossRef]
- Simon, F.; Fernandez, R. Early lipopolysaccharide-induced reactive oxygen species production evokes necrotic cell death in human umbilical vein endothelial cells. J. Hypertens. 2009, 27, 1202–1216. [Google Scholar] [CrossRef]
- Jiang, G.; Jiang, T.; Chen, J.; Yao, H.; Mao, R.; Yang, X.; Chen, Z.; Li, W. Mitochondrial dysfunction and oxidative stress in diabetic wound. J. Biochem. Mol. Toxicol. 2023, 37, e23407. [Google Scholar] [CrossRef]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between mitochondrial membrane potential and ROS formation. Methods Mol. Biol. 2012, 810, 183–205. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Madhavi, Y.V.; Gaikwad, N.; Yerra, V.G.; Kalvala, A.K.; Nanduri, S.; Kumar, A. Targeting AMPK in Diabetes and Diabetic Complications: Energy Homeostasis, Autophagy and Mitochondrial Health. Curr. Med. Chem. 2019, 26, 5207–5229. [Google Scholar] [CrossRef]
- Ren, H.; Shao, Y.; Wu, C.; Ma, X.; Lv, C.; Wang, Q. Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol. Cell. Endocrinol. 2020, 500, 110628. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Guo, H.; Song, A.; Huang, J.; Zhang, Y.; Jin, S.; Li, S.; Zhang, L.; Yang, C.; Yang, P. Progranulin inhibits LPS-induced macrophage M1 polarization via NF-small ka, CyrillicB and MAPK pathways. BMC Immunol. 2020, 21, 32. [Google Scholar] [CrossRef]
- Dong, N.; Li, X.; Xue, C.; Zhang, L.; Wang, C.; Xu, X.; Shan, A. Astragalus polysaccharides alleviates LPS-induced inflammation via the NF-kappaB/MAPK signaling pathway. J. Cell. Physiol. 2020, 235, 5525–5540. [Google Scholar] [CrossRef]
- Fan, C.; Zhang, X.; Zhang, P.; Zhao, J.; Shen, H.; Zhang, Y.; Wu, X.; Jia, Z.; Wang, Y. LPS stimulation during HCV infection induces MMP/TIMP1 imbalance in macrophages. J. Med. Microbiol. 2020, 69, 759–766. [Google Scholar] [CrossRef]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef]
- Galeano, M.; Bitto, A.; Altavilla, D.; Minutoli, L.; Polito, F.; Calo, M.; Lo Cascio, P.; Stagno d’Alcontres, F.; Squadrito, F. Polydeoxyribonucleotide stimulates angiogenesis and wound healing in the genetically diabetic mouse. Wound Repair Regen. 2008, 16, 208–217. [Google Scholar] [CrossRef]
- Tan, T.; Shih, C.; Concha-Moore, K.C.; Diri, M.M.; Hu, B.; Marrero, D.; Zhou, W.; Armstrong, D.G. Disparities in outcomes of patients admitted with diabetic foot infections. PLoS ONE 2019, 14, e0211481. [Google Scholar] [CrossRef]
- Lontchi-Yimagou, E.; Sobngwi, E.; Matsha, T.E.; Kengne, A.P. Diabetes mellitus and inflammation. Curr. Diabetes Rep. 2013, 13, 435–444. [Google Scholar] [CrossRef]
- Tang, G.; Li, S.; Zhang, C.; Chen, H.; Wang, N.; Feng, Y. Clinical efficacies, underlying mechanisms and molecular targets of Chinese medicines for diabetic nephropathy treatment and management. Acta Pharm. Sin. B 2021, 11, 2749–2767. [Google Scholar] [CrossRef]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Veith, A.P.; Henderson, K.; Spencer, A.; Sligar, A.D.; Baker, A.B. Therapeutic strategies for enhancing angiogenesis in wound healing. Adv. Drug Deliv. Rev. 2019, 146, 97–125. [Google Scholar] [CrossRef] [PubMed]
- Okonkwo, U.A.; DiPietro, L.A. Diabetes and Wound Angiogenesis. Int. J. Mol. Sci. 2017, 18, 1419. [Google Scholar] [CrossRef]
- Olgun, M.E.; Altuntas, S.C.; Sert, M.; Tetiker, T. Anemia in Patients with Diabetic Foot Ulcer: Effects on Diabetic Microvascular Complications and Related Conditions. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 985–990. [Google Scholar] [CrossRef]
- McDermott, K.; Fang, M.; Boulton, A.J.M.; Selvin, E.; Hicks, C.W. Etiology, Epidemiology, and Disparities in the Burden of Diabetic Foot Ulcers. Diabetes Care 2023, 46, 209–221. [Google Scholar] [CrossRef]
- Burgess, J.L.; Wyant, W.A.; Abdo Abujamra, B.; Kirsner, R.S.; Jozic, I. Diabetic Wound-Healing Science. Medicina 2021, 57, 1072. [Google Scholar] [CrossRef]
- Everett, E.; Mathioudakis, N. Update on management of diabetic foot ulcers. Ann. N. Y. Acad. Sci. 2018, 1411, 153–165. [Google Scholar] [CrossRef]
- Suntar, I.; Cetinkaya, S.; Panieri, E.; Saha, S.; Buttari, B.; Profumo, E.; Saso, L. Regulatory Role of Nrf2 Signaling Pathway in Wound Healing Process. Molecules 2021, 26, 2424. [Google Scholar] [CrossRef]
- Victor, P.; Sarada, D.; Ramkumar, K.M. Pharmacological activation of Nrf2 promotes wound healing. Eur. J. Pharmacol. 2020, 886, 173395. [Google Scholar] [CrossRef]
- Xu, X.; Sun, J.; Chang, X.; Wang, J.; Luo, M.; Wintergerst, K.A.; Miao, L.; Cai, L. Genetic variants of nuclear factor erythroid-derived 2-like 2 associated with the complications in Han descents with type 2 diabetes mellitus of Northeast China. J. Cell. Mol. Med. 2016, 20, 2078–2088. [Google Scholar] [CrossRef]
- Jang, J.; Wang, Y.; Kim, H.; Lalli, M.A.; Kosik, K.S. Nrf2, a regulator of the proteasome, controls self-renewal and pluripotency in human embryonic stem cells. Stem Cells 2014, 32, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Go, M.Y.; Jeon, C.Y.; Shin, J.U.; Kim, M.; Lim, H.W.; Shin, D.W. Pinitol Improves Diabetic Foot Ulcers in Streptozotocin-Induced Diabetes Rats Through Upregulation of Nrf2/HO-1 Signaling. Antioxidants 2025, 14, 15. https://doi.org/10.3390/antiox14010015
Kim J, Go MY, Jeon CY, Shin JU, Kim M, Lim HW, Shin DW. Pinitol Improves Diabetic Foot Ulcers in Streptozotocin-Induced Diabetes Rats Through Upregulation of Nrf2/HO-1 Signaling. Antioxidants. 2025; 14(1):15. https://doi.org/10.3390/antiox14010015
Chicago/Turabian StyleKim, Jinsick, Min Young Go, Chae Young Jeon, Jung Un Shin, Mujun Kim, Hye Won Lim, and Dong Wook Shin. 2025. "Pinitol Improves Diabetic Foot Ulcers in Streptozotocin-Induced Diabetes Rats Through Upregulation of Nrf2/HO-1 Signaling" Antioxidants 14, no. 1: 15. https://doi.org/10.3390/antiox14010015
APA StyleKim, J., Go, M. Y., Jeon, C. Y., Shin, J. U., Kim, M., Lim, H. W., & Shin, D. W. (2025). Pinitol Improves Diabetic Foot Ulcers in Streptozotocin-Induced Diabetes Rats Through Upregulation of Nrf2/HO-1 Signaling. Antioxidants, 14(1), 15. https://doi.org/10.3390/antiox14010015