


Is Inducible Nitric Oxide Synthase (iNOS) Promising as a New Target Against Pulmonary Hypertension?


Abstract
:1. Introduction
2. Nitric Oxide and Its Role in Cardiopulmonary System
2.1. Nitric Oxide
2.2. The Role of NOS in the Cardiopulmonary System
2.3. iNOS—Protective or Harmful?
3. Pulmonary Hypertension (PH)—Short Characteristics of the Main Clinical Groups
3.1. Pulmonary Arterial Hypertension
3.2. Pulmonary Hypertension Due to Left Heart Disease
3.3. Pulmonary Hypertension Associated with Lung Diseases and/or Hypoxia
3.4. Pulmonary Hypertension Associated with Pulmonary Artery Obstructions
4. Study Selection Criteria
5. iNOS Expression Patterns in Preclinical Models of PH
5.1. A Brief Overlook on Animal Models
5.2. Changes in iNOS Expression in Preclinical Studies

{kind=link}
{kind=link}
{kind=link}
| Disease Entity | Model | Species | Tissue | Changes in iNOS mRNA/Protein Expression | References |
|---|---|---|---|---|---|
| Models of disease entities within group 1 PH | |||||
| PAH | MCT-induced PH | rat | lungs | ↑ | [90,91,92,93,94,136,137] |
| ↔ | [129] | ||||
| alveoli | ↑ | [102] | |||
| PA | ↑ | [102] | |||
| RV (myocardium) | ↑ | [109,112,135] | |||
| RV (fibroblasts) | ↑ | [111] | |||
| Hx-induced PH | rat | lungs | ↑ | [95,96,97,114,115,139,145] | |
| lung macrophages | ↑ | [104] | |||
| alveoli | ↑ | [101] | |||
| PA (endothelium, smooth muscle cells) | ↑ | [100,101,104,106,116,146] | |||
| hearts (whole) | ↑ | [108] | |||
| RV myocardium | ↑ | [110] | |||
| LV myocardium | ↑ | [110] | |||
| mouse | lungs | ↑ | [98] | ||
| Sugen-Hx-induced PH | rat | pulmonary vessels | ↑ | [105] | |
| RV myocardium | ↑ | [105] | |||
| Ren2 rat model | rat | lungs | ↔ | [130] | |
| portal PH | portal vein ligation | rat | lungs | ↑ | [143] |
| PPHN | Hx-induced PH | rat ♀ (pregnant) | lungs | ↔ | [132] |
| pig (newborn) | lungs | ↔ | [131] | ||
| PA | ↔ (membrane fraction) | [134] | |||
| ↓ (cytosolic fraction) | |||||
| guinea pig ♀ (pregnant) | LV | ↑ | [113] | ||
| Models of disease entities withingroup 2 PH | |||||
| HFpEF | TAC | mouse | lungs | ↑ | [89] |
| RV myocardium | ↑ | [144,147] | |||
| metabolic (obesity)-HF | rat | lungs | ↔ | [133] | |
| PA | ↑ | [107] | |||
| Models of disease entities within group 3 PH | |||||
| COPD | elastase-induced lung emphysema | mouse | lungs (macrophages, alveolar wall, alveolar epithelium) | ↑ | [103,141] |
| cigarette smoke-induced lung injury | rat | trachea, intraparenchymal airways | ↑ | [99] | |
| PA | ↑ | [99] | |||
| aorta | ↑ | [120] | |||
| mouse | lungs | ↑ | [119,140,148] | ||
| bronchi | ↑ | [119,148] | |||
| guinea pig | lungs | ↑ | [149] | ||
| IPF | bleomycin-induced pulmonary fibrosis | rat | lungs | ↑ | [121,122] |
| PA | ↑ | [93] | |||
| mouse | lungs | ↑ | [123,124,125,126,150] | ||
| Models of disease entities within group 4 PH | |||||
| chronic thromboembolic PH | administration of embolizing particles | broiler chicken | lungs | ↑ | [127] |
| rabbit | pulmonary vessels, alveoli | ↑ | [128] | ||
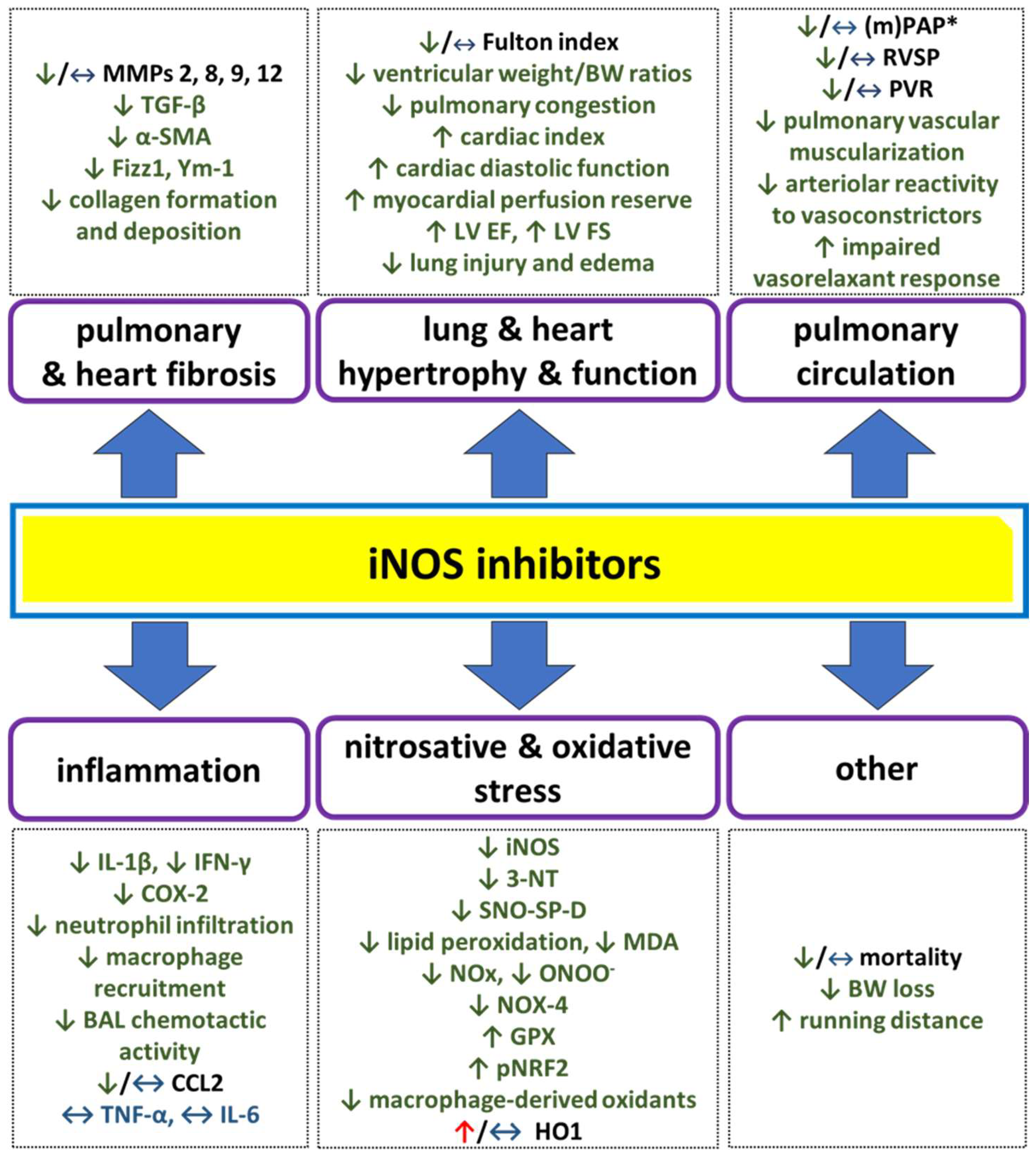
6. Pharmacological iNOS Inhibition—Promising or Discouraging Way to Treat PH?
| Disease Entity | Model(s) | Species | Selective and Non-Selective iNOS Inhibitor(s) | Acute/Chronic/ In Vitro | Preventive (P)/Curative (C) Treatment | Effects | References |
|---|---|---|---|---|---|---|---|
| Models of disease entities within group 1 PH | |||||||
| PAH | MCT- and Hx-induced PH | rat | L-NAME | in vitro | - | isolated lungs: MCT: ↑ basal PAP; ↔ ampl. of HPV Hx: ↔ basal PAP; ↔ ampl. of HPV | [151] |
| isolated PA (MCT and Hx): slight ↑ basal tension slight ↑ of Phe-induced constriction | [152] | ||||||
| Hx-induced PH | mouse | L-NAME | in vitro | - | isolated PA: (-) Ach-induced relaxation ↔ PGF2α-induced contraction | [153] | |
| rat | L-NIL L-NNA | in vitro | - | isolated lungs: ↔ (L-NIL, L-NNA) basal PVR, arterial and venous resistance ↔ (L-NIL)/↑ (L-NNA) of arterial and (weak) venous constrictor responses to TXA2 analogue | [115] | ||
| PPHN | Hx-induced PH | pig | L-NAME AG | in vitro | - | isolated PA: ↓ (L-NAME)/↔ (AG) PA diameter | [154] |
| rat ♀ | L-NNA | in vitro | - | isolated lungs (from Hx neonates): ↑ PVR, ↑ reactivity to TXA2 analogue ↔ pulmonary venous tone | [132] | ||
| PAH | Sugen-Hx-induced PH | rat | L-canavanine | acute | - | 8 weeks after Sugen-Hx: ↔ RVSP, ↔ LVSP, ↔ PVRI, ↔ SVRI | [155] |
| L-NAME | acute | - | 3, 5, and 8 weeks after Sugen-Hx: ↑ RVSP, ↑ LVSP, ↑ PVRI, ↑ SVRI, ↓ CI | ||||
| Hx-induced PH | rat | S-MIT L-canavanine | acute | - | ↔ PAP, ↔ SAP slight ↓ PAs (mainly muscular) diameter | [146] | |
| L-NAME L-NMMA | acute | - | ↑ PAP, ↑ SAP ↓ PAs (muscular and elastic) diameter | ||||
| ONO-1714 | acute | - | ↑ mPAP (slight and transient) | [145] | |||
| Hx-induced PH | rat | L-NIL, L-NAME: (1) 3 days before + during a 1-week Hx; (2) 3 days before + during 1 week of a 3-week Hx; (3) during the final 10 days of a 3-week Hx | chronic | P (1,2); C (3) | L-NIL: ↔ SAP (1,2,3), ↓ PAP (1,2,3), ↓ exhaled NO (1,3), ↔ RV weight (1,3), ↓ RV weight (2), ↔ FI (1,3), ↓ FI (2) L-NAME: ↑ SAP (1,3), ↓ PAP (1,2) ↑ PAP (3), ↓ exhaled NO (1,3), ↓ RV weight (3) | [100] | |
| rat | ONO-1714 (10 days) | chronic | P | ↔ mPAP, ↔ Hx-induced changes in vascular structure, ↔ FI | [145] | ||
| L-NAME (4 weeks) | chronic | P | ↓ PAP, ↑ SAP ↓ RV/BW, ↓ LV/BW | [110] | |||
| MCT-induced PH | rat | AG (4 weeks) | chronic | P | ↓ RVP restoration of Ach-induced relaxation (in PAs and systemic arteries) | [156] | |
| PPHN | Hx-induced PH | pig ♀ | L-NIL (10 days, 4 days after Hx onset) | chronic | C | in fetal hearts: anti-nitrative: ↓ 3-NT anti-oxidative: ↓ MDA anti-fibrotic: ↓ MMP-9, collagen other: ↓ cGMP levels | [142] |
| Models of disease entities within group 2 PH | |||||||
| HFpEF | metabolic (obesity)-HF | rat | 1400W | in vitro | - | isolated PA: modest ↑ Phe-induced vasoconstriction (only to its highest concentration) | [133] |
| TAC | mouse | 1400W (2 weeks) | chronic | C | ↔ LV systolic pressure, ↔ HR LV hypertrophy and dysfunction: ↓ ventricular weight/BW ratio ↓ LV end-systolic diameter, ↑ LV ejection fraction, ↑ LV fractional shortening ↓ LV fibrosis pulmonary congestion: ↓ lung weight/BW ratio | [147] | |
| high-fat diet + L-NAME | mouse | L-NIL (3 days) | chronic | C | ↔ HR, ↔ SBP, ↔ DBP, ↑ cardiac diastolic function (↓ E/A and E/E’ ratios), ↔ ejection fraction, ↑ running distance, ↔ lung edema (wet weight/dry weight ratio), ↔ heart weight/tibia length ratio oxidative status: ↓ MDA, ↑ GPX, ↓ NOX-4 ↑ pNRF2, ↔ SOD2, ↑ HO1 in hearts | [28] | |
| high-fat high-sucrose diet (HFHSD) | mouse | 1400W (8 weeks) | chronic | C | ↓ cardiovascular oxidative stress ↑ myocardial perfusion reserve ↓ arteriolar reactivity (-) HFHSD-induced ↓ in EF and changes in systolic and diastolic strain | [22] | |
| Models of disease entities within group 3 PH | |||||||
| COPD | cigarette smoke-induced lung injury | sheep ♀ | MEG | acute | - | ↓ PVRI, SVRI, ↑ CI, ↓ lung weight ↔ PAP, MAP | [157] |
| mouse | L-NIL: (1) parallel to smoke exposure (8 months) (2) after 8 months of smoke exposure (3 months) | chronic | (1) P (2) C | ↓ RVSP anti-hypertrophic: ↓ FI anti-emphysematic: ↓ mean linear intercept ↓ air space, ↑ septal wall thickness ↓ alveoli/vessels ratio | [119] | ||
| guinea pig | L-NIL: (1) 7 days before smoke exposure (2) 60 days from the 29th day after smoke exposure | chronic | P | anti-emphysematic: ↓ mean linear intercept ↓ destructive index anti-oxidative: ↓ protein nitration and oxidation (lungs) anti-inflammatory: ↓ leukocyte infiltration, IL-1β, IL-8, TGF-β, IL-4 (BAL) ↓ total NOx (heart, liver, BAL) | [149] | ||
| C | ↔ mean linear intercept, ↔ destructive index | ||||||
| elastase-induced lung emphysema | mouse | L-NIL (12 weeks, 3 weeks after elastase instillation) | chronic | C | ↓ RVSP, ↔ SAP, ↔ FI anti-nitrative (lungs): ↓ 3-NT ↓ iNOS anti-inflammatory: ↓ immune cells (CD45+), ↔ TNF-α, ↔ MMP-8, 9, 12 lung structure and function: ↓ pulmonary vascular muscularization | [141] | |
| 1400W (20 days, 1 day before elastase instillation) | chronic | P | anti-nitrative: ↓ 3-NT (lungs) pro-oxidative: ↑ protein carbonyls other: ↔ mean chord length of alveoli ↔ HO1, MMPs, CCL-2, CXCL2, TNF-α, and IL-6 (lungs) ↔ inflammatory cell counts, CCL-2, MMP-2, MMP-9 protein (BAL) ↔ alveolar cell proliferation | [103] | |||
| COPD/IPF | SP-D deficiency-related emphysema | mouse | 1400W (7 weeks from 3 weeks of age) | chronic | C | anti-oxidative: ↓ % of oxidants-producing macrophages anti-inflammatory (time-dependent): ↓ cellular infiltration, ↓ total BAL cell count, ↓ IFN-γ in BAL, ↓ macrophage recruitment anti-fibrotic: ↓ MMP-2, MMP-9 | [150] |
| IPF | bleomycin (BLM)-induced lung injury | mouse | 1400W (6 days before BLM instillation) | chronic | P | anti-nitrative: ↓ SNO-SP-D (BAL) anti-inflammatory: ↓ BAL chemotactic activity ↓ IL-1β, COX-2, CCL2 anti-fibrotic: ↓ Fizz1, TGF-β, Ym-1 | [158] |
| GW274150 (14 days, 1 day after BLM instillation) | chronic | P | anti-oxidative (lungs): ↓ lipid peroxidation anti-inflammatory (lungs): ↓ neutrophils infiltration anti-fibrotic (lungs): ↓ collagen formation and deposition ↓ TGF-β expression other: ↓ lung injury, ↓ edema formation, ↓ mortality rate; ↓ BW loss | [159] | |||
| rat | AG (13 days, 1 day after BLM instillation) | chronic | anti-oxidative: ↓ MDA (pulmonary blood) * anti-nitrative: ↓ NOx (plasma) ‡ ↓ ONOO- formation †,‡ anti-fibrotic: ↓ α-SMA and myofibroblast number ‡ ↓ type I ‡ and III † collagen lung deposition | [160] *, [161] †, [162] ‡ | |||
| Models of disease entities within group 4 PH | |||||||
| CTEPH | application of embolizing particles | chicken | AG | acute | - | ↔ PAP, ↔ PVR, ↔ mortality | [163] |
| L-NAME | acute | - | ↑ PAP, ↑ PVR, ↑ mortality | ||||
| dog ♀ | AG | acute | - | ↓ mPAP, ↓ PVRI | [164] | ||
| S-MIT | acute | - | S-MIT: ↔ mPAP, ↔ PVRI (-) embolization-induced ↑ NOx, MDA, TBARS (plasma) | [165] | |||
| S-MIT with sildenafil: ↓ mPAP, ↓ PVRI, but effect of sildenafil ↔ | [164] | ||||||
| L-NAME | acute | - | ↑ mPAP, ↑ PVRI, ↑ mortality | [165] | |||
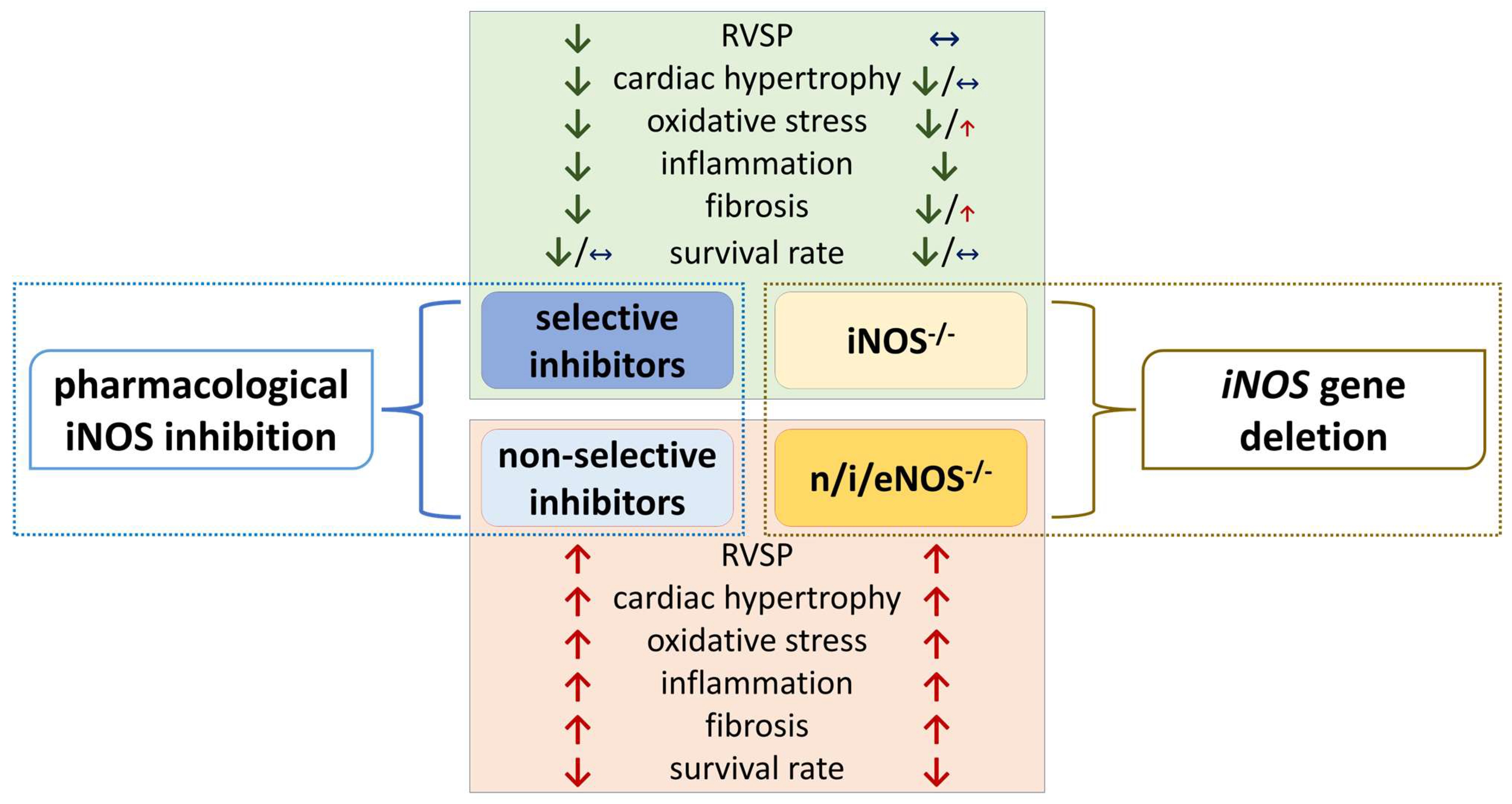
7. Is iNOS Gene Deletion Protective in PH? Insights from Knock-Out Studies
| Disease Entity | Model | Effects | References | |||
|---|---|---|---|---|---|---|
| Disease models within group 1 PH | ||||||
| PAH | Hx-induced PH | iNOS−/− vs. WT: | eNOS−/− vs. WT: | triple n/i/eNOS−/− vs. WT: | [138] | |
| ↔ RVSP; ↔ FI ↔ PA medial thickness ↔ survival rate | moderate ↑ RVSP; ↑ FI ↑ PA medial thickness ↓ survival rate | highest↑ RVSP; ↑ FI↑ PA medial thickness↓ survival rate | ||||
| myeloid-cell-specific iNOS−/− vs. WT: ↔ RVSP, ↔ FI, ↔ TAPSE, ↔ pulmonary vascular remodeling (small vessels) | [68] | |||||
| Disease models within group 2 PH | ||||||
| HFpEF | TAC | iNOS–/–vs. WT: anti-oxidative: ↓ 4-HNE, ↓ 3-NT, ↓ PRMT1, ↓ DDAH1 in LV myocardium anti-hypertrophic: ↓ ventricular weight/BW ratio, ↓ myocyte cross-sectional area, ↓ myocyte diameter, ↓ MMP-2 and collagen-1 in LV myocardium ↓ cardiac dysfunction: ↑ LV ejection fraction, ↑ LV fractional shortening, ↑ LV diastolic wall thickness, ↑ LV diameter in end systole and diastole, ↓ cardiac ANP and BNP levels ↓ pulmonary congestion: ↓ lung weight/BW ratio other effects: ↔ mortality rate | [147] | |||
| iNOS–/–vs. WT: anti-inflammatory: ↓ cardiac IL-1β, IL-6 expression, CD68+ M1 macrophage count ↓ cardiac cytosolic mtDNA levels anti-fibrotic effects: ↓ fibrosis area ↓ cardiac remodeling and hypertrophy: ↓ myocyte cross-sectional area, ↓ heart weight/tibia length ratio ↓ cardiac dysfunction: ↓ cardiac ANP and BNP levels, ↑ LV ejection fraction, ↑ LV fractional shortening | [144] | |||||
| High-fat diet + L-NAME | iNOS–/–vs. WT: (mostly) anti-oxidative: ↓ MDA, ↑ GPX, ↓ NOX-4, ↑ pNRF2, ↔ SOD2, ↑ HO1 in hearts ↓ cardiac dysfunction: ↑ cardiac diastolic function (↓ mitral E/E’ ratio) ↓ pulmonary congestion: ↓ lung edema (wet weight/dry weight ratio) other effects: ↑ running distance, ↔ heart weight/tibia length ratio, ↔ HR, ↔ ejection fraction, ↔ systolic BP, ↔ diastolic BP | [28] | ||||
| HFHSD | iNOS–/–vs. WT: ↓ cardiovascular oxidative stress, ↑ cardiac stress perfusion ↑ vasodilatation to adenosine (in coronary arteries) ↓ HFHSD-induced changes in systolic and diastolic strain, ↔ cardiac rest perfusion, ↔ myocardial perfusion reserve, ↔ arteriolar reactivity | [22] | ||||
| Disease models within group 3 PH | ||||||
| COPD | cigarette smoke-induced lung injury | myeloid-cell-specific iNOS−/−vs. WT: ↓ development of PH, but not emphysema ↓ RV hypertrophy, ↓ pulmonary vascular remodeling | [68] | |||
| iNOS−/−vs. WT: ferroptosis-related proteins: ↓ ACSL4, ↑ GPX4, xCT, FTL, FTH1 | [148] | |||||
| iNOS−/−vs. WT: protection against PH and emphysema | [119] | |||||
| iNOS−/−specific for bone marrow-derived cells *** vs. controls ****: ↓ PH: ↓ RVSP ↔ emphysema: ↔ number of alveoli | iNOS−/−except for bone marrow-derived cells ***** vs. controls ****: ↔ PH: ↔ RVSP ↓ emphysema: ↓ number of alveoli | |||||
| elastase-induced lung emphysema | iNOS−/−vs. WT: anti-nitrative: ↓ 3-NT positive cells pro-oxidative: ↑ protein carbonyls other: ↔ HO1, CCL2, CXCL2, TNF-α, and IL-6 (lungs) ↔ mean chord length of alveoli | [103] | ||||
| AECII-specific iNOS−/−induced by doxycyclin vs. doxycyclin-naïve mice: ↔ RVSP, ↔ RV hypertrophy, ↔ FI, ↔ RVWT RV function: ↔ PAT/PET, ↔ TAPSE, ↔ pulmonary vascular muscularization emphysema development: ↔ lung compliance, ↔ mean linear intercept, ↔ lung airspace | [69] | |||||
| IPF | bleomycin (BLM)-induced lung injury | iNOS−/−vs. WT: anti-fibrotic: ↓ fibrosis score, ↓ TIMP-1, ↓ CCL-2, ↓ hydroxyproline content, ↓ α-SMA (lungs) other: ↔ lung compliance, ↓ mortality | [123,124] | |||
| iNOS−/−vs. WT: anti-nitrative: ↓ SNO-SP-D anti-inflammatory: ↓ chemotactic activity (BAL) ↓ IL-1β, COX-2, CCL-2 pro-fibrotic: ↑ Fizz1, TGF-β, Ym-1 | [158] | |||||
| iNOS−/−vs. WT: anti-oxidative: ↓ lipid peroxidation anti-inflammatory (lungs): ↓ neutrophil infiltration anti-fibrotic (lungs): ↓ collagen formation and deposition; ↓ TGF-β expression other: ↓ mortality rate, (-) loss of body weight, ↓ lung injury, ↓ edema formation | [159] | |||||
| iNOS−/−vs. WT: anti-inflammatory: ↓ TNF-α, CCL-2, lymphocyte count, protein conc. (BAL), ↔ total inflammatory cells (BAL) anti-fibrotic (lungs): ↔ fibrotic area ↓ TGF-β1 ↓ collagen 1 other: ↔ BW, IL-1β, IL-6, IFN-γ, CTGF (BAL) | eNOS−/−vs. WT: inflammatory parameters: ↔ TNF-α, CCL-2, IL-1β, IL-6, IFN-γ, CTGF, protein conc., lymphocyte count, total inflammatory cells (BAL) fibrotic parameters: ↔ lung fibrotic area ↔ TGF-β1 and collagen 1 in lungs other: ↔ BW | triple n/i/eNOS−/−vs. WT: pro-inflammatory: ↑ TNF-α, CCL-2, IL-1β, IL-6, IFN-γ, lymphocyte count and protein conc. (BAL) ↑ total inflammatory cells (BAL) pro-fibrotic: ↑ lung fibrotic area, ↑ TGF-β1 ,↑ collagen 1, ↑ CTGF in lungs other: ↓ BW | [167] | |||
8. iNOS Expression in Patients Affected by Diseases Associated with PH
| Disease Entity | Size of Sample | Number of Control Patients | Development of PH Confirmed? (+/-) | Changes in iNOS Expression (If Any) Versus Respective Control Group; Alteration of eNOS (If Studied) | References |
|---|---|---|---|---|---|
| Group 1 PH | |||||
| PAH associated with congenital heart disease (CHD) | 18 (flow-associated PH) 6 (congestive vasculopathy) 10 (increased pulmonary blood flow but normal PAP) | 4 | + | ↑ in PA ↔ eNOS | [193] |
| 26 (septal defects) | 8 | + | ↑ in PA endothelium ↑ eNOS | [194] | |
| 24 (VSD, including 10 surgically corrected) | - | + | ↔ in lungs * ↓ eNOS | [195] | |
| 7 (TOF); 8 (VSD) | - | TOF: -; VSD: + | detected in RA and RV myocardium | [196] | |
| 19 (CHD) | 10 | - | ↑ in LV myocardium ** | [170] | |
| rapid persistent PH of the newborn | 2 neonates | 3 neonates | + | ↔ in lungs (PA endothelium, PA smooth muscle cells, macrophages, epithelium) | [189] |
| PH associated with congenital diaphragmatic hernia (CDH) | 33 (10 ECMO-treated and 23 not treated by ECMO) | 11 | + | in small PA endothelium: ↔ treated by ECMO, ↓ not treated by ECMO ↔ eNOS | [197] |
| 13 (PH-CDH); 20 (lung hypoplasia due to other causes) | 33 | - | ↔ in lung vasculature | [171] | |
| Group 2 PH | |||||
| PH associated with left heart disease (PH-LHD) | 43 | 15 | + | ↑ in monocytes | [198] |
| 20 | 15 | + | ↑ in PBMC | [199] | |
| 15 (decompensated HF) | 6 | - | ↑ in venous endothelium ↔ eNOS | [172] | |
| 24 (DCM); 17 (IHD); 10 (VHD) | 11 | - | ↑ in heart | [173] | |
| 9 (HF—transplant group); 10 (LVAD); 11 (post-LVAD transplantation) | 7 | - | ↑ in heart (HF-transplant and LVAD groups) | [174] | |
| 28 | 4 | - | ↑ in heart ↑ in macrophages ↑ eNOS in cardiomyocytes and subendocardial areas | [175] | |
| 18 (DCM); 7 (ischemic cardiopathy and severe ventricular dysfunction); 4 (AMI) | 11 | - | ↑ in myocardium | [176] | |
| 8 (DCM); 14 (IHD) | - | - | ↑ in myocardium ↑ in endothelium, vascular smooth muscle cells | [177] | |
| 14 (DCM); 9 (ICM); 7 (PCM) | 5 | - | ↔ in myocardium ↑ eNOS | [178] | |
| 24 (end-stage HF) | 5 | - | ↑ in LV ↓ eNOS | [179] | |
| 10 (HF due to CAD) | 10 | - | ↑ in RA ↓ eNOS | [180] | |
| 19 | 20 | - | ↑ in macrophages | [181] | |
| 25 (acute congestive HF) | ? | - | ↑ in plasma | [182] | |
| 10 | ? | - | ↑ in plasma | [183] | |
| 40 | 20 | - | ↑ in plasma | [184] | |
| 42 (HFpEF) 38 (HFrEF) | - | - | ↑ in serum (HFpEF) ↑ eNOS (HFrEF) | [185] | |
| 23 (LVAD implantation) 36 (elective heart transplantation) | - | - | detected in heart and blood vessels | [186] | |
| 7 (end-stage HF) | - | - | detected in LV myocardium | [187] | |
| Group 3 PH | |||||
| PH associated with chronic obstructive pulmonary disease (COPD) | 11 (severe COPD) | 13 *** | - | ↑ in lungs (alveolar wall, alveolar macrophages, bronchial wall, adventitia of PAs, smooth muscle cells) | [188] |
| 10 | 10 | - | ↑ in lungs | [148] | |
| 10 | 10 | - | ↑ in pulmonary macrophages | [68] | |
| 10 | 11 | - | ↑ in airway inflammatory cells | [190] | |
| 13 (severe COPD); 14 (mild/moderate COPD) | 13 smokers, 11 non-smokers | - | ↑ in bronchial submucosa and bronchoalveolar lavage (smokers) | [191] | |
| 7 (normal BMI); 7 (low BMI) | - | - | ↑ in skeletal muscles (low BMI) | [192] | |
| PH associated with idiopathic pulmonary fibrosis (IPF) | 17 | 21 | + | ↑ in PAs ↓ eNOS | [200] |
| 17 | 10 | - | ↑ in lungs | [123] | |
| 12 | 6 | - | ↑ in lungs (fibrotic scars, thickened septa, fibroblast foci) | [124] | |
| 48 | 21 | + | ↑ in lungs (macrophages, neutrophiles, alveolar epithelium, PA endothelium, PA smooth muscle cells) ↓ eNOS | [201] | |
9. Limitations and Perspectives
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 1400W | N-(3-(aminomethyl)benzyl)acetamidine |
| AG | aminoguanidine |
| BMPR2 | bone morphogenetic protein receptor 2 |
| cGMP | cyclic guanosine-3′,5′-monophosphate |
| CHD | congenital heart disease |
| COPD | chronic obstructive pulmonary disease |
| COX-2 | cyclooxygenase-2 |
| CTEPH | chronic thromboembolic pulmonary hypertension |
| ECMO | extracorporeal membrane oxygenation |
| EMA | European Medicines Agency |
| eNOS (NOS3) | endothelial nitric oxide synthase |
| FDA | Food and Drug Administration |
| FI | Fulton index |
| HF | heart failure |
| HFpEF | heart failure with preserved ejection fraction |
| HIF-1α | hypoxia-inducible factor 1 alpha |
| HO1 | heme oxygenase 1 |
| HPV | hypoxic pulmonary vasoconstriction |
| Hx | chronic hypoxia |
| IFN-γ | interferon gamma |
| IL-1β | interleukin-1 beta |
| iNANC | inducible non-adrenergic-non-cholinergic autonomic system |
| iNOS (NOS2) | inducible nitric oxide synthase |
| IPF | idiopathic pulmonary fibrosis |
| L-NAME | Nω-nitro-L-arginine methyl ester |
| L-NIL | L-Nω-(1-iminoethyl)lysine |
| L-NMMA | Nω-monomethyl-L-arginine |
| L-NNA | Nω–nitro-L-arginine |
| LPS | lipopolysaccharide |
| LV | left ventricle |
| MCT | monocrotaline |
| MEG | mercaptoethylguanidine |
| (m)PAP | (mean) pulmonary artery pressure |
| nNOS (NOS1) | neuronal nitric oxide synthase |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| NYHA | New York Heart Association |
| PA(s) | pulmonary artery(-ies) |
| P(A)H | pulmonary (arterial) hypertension |
| PH-LHD | pulmonary hypertension due to left heart disease |
| PVR(I) | pulmonary vascular resistance (index) |
| qPCR | quantitative polymerase chain reaction |
| RT-PCR | reverse transcription polymerase chain reaction |
| RV(S)P | right ventricular (systolic) pressure |
| RV | right ventricle |
| sGC | soluble guanylate cyclase |
| S-MIT | S-methylisothiourea |
| TAC | transverse aortic constriction |
| TGF-β | transforming growth factor beta |
| TNF-α | tumor necrosis factor alpha |
| TOF | tetralogy of Fallot |
| VEGFR | vascular endothelial growth factor receptor |
| VSD | ventricular septal defect |
| WT | wild type |
References
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A. Revised definition of pulmonary hypertension and approach to management: A clinical primer. J. Am. Heart Assoc. 2023, 12, e029024. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Guignabert, C.; Savale, L.; Boucly, A.; Gallant-Dewavrin, M.; McLaughlin, V.; Hoeper, M.M.; Weatherald, J. Treatment of pulmonary arterial hypertension: Recent progress and a look to the future. Lancet Respir. Med. 2023, 11, 804–819. [Google Scholar] [CrossRef]
- Shah, A.J.; Beckmann, T.; Vorla, M.; Kalra, D.K. New drugs and therapies in pulmonary arterial hypertension. Int. J. Mol. Sci. 2023, 24, 5850. [Google Scholar] [CrossRef]
- Zhao, H.; Song, J.; Li, X.; Xia, Z.; Wang, Q.; Fu, J.; Miao, Y.; Wang, D.; Wang, X. The role of immune cells and inflammation in pulmonary hypertension: Mechanisms and implications. Front. Immunol. 2024, 15, 1374506. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Antosova, M.; Mokra, D.; Pepucha, L.; Plevkova, J.; Buday, T.; Sterusky, M.; Bencova, A. Physiology of nitric oxide in the respiratory system. Physiol. Res. 2017, 66 (Suppl. S2), S159–S172. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Minhas, R.; Bansal, Y.; Bansal, G. Inducible nitric oxide synthase inhibitors: A comprehensive update. Med. Res. Rev. 2020, 40, 823–855. [Google Scholar] [CrossRef]
- Anavi, S.; Tirosh, O. iNOS as a metabolic enzyme under stress conditions. Free Radic. Biol. Med. 2020, 146, 16–35. [Google Scholar] [CrossRef] [PubMed]
- Tabima, D.M.; Frizzell, S.; Gladwin, M.T. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic. Biol. Med. 2012, 52, 1970–1986. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.; Hayes, A.; Caprnda, M.; Petrovic, D.; Rodrigo, L.; Kruzliak, P.; Zulli, A. Inducible nitric oxide synthase: Good or bad? Biomed. Pharmacother. 2017, 93, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Jaitovich, A.; Jourd’heuil, D. A brief overview of nitric oxide and reactive oxygen species signaling in hypoxia-induced pulmonary hypertension. Adv. Exp. Med. Biol. 2017, 967, 71–81. [Google Scholar] [CrossRef]
- Chakravortty, D.; Hensel, M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infect. 2003, 5, 621–627. [Google Scholar] [CrossRef]
- Shah, A.J.; Vorla, M.; Kalra, D.K. Molecular pathways in pulmonary arterial hypertension. Int. J. Mol. Sci. 2022, 23, 10001. [Google Scholar] [CrossRef]
- Pechkovsky, D.V.; Zissel, G.; Stamme, C.; Goldmann, T.; Ari Jaffe, H.; Einhaus, M.; Taube, C.; Magnussen, H.; Schlaak, M.; Müller-Quernheim, J. Human alveolar epithelial cells induce nitric oxide synthase-2 expression in alveolar macrophages. Eur. Respir. J. 2002, 19, 672–683. [Google Scholar] [CrossRef]
- Grasemann, H.; Storm van’s Gravesande, K.; Buscher, R.; Knauer, N.; Silverman, E.S.; Palmer, L.J.; Drazen, J.M.; Ratjen, F. Endothelial nitric oxide synthase variants in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2003, 167, 390–394. [Google Scholar] [CrossRef]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Bui, I.; Baritaki, S.; Libra, M.; Zaravinos, A.; Bonavida, B. Cancer resistance is mediated by the upregulation of several anti-apoptotic gene products via the inducible nitric oxide synthase/nitric oxide pathway: Therapeutic implications. Antioxid. Redox Signal. 2023, 39, 853–889. [Google Scholar] [CrossRef]
- Kruglyakov, D.; Ojha, S.K.; Kartawy, M.; Tripathi, M.K.; Hamoudi, W.; Bazbaz, W.; Khaliulin, I.; Amal, H. Nitric oxide synthase inhibition prevents cell proliferation in glioblastoma. J. Mol. Neurosci. 2023, 73, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Reagan, C.E.; Bresticker, J.E.; Wolpe, A.G.; Good, M.E.; Macal, E.H.; Billcheck, H.O.; Bradley, L.A.; French, B.A.; Isakson, B.E.; et al. Obesity-induced coronary microvascular disease is prevented by iNOS deletion and reversed by iNOS inhibition. JACC Basic Transl. Sci. 2023, 8, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Zanotto, T.M.; Quaresma, P.G.F.; Guadagnini, D.; Weissmann, L.; Santos, A.C.; Vecina, J.F.; Calisto, K.; Santos, A.; Prada, P.O.; Saad, M.J.A. Blocking iNOS and endoplasmic reticulum stress synergistically improves insulin resistance in mice. Mol. Metab. 2016, 6, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Soskić, S.S.; Dobutović, B.D.; Sudar, E.M.; Obradović, M.M.; Nikolić, D.M.; Djordjevic, J.D.; Radak, D.J.; Mikhailidis, D.P.; Isenović, E.R. Regulation of inducible nitric oxide synthase (iNOS) and its potential role in insulin resistance, diabetes and heart failure. Open Cardiovasc. Med. J. 2011, 5, 153–163. [Google Scholar] [CrossRef]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef]
- Wilmes, V.; Scheiper, S.; Roehr, W.; Niess, C.; Kippenberger, S.; Steinhorst, K.; Verhoff, M.A.; Kauferstein, S. Increased inducible nitric oxide synthase (iNOS) expression in human myocardial infarction. Int. J. Leg. Med. 2020, 134, 575–581. [Google Scholar] [CrossRef]
- Golden, T.N.; Venosa, A.; Gow, A.J. Cell origin and iNOS function are critical to macrophage activation following acute lung injury. Front. Pharmacol. 2022, 12, 761496. [Google Scholar] [CrossRef]
- Guo, Y.; Wen, J.; He, A.; Qu, C.; Peng, Y.; Luo, S.; Wang, X. iNOS contributes to heart failure with preserved ejection fraction through mitochondrial dysfunction and Akt S-nitrosylation. J. Adv. Res. 2023, 43, 175–186. [Google Scholar] [CrossRef]
- Prado, C.M.; Yano, L.; Rocha, G.; Starling, C.M.; Capelozzi, V.L.; Leick-Maldonado, E.A.; Martins, M.d.A.; Tibério, I.F. Effects of inducible nitric oxide synthase inhibition in bronchial vascular remodeling-induced by chronic allergic pulmonary inflammation. Exp. Lung Res. 2011, 37, 259–268. [Google Scholar] [CrossRef]
- Prado, C.M.; Righetti, R.F.; Lopes, F.D.T.Q.D.S.; Leick, E.A.; Arantes-Costa, F.M.; de Almeida, F.M.; Saldiva, P.H.N.; Mauad, T.; Tibério, I.F.L.C.; Martins, M.A. iNOS inhibition reduces lung mechanical alterations and remodeling induced by particulate matter in mice. Pulm. Med. 2019, 2019, 4781528. [Google Scholar] [CrossRef]
- Yu, X.; Ge, L.; Niu, L.; Lian, X.; Ma, H.; Pang, L. The dual role of inducible nitric oxide synthase in myocardial ischemia/reperfusion injury: Friend or foe? Oxid. Med. Cell. Longev. 2018, 2018, 8364848. [Google Scholar] [CrossRef]
- Manoury, B.; Montiel, V.; Balligand, J.L. Nitric oxide synthase in post-ischaemic remodelling: New pathways and mechanisms. Cardiovasc. Res. 2012, 94, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Sparacino-Watkins, C.E.; Lai, Y.C.; Gladwin, M.T. Nitrate-nitrite-nitric oxide pathway in pulmonary arterial hypertension therapeutics. Circulation. 2012, 125, 2824–2826. [Google Scholar] [CrossRef] [PubMed]
- Karyofyllis, P.; Demerouti, E.; Habibis, P.; Apostolopoulou, S.; Tsetika, E.G.; Tsiapras, D. Should we change the target of therapy in pulmonary hypertension? Life 2023, 13, 1202. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, C.; Yan, Q.; Wang, S.; Tan, Y.; Long, J.; Lin, Y.; Ning, S.; Wang, J.; Zhang, S.; et al. A peripheral system disease-Pulmonary hypertension. Biomed. Pharmacother. 2024, 175, 116787. [Google Scholar] [CrossRef]
- Hudson, J.; Farkas, L. Epigenetic regulation of endothelial dysfunction and inflammation in pulmonary arterial hypertension. Int. J. Mol. Sci. 2021, 22, 12098. [Google Scholar] [CrossRef]
- Cober, N.D.; VandenBroek, M.M.; Ormiston, M.L.; Stewart, D.J. Evolving concepts in endothelial pathobiology of pulmonary arterial hypertension. Hypertension. 2022, 79, 1580–1590. [Google Scholar] [CrossRef]
- Chester, A.H.; Yacoub, M.H. The role of endothelin-1 in pulmonary arterial hypertension. Glob. Cardiol. Sci. Pract. 2014, 2014, 62–78. [Google Scholar] [CrossRef]
- Iyinikkel, J.; Murray, F. GPCRs in pulmonary arterial hypertension: Tipping the balance. Br. J. Pharmacol. 2018, 175, 3063–3079. [Google Scholar] [CrossRef]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ. 2018, 360, j5492. [Google Scholar] [CrossRef]
- Kuhr, F.K.; Smith, K.A.; Song, M.Y.; Levitan, I.; Yuan, J.X. New mechanisms of pulmonary arterial hypertension: Role of Ca2+ signaling. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1546–H1562. [Google Scholar] [CrossRef] [PubMed]
- Condon, D.F.; Agarwal, S.; Chakraborty, A.; Auer, N.; Vazquez, R.; Patel, H.; Zamanian, R.T.; de Jesus Perez, V.A. Novel mechanisms targeted by drug trials in pulmonary arterial hypertension. Chest 2022, 161, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Mamazhakypov, A.; Viswanathan, G.; Lawrie, A.; Schermuly, R.T.; Rajagopal, S. The role of chemokines and chemokine receptors in pulmonary arterial hypertension. Br. J. Pharmacol. 2021, 178, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.J.R.; Li, X.; Bordan, Z.; Haigh, S.; Bentley, A.; Chen, F.; Barman, S.A. Reactive oxygen and nitrogen species in the development of pulmonary hypertension. Antioxidants 2017, 6, 54. [Google Scholar] [CrossRef]
- Klouda, T.; Yuan, K. Inflammation in pulmonary arterial hypertension. Adv. Exp. Med. Biol. 2021, 1303, 351–372. [Google Scholar] [CrossRef]
- Peng, H.; Xiao, Y.; Deng, X.; Luo, J.; Hong, C.; Qin, X. The Warburg effect: A new story in pulmonary arterial hypertension. Clin. Chim. Acta 2016, 461, 53–58. [Google Scholar] [CrossRef]
- Han, S.; Chandel, N.S. Lessons from cancer metabolism for pulmonary arterial hypertension and fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 65, 134–145. [Google Scholar] [CrossRef]
- Cullivan, S.; Murphy, C.A.; Weiss, L.; Comer, S.P.; Kevane, B.; McCullagh, B.; Maguire, P.B.; Ní Ainle, F.; Gaine, S.P. Platelets, extracellular vesicles and coagulation in pulmonary arterial hypertension. Pulm. Circ. 2021, 11, 20458940211021036. [Google Scholar] [CrossRef]
- Cuthbertson, I.; Morrell, N.W.; Caruso, P. BMPR2 mutation and metabolic reprogramming in pulmonary arterial hypertension. Circ. Res. 2023, 132, 109–126. [Google Scholar] [CrossRef]
- Tatius, B.; Wasityastuti, W.; Astarini, F.D.; Nugrahaningsih, D.A.A. Significance of BMPR2 mutations in pulmonary arterial hypertension. Respir. Investig. 2021, 59, 397–407. [Google Scholar] [CrossRef]
- Andersson, K.E. PDE5 inhibitors—Pharmacology and clinical applications 20 years after sildenafil discovery. Br. J. Pharmacol. 2018, 175, 2554–2565. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R.; Elliott, C.G.; Levine, D.J.; Bossone, E.; Duvall, L.; Fagan, K.; Frantsve-Hawley, J.; Kawut, S.M.; Ryan, J.J.; Rosenzweig, E.B.; et al. Therapy for pulmonary arterial hypertension in adults: Update of the CHEST guideline and expert panel report. Chest 2019, 155, 565–586. [Google Scholar] [CrossRef] [PubMed]
- Mandras, S.; Kovacs, G.; Olschewski, H.; Broderick, M.; Nelsen, A.; Shen, E.; Champion, H. Combination therapy in pulmonary arterial hypertension-targeting the nitric oxide and prostacyclin pathways. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Ghofrani, H.A.; Pak, O.; Bonnet, S.; Provencher, S.; Sitbon, O.; Rosenkranz, S.; Hoeper, M.M.; Kiely, D.G. Current and future treatments of pulmonary arterial hypertension. Br. J. Pharmacol. 2021, 178, 6–30. [Google Scholar] [CrossRef]
- Alamri, A.K.; Ma, C.L.; Ryan, J.J. Novel drugs for the treatment of pulmonary arterial hypertension: Where are we going? Drugs 2023, 83, 577–585. [Google Scholar] [CrossRef]
- Omote, K.; Sorimachi, H.; Obokata, M.; Reddy, Y.N.V.; Verbrugge, F.H.; Omar, M.; DuBrock, H.M.; Redfield, M.M.; Borlaug, B.A. Pulmonary vascular disease in pulmonary hypertension due to left heart disease: Pathophysiologic implications. Eur. Heart J. 2022, 43, 3417–3431. [Google Scholar] [CrossRef]
- Wissmüller, M.; Dohr, J.; Adler, J.; Ochs, L.; Tichelbäcker, T.; Hohmann, C.; Baldus, S.; Rosenkranz, S. Pulmonary hypertension associated with left heart disease. Herz 2023, 48, 266–273. [Google Scholar] [CrossRef]
- Vachiéry, J.L.; Tedford, R.J.; Rosenkranz, S.; Palazzini, M.; Lang, I.; Guazzi, M.; Coghlan, G.; Chazova, I.; De Marco, T. Pulmonary hypertension due to left heart disease. Eur. Respir. J. 2019, 53, 1801897. [Google Scholar] [CrossRef]
- Aras, M.A.; Psotka, M.A.; De Marco, T. Pulmonary hypertension due to left heart disease: An update. Curr. Cardiol. Rep. 2019, 21, 62. [Google Scholar] [CrossRef]
- Marra, A.M.; Benjamin, N.; Cittadini, A.; Bossone, E.; Grünig, E. When pulmonary hypertension complicates heart failure. Cardiol. Clin. 2022, 40, 191–198. [Google Scholar] [CrossRef]
- Huston, J.H.; Shah, S.J. Understanding the pathobiology of pulmonary hypertension due to left heart disease. Circ. Res. 2022, 130, 1382–1403. [Google Scholar] [CrossRef] [PubMed]
- Macera, F.; Vachiéry, J.L. Management of pulmonary hypertension in left heart disease. Methodist Debakey Cardiovasc. J. 2021, 17, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Ltaief, Z.; Yerly, P.; Liaudet, L. Pulmonary hypertension in left heart diseases: Pathophysiology, hemodynamic assessment and therapeutic management. Int. J. Mol. Sci. 2023, 24, 9971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Zhao, S.; Wang, Z.; Zhang, M.; Zhang, S.; Wang, X.; Zhang, S.; Zhang, W.; Hao, L.; et al. The incidence and prevalence of pulmonary hypertension in the COPD population: A systematic review and meta-analysis. Int. J. Chron. Obs. Pulmon Dis. 2022, 17, 1365–1379. [Google Scholar] [CrossRef]
- Rajagopal, K.; Bryant, A.J.; Sahay, S.; Wareing, N.; Zhou, Y.; Pandit, L.M.; Karmouty-Quintana, H. Idiopathic pulmonary fibrosis and pulmonary hypertension: Heracles meets the Hydra. Br. J. Pharmacol. 2021, 178, 172–186. [Google Scholar] [CrossRef]
- Christenson, S.A.; Smith, B.M.; Bafadhel, M.; Putcha, N. Chronic obstructive pulmonary disease. Lancet 2022, 399, 2227–2242. [Google Scholar] [CrossRef]
- Cassady, S.J.; Reed, R.M. Pulmonary hypertension in COPD: A case study and review of the literature. Medicina 2019, 55, 432. [Google Scholar] [CrossRef]
- Gredic, M.; Wu, C.Y.; Hadzic, S.; Pak, O.; Savai, R.; Kojonazarov, B.; Doswada, S.; Weiss, A.; Weigert, A.; Guenther, A.; et al. Myeloid-cell-specific deletion of inducible nitric oxide synthase protects against smoke-induced pulmonary hypertension in mice. Eur. Respir. J. 2022, 59, 2101153. [Google Scholar] [CrossRef]
- Gredic, M.; Sharma, V.; Hadzic, S.; Wu, C.Y.; Pak, O.; Kojonazarov, B.; Duerr, J.; Mall, M.A.; Guenther, A.; Schermuly, R.T.; et al. iNOS deletion in alveolar epithelium cannot reverse the elastase-induced emphysema in mice. Cells 2022, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Kropski, J.A.; Jones, M.G.; Lee, J.S.; Rossi, G.; Karampitsakos, T.; Maher, T.M.; Tzouvelekis, A.; Ryerson, C.J. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol. Ther. 2021, 222, 107798. [Google Scholar] [CrossRef]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2022, 17, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Collum, S.D.; Amione-Guerra, J.; Cruz-Solbes, A.S.; DiFrancesco, A.; Hernandez, A.M.; Hanmandlu, A.; Youker, K.; Guha, A.; Karmouty-Quintana, H. Pulmonary hypertension associated with idiopathic pulmonary fibrosis: Current and future perspectives. Can. Respir. J. 2017, 2017, 1430350. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Snary, K.J.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.G.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic pulmonary vasoconstriction: From molecular mechanisms to medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, J.; Freytag, J.; Schiefer, L.M.; Macholz, F.; Sareban, M.; Schmidt-Hutten, L.; Stang, H.; Schwedhelm, E.; Swenson, E.R.; Böger, R.; et al. Asymmetric and symmetric dimethylarginine in high altitude pulmonary hypertension (HAPH) and high altitude pulmonary edema (HAPE). Front. Physiol. 2023, 14, 1297636. [Google Scholar] [CrossRef]
- Stam, K.; van Duin, R.W.B.; Uitterdijk, A.; Cai, Z.; Duncker, D.J.; Merkus, D. Exercise facilitates early recognition of cardiac and vascular remodeling in chronic thromboembolic pulmonary hypertension in swine. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H627–H642. [Google Scholar] [CrossRef]
- Alba, G.A.; Atri, D.; Darbha, S.; Singh, I.; Tapson, V.F.; Lewis, M.I.; Chun, H.J.; Yu, Y.R.; Maron, B.A.; Rajagopal, S. Chronic thromboembolic pulmonary hypertension: The bench. Curr. Cardiol. Rep. 2021, 23, 141. [Google Scholar] [CrossRef]
- Yang, J.; Madani, M.M.; Mahmud, E.; Kim, N.H. Evaluation and management of chronic thromboembolic pulmonary hypertension. Chest 2023, 164, 490–502. [Google Scholar] [CrossRef]
- Dignam, J.P.; Scott, T.E.; Kemp-Harper, B.K.; Hobbs, A.J. Animal models of pulmonary hypertension: Getting to the heart of the problem. Br. J. Pharmacol. 2022, 179, 811–837. [Google Scholar] [CrossRef]
- Wu, X.H.; Ma, J.L.; Ding, D.; Ma, Y.J.; Wei, Y.P.; Jing, Z.C. Experimental animal models of pulmonary hypertension: Development and challenges. Anim. Model. Exp. Med. 2022, 5, 207–216. [Google Scholar] [CrossRef]
- Liu, S.F.; Yan, Y. Animal models of pulmonary hypertension due to left heart disease. Anim. Model. Exp. Med. 2022, 5, 197–206. [Google Scholar] [CrossRef]
- Jasińska-Stroschein, M. An updated review of experimental rodent models of pulmonary hypertension and left heart disease. Front. Pharmacol. 2024, 14, 1308095. [Google Scholar] [CrossRef]
- Boucherat, O.; Agrawal, V.; Lawrie, A.; Bonnet, S. The latest in animal models of pulmonary hypertension and right ventricular failure. Circ. Res. 2022, 130, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, J.; Rubio, G.A.; Limper, A.H.; Williams, K.; Elliot, S.J.; Ninou, I.; Aidinis, V.; Tzouvelekis, A.; Glassberg, M.K. Exploring animal models that resemble idiopathic pulmonary fibrosis. Front. Med. 2017, 4, 118. [Google Scholar] [CrossRef]
- Karpov, A.A.; Vaulina, D.D.; Smirnov, S.S.; Moiseeva, O.M.; Galagudza, M.M. Rodent models of pulmonary embolism and chronic thromboembolic pulmonary hypertension. Heliyon 2022, 8, e09014. [Google Scholar] [CrossRef]
- Bueno-Beti, C.; Sassi, Y.; Hajjar, R.J.; Hadri, L. Pulmonary artery hypertension model in rats by monocrotaline administration. Methods Mol. Biol. 2018, 1816, 233–241. [Google Scholar] [CrossRef]
- Gomez-Arroyo, J.G.; Farkas, L.; Alhussaini, A.A.; Farkas, D.; Kraskauskas, D.; Voelkel, N.F.; Bogaard, H.J. The monocrotaline model of pulmonary hypertension in perspective. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L363–L369. [Google Scholar] [CrossRef]
- Wang, L.; Halliday, G.; Huot, J.R.; Satoh, T.; Baust, J.J.; Fisher, A.; Cook, T.; Hu, J.; Avolio, T.; Goncharov, D.A.; et al. Treatment with treprostinil and metformin normalizes hyperglycemia and improves cardiac function in pulmonary hypertension associated with heart failure with preserved ejection fraction. Arter. Thromb. Vasc. Biol. 2020, 40, 1543–1558. [Google Scholar] [CrossRef]
- Tanner, L.; Single, A.B. Animal models reflecting chronic obstructive pulmonary disease and related respiratory disorders: Translating pre-clinical data into clinical relevance. J. Innate Immun. 2020, 12, 203–225. [Google Scholar] [CrossRef]
- Lu, H.I.; Huang, T.H.; Sung, P.H.; Chen, Y.L.; Chua, S.; Chai, H.Y.; Chung, S.Y.; Liu, C.F.; Sun, C.K.; Chang, H.W.; et al. Administration of antioxidant peptide SS-31 attenuates transverse aortic constriction-induced pulmonary arterial hypertension in mice. Acta Pharmacol. Sin. 2016, 37, 589–603. [Google Scholar] [CrossRef]
- Rampa, D.R.; Murugesan, P.; Chao, H.; Feng, H.; Dai, W.; Lee, D.; Pekcec, A.; Doods, H.; Wu, D. Reversal of pulmonary arterial hypertension and neointimal formation by kinin B1 receptor blockade. Respir. Res. 2021, 22, 281. [Google Scholar] [CrossRef]
- Soltani Hekmat, A.; Amini, F.; Javanmardi, K. Effects of alamandine on monocrotaline-induced pulmonary hypertension in rats. Iran. J. Basic. Med. Sci. 2024, 27, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.P.; Dong, W.P.; Yang, Y.C.; Zeng, Y.Y.; Liu, Y.; Dong, Z.; Ma, X.M.; Cao, Y.Q.; Bai, Y.Z.; Yang, B.; et al. Tetramethylpyrazine improves monocrotaline-induced pulmonary hypertension through the ROS/iNOS/PKG-1 axis. J. Healthc. Eng. 2022, 2022, 1890892. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.J.; Han, J.Y.; Lee, S.G.; Jeon, B.T.; Choi, W.S.; Hwang, Y.S.; Roh, G.S.; Lee, J.D. Temporal changes of angiopoietins and Tie2 expression in rat lungs after monocrotaline-induced pulmonary hypertension. Comp. Med. 2009, 59, 350–356. [Google Scholar] [PubMed]
- Zhang, T.T.; Cui, B.; Dai, D.Z.; Su, W. CPU 86017, p-chlorobenzyltetrahydroberberine chloride, attenuates monocrotaline-induced pulmonary hypertension by suppressing endothelin pathway. Acta Pharmacol. Sin. 2005, 26, 1309–1316. [Google Scholar] [CrossRef]
- Zhou, Y.; Tian, X.; Wang, X.; Wang, Y.; Fan, R.; Wang, Y.; Feng, N.; Zhang, S.; Guo, H.; Gu, X.; et al. Quaternary ammonium salt of U50,488H elicits protective effects against hypoxic pulmonary hypertension. Eur. J. Pharmacol. 2018, 832, 129–137. [Google Scholar] [CrossRef]
- Hung, M.W.; Yeung, H.M.; Lau, C.F.; Poon, A.M.S.; Tipoe, G.L.; Fung, M.L. Melatonin attenuates pulmonary hypertension in chronically hypoxic rats. Int. J. Mol. Sci. 2017, 18, 1125. [Google Scholar] [CrossRef]
- Teng, X.; Li, D.; Catravas, J.D.; Johns, R.A. C/EBP-beta mediates iNOS induction by hypoxia in rat pulmonary microvascular smooth muscle cells. Circ. Res. 2002, 90, 125–127. [Google Scholar] [CrossRef]
- Fagan, K.A.; Morrissey, B.; Fouty, B.W.; Sato, K.; Harral, J.W.; Morris, K.G., Jr.; Hoedt-Miller, M.; Vidmar, S.; McMurtry, I.F.; Rodman, D.M. Upregulation of nitric oxide synthase in mice with severe hypoxia-induced pulmonary hypertension. Respir. Res. 2001, 2, 306–313. [Google Scholar] [CrossRef]
- Wright, J.L.; Tai, H.; Dai, J.; Churg, A. Cigarette smoke induces rapid changes in gene expression in pulmonary arteries. Lab. Investig. 2002, 82, 1391–1398. [Google Scholar] [CrossRef]
- Hampl, V.; Bíbová, J.; Banasová, A.; Uhlík, J.; Miková, D.; Hnilicková, O.; Lachmanová, V.; Herget, J. Pulmonary vascular iNOS induction participates in the onset of chronic hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L11–L20. [Google Scholar] [CrossRef]
- Palmer, L.A.; Semenza, G.L.; Stoler, M.H.; Johns, R.A. Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. Am. J. Physiol. 1998, 274, L212–L219. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Chen, Y.F.; Zhao, X.; Creighton, J.R.; Guo, Y.; Hage, F.G.; Oparil, S.; Xing, D.D. Targeted delivery of pulmonary arterial endothelial cells overexpressing interleukin-8 receptors attenuates monocrotaline-induced pulmonary vascular remodeling. Arter. Thromb. Vasc. Biol. 2014, 34, 1539–1547. [Google Scholar] [CrossRef]
- Boyer, L.; Plantier, L.; Dagouassat, M.; Lanone, S.; Goven, D.; Caramelle, P.; Berrehar, F.; Kerbrat, S.; Dinh-Xuan, A.T.; Crestani, B.; et al. Role of nitric oxide synthases in elastase-induced emphysema. Lab. Investig. 2011, 91, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Johns, R.A. Upregulation of nitric oxide synthase correlates temporally with onset of pulmonary vascular remodeling in the hypoxic rat. Hypertension 1996, 28, 743–753. [Google Scholar] [CrossRef]
- Silva, G.F.; da Silva, J.S.; de Alencar, A.K.N.; de Moraes Carvalho da Silva, M.; Montagnoli, T.L.; de Souza Rocha, B.; de Freitas, R.H.C.N.; Sudo, R.T.; Fraga, C.A.M.; Zapata-Sudo, G. Novel p38 mitogen-activated protein kinase inhibitor reverses hypoxia-induced pulmonary arterial hypertension in rats. Pharmaceuticals 2022, 15, 900. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.D.; Xu, Z.J.; Hu, X.G.; Wu, C.Y.; Dai, Y.R.; Yang, L. Impaired iNOS-sGC-cGMP signalling contributes to chronic hypoxic and hypercapnic pulmonary hypertension in rat. Cell Biochem. Funct. 2012, 30, 279–285. [Google Scholar] [CrossRef]
- Moral-Sanz, J.; Menendez, C.; Moreno, L.; Moreno, E.; Cogolludo, A.; Perez-Vizcaino, F. Pulmonary arterial dysfunction in insulin resistant obese Zucker rats. Respir. Res. 2011, 12, 51. [Google Scholar] [CrossRef]
- Chiş, I.C.; Baltaru, D.; Dumitrovici, A.; Coseriu, A.; Radu, B.C.; Moldovan, R.; Mureşan, A. Protective effects of quercetin from oxidative/nitrosative stress under intermittent hypobaric hypoxia exposure in the rat’s heart. Physiol. Int. 2018, 105, 233–246. [Google Scholar] [CrossRef]
- Gong, Y.; Yang, Y.; Wu, Q.; Gao, G.; Liu, Y.; Xiong, Y.; Huang, C.; Wu, S. Activation of LXRα improves cardiac remodeling induced by pulmonary artery hypertension in rats. Sci. Rep. 2017, 7, 6169. [Google Scholar] [CrossRef]
- Rouet-Benzineb, P.; Eddahibi, S.; Raffestin, B.; Laplace, M.; Depond, S.; Adnot, S.; Crozatier, B. Induction of cardiac nitric oxide synthase 2 in rats exposed to chronic hypoxia. J. Mol. Cell. Cardiol. 1999, 31, 1697–1708. [Google Scholar] [CrossRef]
- Imoto, K.; Okada, M.; Yamawaki, H. Periostin mediates right ventricular failure through induction of inducible nitric oxide synthase expression in right ventricular fibroblasts from monocrotaline-induced pulmonary arterial hypertensive rats. Int. J. Mol. Sci. 2018, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Luo, H.; Yin, X.; Wang, L.; Zhang, J.; Cao, Y.; Zhang, Z.; Ye, Z.; Wang, E. Effects of sevoflurane on hemodynamics and inducible nitric oxide synthase/soluble guanylate cyclase signaling pathway in a rat model of pulmonary arterial hypertension. Anesth. Analg. 2017, 125, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.; Dong, Y.; Evans, L. Chronic hypoxia increases inducible NOS-derived nitric oxide in fetal guinea pig hearts. Pediatr. Res. 2009, 65, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Rengasamy, A.; Le Cras, T.D.; Koberna, P.A.; Dailey, G.C.; Johns, R.A. Distribution of NOS in normoxic vs. hypoxic rat lung: Upregulation of NOS by chronic hypoxia. Am. J. Physiol 1994, 267, L667–L678. [Google Scholar] [CrossRef] [PubMed]
- Resta, T.C.; O’Donaughy, T.L.; Earley, S.; Chicoine, L.G.; Walker, B.R. Unaltered vasoconstrictor responsiveness after iNOS inhibition in lungs from chronically hypoxic rats. Am. J. Physiol. 1999, 276, L122–L130. [Google Scholar] [CrossRef]
- Hu, R.; Dai, A.; Tan, S. Hypoxia-inducible factor 1 alpha upregulates the expression of inducible nitric oxide synthase gene in pulmonary arteries of hyposic rat. Chin. Med. J. 2002, 115, 1833–1837. [Google Scholar]
- Rairigh, R.L.; Storme, L.; Parker, T.A.; le Cras, T.D.; Kinsella, J.P.; Jakkula, M.; Abman, S.H. Inducible NO synthase inhibition attenuates shear stress-induced pulmonary vasodilation in the ovine fetus. Am. J. Physiol. 1999, 276, L513–L521. [Google Scholar] [CrossRef]
- Gosgnach, W.; Messika-Zeitoun, D.; Gonzalez, W.; Philipe, M.; Michel, J.B. Shear stress induces iNOS expression in cultured smooth muscle cells: Role of oxidative stress. Am. J. Physiol. Cell Physiol. 2000, 279, C1880–C1888. [Google Scholar] [CrossRef]
- Seimetz, M.; Parajuli, N.; Pichl, A.; Veit, F.; Kwapiszewska, G.; Weisel, F.C.; Milger, K.; Egemnazarov, B.; Turowska, A.; Fuchs, B.; et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 2011, 147, 293–305. [Google Scholar] [CrossRef]
- Liao, K.; Lv, D.Y.; Yu, H.L.; Chen, H.; Luo, S.X. iNOS regulates activation of the NLRP3 inflammasome through the sGC/cGMP/PKG/TACE/TNF-α axis in response to cigarette smoke resulting in aortic endothelial pyroptosis and vascular dysfunction. Int. Immunopharmacol. 2021, 101, 108334. [Google Scholar] [CrossRef]
- Kseibati, M.O.; Shehatou, G.S.G.; Sharawy, M.H.; Eladl, A.E.; Salem, H.A. Nicorandil ameliorates bleomycin-induced pulmonary fibrosis in rats through modulating eNOS, iNOS, TXNIP and HIF-1α levels. Life Sci. 2020, 246, 117423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Ma, A.Q.; Yang, L.; Dang, X.M. Atorvastatin attenuates bleomycin-induced pulmonary fibrosis via suppressing iNOS expression and the CTGF (CCN2)/ERK signaling pathway. Int. J. Mol. Sci. 2013, 14, 24476–24491. [Google Scholar] [CrossRef]
- Cinar, R.; Gochuico, B.R.; Iyer, M.R.; Jourdan, T.; Yokoyama, T.; Park, J.K.; Coffey, N.J.; Pri-Chen, H.; Szanda, G.; Liu, Z.; et al. Cannabinoid CB1 receptor overactivity contributes to the pathogenesis of idiopathic pulmonary fibrosis. JCI Insight 2017, 2, e92281. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Savai, R.; Dumitrascu, R.; Dahal, B.K.; Wilhelm, J.; Konigshoff, M.; Zakrzewicz, D.; Ghofrani, H.A.; Weissmann, N.; Eickelberg, O.; et al. The role of dimethylarginine dimethylaminohydrolase in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2011, 3, 87ra53. [Google Scholar] [CrossRef] [PubMed]
- Cinar, R.; Park, J.K.; Zawatsky, C.N.; Coffey, N.J.; Bodine, S.P.; Abdalla, J.; Yokoyama, T.; Jourdan, T.; Jay, L.; Zuo, M.X.G.; et al. CB1 R and iNOS are distinct players promoting pulmonary fibrosis in Hermansky-Pudlak syndrome. Clin. Transl. Med. 2021, 11, e471. [Google Scholar] [CrossRef]
- Iyer, A.K.; Ramesh, V.; Castro, C.A.; Kaushik, V.; Kulkarni, Y.M.; Wright, C.A.; Venkatadri, R.; Rojanasakul, Y.; Azad, N. Nitric oxide mediates bleomycin-induced angiogenesis and pulmonary fibrosis via regulation of VEGF. J. Cell. Biochem. 2015, 116, 2484–2493. [Google Scholar] [CrossRef]
- Hamal, K.R.; Wideman, R.; Anthony, N.; Erf, G.F. Expression of inducible nitric oxide synthase in lungs of broiler chickens following intravenous cellulose microparticle injection. Poult. Sci. 2008, 87, 636–644. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, D.; Yu, Y.; Liu, X.; Hu, L.; Gu, Y. Association between inflammatory mediators and pulmonary blood flow in a rabbit model of acute pulmonary embolism combined with shock. Front. Physiol. 2020, 11, 1051. [Google Scholar] [CrossRef]
- Koo, H.S.; Kim, K.C.; Hong, Y.M. Gene expressions of nitric oxide synthase and matrix metalloproteinase-2 in monocrotaline-induced pulmonary hypertension in rats after bosentan treatment. Korean Circ. J. 2011, 41, 83–90. [Google Scholar] [CrossRef]
- DeMarco, V.G.; Habibi, J.; Whaley-Connell, A.T.; Schneider, R.I.; Sowers, J.R.; Andresen, B.T.; Gutweiler, A.A.; Ma, L.; Johnson, M.S.; Ferrario, C.M.; et al. Rosuvastatin ameliorates the development of pulmonary arterial hypertension in the transgenic (mRen2)27 rat. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1128–H1139. [Google Scholar] [CrossRef]
- Camelo, J.S., Jr.; Martins, A.R.; Rosa, E.; Ramos, S.G.; Hehre, D.; Bancalari, E.; Suguihara, C. Angiotensin II type 1 receptor blockade partially attenuates hypoxia-induced pulmonary hypertension in newborn piglets: Relationship with the nitrergic system. Braz. J. Med. Biol. Res. 2012, 45, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Sheak, J.R.; Weise-Cross, L.; deKay, R.J.; Walker, B.R.; Jernigan, N.L.; Resta, T.C. Enhanced NO-dependent pulmonary vasodilation limits increased vasoconstrictor sensitivity in neonatal chronic hypoxia. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H828–H838. [Google Scholar] [CrossRef] [PubMed]
- Morales-Cano, D.; Callejo, M.; Barreira, B.; Mondejar-Parreño, G.; Esquivel-Ruiz, S.; Ramos, S.; Martín, M.Á.; Cogolludo, A.; Moreno, L.; Perez-Vizcaino, F. Elevated pulmonary arterial pressure in Zucker diabetic fatty rats. PLoS ONE 2019, 14, e0211281. [Google Scholar] [CrossRef] [PubMed]
- Berkenbosch, J.W.; Baribeau, J.; Perreault, T. Decreased synthesis and vasodilation to nitric oxide in piglets with hypoxia-induced pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L276–L283. [Google Scholar] [CrossRef] [PubMed]
- Fontoura, D.; Oliveira-Pinto, J.; Tavares-Silva, M.; Leite, S.; Vasques-Nóvoa, F.; Mendes-Ferreira, P.; Lourenço, A.P.; Leite-Moreira, A.F. Myocardial and anti-inflammatory effects of chronic bosentan therapy in monocrotaline-induced pulmonary hypertension. Rev. Port. Cardiol. 2014, 33, 213–222. [Google Scholar] [CrossRef]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, K.C.; Cho, M.S.; Hong, Y.M. Changes of pulmonary pathology and gene expressions after simvastatin treatment in the monocrotaline-induced pulmonary hypertension rat model. Korean Circ. J. 2011, 41, 518–527. [Google Scholar] [CrossRef]
- Ogoshi, T.; Tsutsui, M.; Kido, T.; Sakanashi, M.; Naito, K.; Oda, K.; Ishimoto, H.; Yamada, S.; Wang, K.Y.; Toyohira, Y.; et al. Protective role of myelocytic nitric oxide synthases against hypoxic pulmonary hypertension in mice. Am. J. Respir. Crit. Care Med. 2018, 198, 232–244. [Google Scholar] [CrossRef]
- Le Cras, T.D.; Xue, C.; Rengasamy, A.; Johns, R.A. Chronic hypoxia upregulates endothelial and inducible NO synthase gene and protein expression in rat lung. Am. J. Physiol. 1996, 270, L164–L170. [Google Scholar] [CrossRef]
- Bodas, M.; Silverberg, D.; Walworth, K.; Brucia, K.; Vij, N. Augmentation of S-nitrosoglutathione controls cigarette smoke-induced inflammatory-oxidative stress and chronic obstructive pulmonary disease-emphysema pathogenesis by restoring cystic fibrosis transmembrane conductance regulator function. Antioxid. Redox Signal. 2017, 27, 433–451. [Google Scholar] [CrossRef]
- Fysikopoulos, A.; Seimetz, M.; Hadzic, S.; Knoepp, F.; Wu, C.Y.; Malkmus, K.; Wilhelm, J.; Pichl, A.; Bednorz, M.; Tadele Roxlau, E.; et al. Amelioration of elastase-induced lung emphysema and reversal of pulmonary hypertension by pharmacological iNOS inhibition in mice. Br. J. Pharmacol. 2021, 178, 152–171. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.C.; Liu, H.; Pinkas, G.A.; Thompson, L.P. Chronic hypoxia increases peroxynitrite, MMP9 expression, and collagen accumulation in fetal guinea pig hearts. Pediatr. Res. 2012, 71, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, R.A.; Ewing, C.A.; Sitzmann, J.V.; Kuo, P.C. Pulmonary expression of iNOS and HO-1 protein is upregulated in a rat model of prehepatic portal hypertension. Dig. Dis. Sci. 2000, 45, 2405–2410. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; You, Y.; Shang, F.F.; Wang, X.; Huang, B.; Zhao, B.; Lv, D.; Yang, S.; Xie, M.; Kong, L.; et al. iNOS aggravates pressure overload-induced cardiac dysfunction via activation of the cytosolic-mtDNA-mediated cGAS-STING pathway. Theranostics 2023, 13, 4229–4246. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Maruyama, J.; Yokochi, A.; Mitani, Y.; Maruyama, K. A novel inhibitor of inducible nitric oxide synthase, ONO-1714, does not ameliorate hypoxia-induced pulmonary hypertension in rats. Lung 2007, 185, 303–308. [Google Scholar] [CrossRef]
- Shirai, M.; Pearson, J.T.; Shimouchi, A.; Nagaya, N.; Tsuchimochi, H.; Ninomiya, I.; Mori, H. Changes in functional and histological distributions of nitric oxide synthase caused by chronic hypoxia in rat small pulmonary arteries. Br. J. Pharmacol. 2003, 139, 899–910. [Google Scholar] [CrossRef]
- Zhang, P.; Xu, X.; Hu, X.; van Deel, E.D.; Zhu, G.; Chen, Y. Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ. Res. 2007, 100, 1089–1098. [Google Scholar] [CrossRef]
- Zi, Y.; Wang, X.; Zi, Y.; Yu, H.; Lan, Y.; Fan, Y.; Ren, C.; Liao, K.; Chen, H. Cigarette smoke induces the ROS accumulation and iNOS activation through deactivation of Nrf-2/SIRT3 axis to mediate the human bronchial epithelium ferroptosis. Free Radic. Biol. Med. 2023, 200, 73–86. [Google Scholar] [CrossRef]
- Gupta, I.; Ganguly, S.; Rozanas, C.R.; Stuehr, D.J.; Panda, K. Ascorbate attenuates pulmonary emphysema by inhibiting tobacco smoke and Rtp801-triggered lung protein modification and proteolysis. Proc. Natl. Acad. Sci. USA 2016, 113, E4208–E4217. [Google Scholar] [CrossRef]
- Atochina-Vasserman, E.N.; Beers, M.F.; Kadire, H.; Tomer, Y.; Inch, A.; Scott, P.; Guo, C.J.; Gow, A.J. Selective inhibition of inducible NO synthase activity in vivo reverses inflammatory abnormalities in surfactant protein D-deficient mice. J. Immunol. 2007, 179, 8090–8097. [Google Scholar] [CrossRef]
- Kim, H.J.; Yoo, H.Y. Hypoxic pulmonary vasoconstriction and vascular contractility in monocrotaline-induced pulmonary arterial hypertensive rats. Korean J. Physiol. Pharmacol. 2016, 20, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Mam, V.; Tanbe, A.F.; Vitali, S.H.; Arons, E.; Christou, H.A.; Khalil, R.A. Impaired vasoconstriction and nitric oxide-mediated relaxation in pulmonary arteries of hypoxia- and monocrotaline-induced pulmonary hypertensive rats. J. Pharmacol. Exp. Ther. 2010, 332, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Fresquet, F.; Pourageaud, F.; Leblais, V.; Brandes, R.P.; Savineau, J.P.; Marthan, R.; Muller, B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br. J. Pharmacol. 2006, 148, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Fike, C.D.; Aschner, J.L.; Zhang, Y.; Kaplowitz, M.R. Impaired NO signaling in small pulmonary arteries of chronically hypoxic newborn piglets. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L1244–L1254. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Abe, K.; Oka, M.; Saku, K.; Yoshida, K.; Ishikawa, T.; McMurtry, I.F.; Sunagawa, K.; Hoka, S.; Tsutsui, H. Inhibition of nitric oxide synthase unmasks vigorous vasoconstriction in established pulmonary arterial hypertension. Physiol. Rep. 2017, 5, e13537. [Google Scholar] [CrossRef]
- Medvedeva, N.A.; Bonartsev, A.P.; Slavutskaya, A.V.; Diakonov, K.B.; Postnikov, A.B. Aminoguanidine restores reactivity of pulmonary and systemic vessels in rats with monocrotaline-induced pulmonary hypertension. Am. J. Hypertens. 2004, 17, 72A–73A. [Google Scholar] [CrossRef]
- Soejima, K.; McGuire, R.; Snyder, N.; Uchida, T.; Szabó, C.; Salzman, A.; Traber, L.D.; Traber, D.L. The effect of inducible nitric oxide synthase (iNOS) inhibition on smoke inhalation injury in sheep. Shock 2000, 13, 261–266. [Google Scholar] [CrossRef]
- Guo, C.; Atochina-Vasserman, E.; Abramova, H.; George, B.; Manoj, V.; Scott, P.; Gow, A. Role of NOS2 in pulmonary injury and repair in response to bleomycin. Free Radic. Biol. Med. 2016, 91, 293–301. [Google Scholar] [CrossRef]
- Genovese, T.; Cuzzocrea, S.; Di Paola, R.; Failla, M.; Mazzon, E.; Sortino, M.A.; Frasca, G.; Gili, E.; Crimi, N.; Caputi, A.P.; et al. Inhibition or knock out of inducible nitric oxide synthase result in resistance to bleomycin-induced lung injury. Respir. Res. 2005, 6, 58. [Google Scholar] [CrossRef]
- Chen, X.L.; Huang, S.S.; Li, W.B.; Wang, D.H.; Wang, X.L. Inhibitory effect of aminoguanidine on bleomycin-induced pulmonary toxicity in rat. Acta Pharmacol. Sin. 2001, 22, 711–715. [Google Scholar]
- Chen, X.L.; Li, W.B.; Zhou, A.M.; Ai, J.; Huang, S.S. Role of endogenous peroxynitrite in pulmonary injury and fibrosis induced by bleomycin A5 in rats. Acta Pharmacol. Sin. 2003, 24, 697–702. [Google Scholar] [PubMed]
- Chen, C.; Yun, X.J.; Liu, L.Z.; Guo, H.; Liu, L.F.; Chen, X.L. Exogenous nitric oxide enhances the prophylactic effect of aminoguanidine, a preferred iNOS inhibitor, on bleomycin-induced fibrosis in the lung: Implications for the direct roles of the NO molecule in vivo. Nitric Oxide 2017, 70, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Wideman, R.F.; Bowen, O.T.; Erf, G.F.; Chapman, M.E. Influence of aminoguanidine, an inhibitor of inducible nitric oxide synthase, on the pulmonary hypertensive response to microparticle injections in broilers. Poult. Sci. 2006, 85, 511–527. [Google Scholar] [CrossRef] [PubMed]
- Dias-Junior, C.A.; Sertorio, J.T.; Tanus-Santos, J.E. Aminoguanidine produces beneficial haemodynamic effects in a canine model of acute pulmonary thromboembolism. Acta Physiol. 2007, 191, 189–196. [Google Scholar] [CrossRef]
- Dias-Junior, C.A.; Neto-Neves, E.M.; Montenegro, M.F.; Tanus-Santos, J.E. Hemodynamic effects of inducible nitric oxide synthase inhibition combined with sildenafil during acute pulmonary embolism. Nitric Oxide 2010, 23, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Yatera, K.; Mukae, H.; Tsutsui, M. Role of nitric oxide synthases in respiratory health and disease: Insights from triple nitric oxide synthases knockout mice. Int. J. Mol. Sci. 2024, 25, 9317. [Google Scholar] [CrossRef]
- Noguchi, S.; Yatera, K.; Wang, K.Y.; Oda, K.; Akata, K.; Yamasaki, K.; Kawanami, T.; Ishimoto, H.; Toyohira, Y.; Shimokawa, H.; et al. Nitric oxide exerts protective effects against bleomycin-induced pulmonary fibrosis in mice. Respir. Res. 2014, 15, 92. [Google Scholar] [CrossRef]
- Roy, R.; Wilcox, J.; Webb, A.J.; O’Gallagher, K. Dysfunctional and dysregulated nitric oxide synthases in cardiovascular disease: Mechanisms and therapeutic potential. Int. J. Mol. Sci. 2023, 24, 15200. [Google Scholar] [CrossRef]
- Tsutsui, M.; Shimokawa, H.; Morishita, T.; Nakashima, Y.; Yanagihara, N. Development of genetically engineered mice lacking all three nitric oxide synthases. J. Pharmacol. Sci. 2006, 102, 147–154. [Google Scholar] [CrossRef]
- Yin, H.L.; Luo, C.W.; Dai, Z.K.; Shaw, K.P.; Chai, C.Y.; Wu, C.C. Hypoxia-inducible factor-1α, vascular endothelial growth factor, inducible nitric oxide synthase, and endothelin-1 expression correlates with angiogenesis in congenital heart disease. Kaohsiung J. Med. Sci. 2016, 32, 348–355. [Google Scholar] [CrossRef]
- de Rooij, J.D.; Hösgör, M.; Ijzendoorn, Y.; Rottier, R.; Groenman, F.A.; Tibboel, D.; de Krijger, R.R. Expression of angiogenesis-related factors in lungs of patients with congenital diaphragmatic hernia and pulmonary hypoplasia of other causes. Pediatr. Dev. Pathol. 2004, 7, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Colombo, P.C.; Banchs, J.E.; Celaj, S.; Talreja, A.; Lachmann, J.; Malla, S.; DuBois, N.B.; Ashton, A.W.; Latif, F.; Jorde, U.P.; et al. Endothelial cell activation in patients with decompensated heart failure. Circulation 2005, 111, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Haywood, G.A.; Tsao, P.S.; von der Leyen, H.E.; Mann, M.J.; Keeling, P.J.; Trindade, P.T.; Lewis, N.P.; Byrne, C.D.; Rickenbacher, P.R.; Bishopric, N.H.; et al. Expression of inducible nitric oxide synthase in human heart failure. Circulation 1996, 93, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Patten, R.D.; Denofrio, D.; El-Zaru, M.; Kakkar, R.; Saunders, J.; Celestin, F.; Warner, K.; Rastegar, H.; Khabbaz, K.R.; Udelson, J.E.; et al. Ventricular assist device therapy normalizes inducible nitric oxide synthase expression and reduces cardiomyocyte apoptosis in the failing human heart. J. Am. Coll. Cardiol. 2005, 45, 1419–1424. [Google Scholar] [CrossRef]
- Fukuchi, M.; Hussain, S.N.; Giaid, A. Heterogeneous expression and activity of endothelial and inducible nitric oxide synthases in end-stage human heart failure: Their relation to lesion site and beta-adrenergic receptor therapy. Circulation 1998, 98, 132–139. [Google Scholar] [CrossRef]
- Orús, J.; Heras, M.; Morales-Ruiz, M.; Leivas, A.; Roig, E.; Rigol, M.; Rivera, F.; Sanz, G.; Jiménez, W. Nitric oxide synthase II mRNA expression in cardiac tissue of patients with heart failure undergoing cardiac transplantation. J. Heart Lung Transplant. 2000, 19, 139–144. [Google Scholar] [CrossRef]
- Vejlstrup, N.G.; Bouloumie, A.; Boesgaard, S.; Andersen, C.B.; Nielsen-Kudsk, J.E.; Mortensen, S.A.; Kent, J.D.; Harrison, D.G.; Busse, R.; Aldershvile, J. Inducible nitric oxide synthase (iNOS) in the human heart: Expression and localization in congestive heart failure. J. Mol. Cell. Cardiol. 1998, 30, 1215–1223. [Google Scholar] [CrossRef]
- Stein, B.; Eschenhagen, T.; Rüdiger, J.; Scholz, H.; Förstermann, U.; Gath, I. Increased expression of constitutive nitric oxide synthase III, but not inducible nitric oxide synthase II, in human heart failure. J. Am. Coll. Cardiol. 1998, 32, 1179–1186. [Google Scholar] [CrossRef]
- Drexler, H.; Kästner, S.; Strobel, A.; Studer, R.; Brodde, O.E.; Hasenfuss, G. Expression, activity and functional significance of inducible nitric oxide synthase in the failing human heart. J. Am. Coll. Cardiol. 1998, 32, 955–963. [Google Scholar] [CrossRef]
- Ferreiro, C.R.; Chagas, A.C.; Carvalho, M.H.; Dantas, A.P.; Scavone, C.; Souza, L.C.; Buffolo, E.; da Luz, P.L. Expression of inducible nitric oxide synthase is increased in patients with heart failure due to ischemic disease. Braz. J. Med. Biol. Res. 2004, 37, 1313–1320. [Google Scholar] [CrossRef]
- Łabuzek, K.; Liber, S.; Bułdak, Ł.; Krupej-Kędzierska, J.; Machnik, G.; Bobrzyk, M.; Okopień, B. Eplerenone mimics features of the alternative activation in macrophages obtained from patients with heart failure and healthy volunteers. Eur. J. Pharmacol. 2014, 726, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Speranza, L.; Franceschelli, S.; Pesce, M.; Ferrone, A.; Patruno, A.; Riccioni, G.; De Lutiis, M.A.; Felaco, M.; Grilli, A. Negative feedback interaction of HO-1/INOS in PBMC of acute congestive heart failure patients. J. Biol. Regul. Homeost. Agents 2013, 27, 739–748. [Google Scholar] [PubMed]
- Mohammadi, A.; Balizadeh Karami, A.R.; Dehghan Mashtani, V.; Sahraei, T.; Bandani Tarashoki, Z.; Khattavian, E.; Mobarak, S.; Moradi Kazerouni, H.; Radmanesh, E. Evaluation of oxidative stress, apoptosis, and expression of microRNA-208a and microRNA-1 in cardiovascular patients. Rep. Biochem. Mol. Biol. 2021, 10, 183–196. [Google Scholar] [CrossRef]
- Chen, C.; Zong, M.; Lu, Y.; Guo, Y.; Lv, H.; Xie, L.; Fu, Z.; Cheng, Y.; Si, Y.; Ye, B.; et al. Differentially expressed lnc-NOS2P3-miR-939-5p axis in chronic heart failure inhibits myocardial and endothelial cells apoptosis via iNOS/TNFα pathway. J. Cell. Mol. Med. 2020, 24, 11381–11396. [Google Scholar] [CrossRef] [PubMed]
- Momot, K.; Wojciechowska, M.; Krauz, K.; Czarzasta, K.; Puchalska, L.; Zarębiński, M.; Cudnoch-Jędrzejewska, A. Endoplasmic reticulum stress and expression of nitric oxide synthases in heart failure with preserved and with reduced ejection fraction—Pilot study. Cardiol. J. 2024, 31, 885–894. [Google Scholar] [CrossRef]
- Birks, E.J.; Latif, N.; Owen, V.; Bowles, C.; Felkin, L.E.; Mullen, A.J.; Khaghani, A.; Barton, P.J.; Polak, J.M.; Pepper, J.R.; et al. Quantitative myocardial cytokine expression and activation of the apoptotic pathway in patients who require left ventricular assist devices. Circulation 2001, 104, I233–I240. [Google Scholar] [CrossRef]
- Chen, Y.; Park, S.; Li, Y.; Missov, E.; Hou, M.; Han, X.; Hall, J.L.; Miller, L.W.; Bache, R.J. Alterations of gene expression in failing myocardium following left ventricular assist device support LVAD. Physiol. Genom. 2003, 14, 251–260. [Google Scholar] [CrossRef]
- Maestrelli, P.; Páska, C.; Saetta, M.; Turato, G.; Nowicki, Y.; Monti, S.; Formichi, B.; Miniati, M.; Fabbri, L.M. Decreased haem oxygenase-1 and increased inducible nitric oxide synthase in the lung of severe COPD patients. Eur. Respir. J. 2003, 21, 971–976. [Google Scholar] [CrossRef]
- Hoehn, T.; Preston, A.A.; McPhaden, A.R.; Stiller, B.; Vogel, M.; Bührer, C.; Wadsworth, R.M. Endothelial nitric oxide synthase (NOS) is upregulated in rapid progressive pulmonary hypertension of the newborn. Intensive Care Med. 2003, 29, 1757–1762. [Google Scholar] [CrossRef]
- Ichinose, M.; Sugiura, H.; Yamagata, S.; Koarai, A.; Shirato, K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am. J. Respir. Crit. Care Med. 2000, 162, 701–706. [Google Scholar] [CrossRef]
- Ricciardolo, F.L.; Caramori, G.; Ito, K.; Capelli, A.; Brun, P.; Abatangelo, G.; Papi, A.; Chung, K.F.; Adcock, I.; Barnes, P.J.; et al. Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2005, 116, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Agustí, A.; Morlá, M.; Sauleda, J.; Saus, C.; Busquets, X. NF-kappaB activation and iNOS upregulation in skeletal muscle of patients with COPD and low body weight. Thorax 2004, 59, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.M.; Geiger, R.; Hess, J.; Bogers, A.J.; Mooi, W.J. Altered arterial expression patterns of inducible and endothelial nitric oxide synthase in pulmonary plexogenic arteriopathy caused by congenital heart disease. Am. J. Respir. Crit. Care Med. 2001, 163, 1493–1499. [Google Scholar] [CrossRef]
- Hoehn, T.; Stiller, B.; McPhaden, A.R.; Wadsworth, R.M. Nitric oxide synthases in infants and children with pulmonary hypertension and congenital heart disease. Respir. Res. 2009, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Zheng, Z.; Hu, S.; Li, S.; Wei, Y.; Zhang, Y.; Cheng, X.; Ma, K. Mechanisms of pulmonary hypertension related to ventricular septal defect in congenital heart disease. Ann. Thorac. Surg. 2011, 92, 2215–2220. [Google Scholar] [CrossRef]
- Qing, M.; Schumacher, K.; Heise, R.; Wöltje, M.; Vazquez-Jimenez, J.F.; Richter, T.; Arranda-Carrero, M.; Hess, J.; von Bernuth, G.; Seghaye, M.C. Intramyocardial synthesis of pro- and anti-inflammatory cytokines in infants with congenital cardiac defects. J. Am. Coll. Cardiol. 2003, 41, 2266–2274. [Google Scholar] [CrossRef]
- Shehata, S.M.; Sharma, H.S.; Mooi, W.J.; Tibboel, D. Pulmonary hypertension in human newborns with congenital diaphragmatic hernia is associated with decreased vascular expression of nitric-oxide synthase. Cell Biochem. Biophys. 2006, 44, 147–155. [Google Scholar] [CrossRef]
- Comini, L.; Bachetti, T.; Agnoletti, L.; Gaia, G.; Curello, S.; Milanesi, B.; Volterrani, M.; Parrinello, G.; Ceconi, C.; Giordano, A.; et al. Induction of functional inducible nitric oxide synthase in monocytes of patients with congestive heart failure. Link with tumour necrosis factor-alpha. Eur. Heart J. 1999, 20, 1503–1513. [Google Scholar] [CrossRef]
- Speranza, L.; Franceschelli, S.; Riccioni, G.; Di Nicola, M.; Ruggeri, B.; Gallina, S.; Felaco, M.; Grilli, A. BNP and iNOS in decompensated chronic heart failure: A linear correlation. Front. Biosci. (Elite Ed.) 2012, 4, 1255–1262. [Google Scholar] [CrossRef]
- Almudéver, P.; Milara, J.; De Diego, A.; Serrano-Mollar, A.; Xaubet, A.; Perez-Vizcaino, F.; Cogolludo, A.; Cortijo, J. Role of tetrahydrobiopterin in pulmonary vascular remodelling associated with pulmonary fibrosis. Thorax 2013, 68, 938–948. [Google Scholar] [CrossRef]
- Saleh, D.; Barnes, P.J.; Giaid, A. Increased production of the potent oxidant peroxynitrite in the lungs of patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Vanderheyden, M.; Bartunek, J.; Knaapen, M.; Kockx, M.; De Bruyne, B.; Goethals, M. Hemodynamic effects of inducible nitric oxide synthase and nitrotyrosine generation in heart failure. J. Heart Lung Transplant. 2004, 23, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.D.; Kansal, M.; Desai, A.A.; Riden, K.; Arwood, M.J.; Yacob, A.A.; Stamos, T.D.; Cavallari, L.H.; Zamanian, R.T.; Shah, S.J.; et al. Endothelial nitric oxide synthase genotype is associated with pulmonary hypertension severity in left heart failure patients. Pulm. Circ. 2018, 8, 2045894018773049. [Google Scholar] [CrossRef] [PubMed]
- Dignam, J.P.; Sharma, S.; Stasinopoulos, I.; MacLean, M.R. Pulmonary arterial hypertension: Sex matters. Br. J. Pharmacol. 2024, 181, 938–966. [Google Scholar] [CrossRef] [PubMed]
- Stam, K.; Clauss, S.; Taverne, Y.J.H.J.; Merkus, D. Chronic thromboembolic pulmonary hypertension—What have we learned from large animal models. Front. Cardiovasc. Med. 2021, 8, 574360. [Google Scholar] [CrossRef]
- Roy, H.S.; Singh, R.; Ghosh, D. Recent advances in nanotherapeutic strategies that target nitric oxide pathway for preventing cartilage degeneration. Nitric Oxide 2021, 109–110, 1–11. [Google Scholar] [CrossRef]
- Maccallini, C.; Budriesi, R.; De Filippis, B.; Amoroso, R. Advancements in the research of new modulators of nitric oxide saynthases activity. Int. J. Mol. Sci. 2024, 25, 8486. [Google Scholar] [CrossRef]
- Ji, D.; Jin, C.; Tao, M.; Sun, Y.; Chen, H.; Li, H.; Qu, X.; Ye, H.; Zhang, L.; Huang, Z.; et al. Design, synthesis, and biological evaluation of novel iNOS inhibitors as potent neuroprotective agents for ischemic stroke. Eur. J. Med. Chem. 2024, 280, 116907. [Google Scholar] [CrossRef]
- Ndongson-Dongmo, B.; Lang, G.P.; Mece, O.; Hechaichi, N.; Lajqi, T.; Hoyer, D.; Brodhun, M.; Heller, R.; Wetzker, R.; Franz, M.; et al. Reduced ambient temperature exacerbates SIRS-induced cardiac autonomic dysregulation and myocardial dysfunction in mice. Basic Res. Cardiol. 2019, 114, 26. [Google Scholar] [CrossRef]
- Ryszkiewicz, P.; Malinowska, B.; Schlicker, E. Polypharmacology: Promises and new drugs in 2022. Pharmacol. Rep. 2023, 75, 755–770. [Google Scholar] [CrossRef]
- Padilha, E.C.; Yang, M.; Shah, P.; Wang, A.Q.; Duan, J.; Park, J.K.; Zawatsky, C.N.; Malicdan, M.C.V.; Kunos, G.; Iyer, M.R.; et al. In vitro and in vivo pharmacokinetic characterization, chiral conversion and PBPK scaling towards human PK simulation of S-MRI-1867, a drug candidate for Hermansky-Pudlak syndrome pulmonary fibrosis. Biomed. Pharmacother. 2023, 168, 115178. [Google Scholar] [CrossRef] [PubMed]
- Hellio le Graverand, M.P.; Clemmer, R.S.; Redifer, P.; Brunell, R.M.; Hayes, C.W.; Brandt, K.D.; Abramson, S.B.; Manning, P.T.; Miller, C.G.; Vignon, E. A 2-year randomised, double-blind, placebo-controlled, multicentre study of oral selective iNOS inhibitor, cindunistat (SD-6010), in patients with symptomatic osteoarthritis of the knee. Ann. Rheum. Dis. 2013, 72, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Seymour, M.; Pétavy, F.; Chiesa, F.; Perry, H.; Lukey, P.T.; Binks, M.; Donatien, P.D.; Freidin, A.J.; Eckersley, R.J.; McClinton, C.; et al. Ultrasonographic measures of synovitis in an early phase clinical trial: A double-blind, randomised, placebo and comparator controlled phase IIa trial of GW274150 (a selective inducible nitric oxide synthase inhibitor) in rheumatoid arthritis. Clin. Exp. Rheumatol. 2012, 30, 254–261. [Google Scholar] [PubMed]
- Høivik, H.O.; Laurijssens, B.E.; Harnisch, L.O.; Twomey, C.K.; Dixon, R.M.; Kirkham, A.J.; Williams, P.M.; Wentz, A.L.; Lunnon, M.W. Lack of efficacy of the selective iNOS inhibitor GW274150 in prophylaxis of migraine headache. Cephalalgia 2010, 30, 1458–1467. [Google Scholar] [CrossRef]

| Clinical Groups | Classification |
|---|---|
| Group 1 pulmonary arterial hypertension (PAH) | 1. Idiopathic: 1.1. Non-responders at vasoreactivity testing 1.2. Acute responders at vasoreactivity testing 2. Heritable 3. Associated with drugs and toxins 4. Associated with: 4.1. Connective tissue disease 4.2. Human immunodeficiency virus (HIV) infection 4.3. Portal hypertension 4.4. Congenital heart disease 4.5. Schistosomiasis 5. PAH with features of venous/capillary involvement 6. Persistent PH of the newborn |
| Group 2 PH associated with left heart disease | 1. Heart failure: 1.1. With preserved ejection fraction 1.2. With reduced or mildly reduced ejection fraction 2. Valvular heart disease 3. Congenital/acquired cardiovascular conditions leading to post-capillary PH |
| Group 3 PH associated with lung diseases and/or hypoxia | 1. Obstructive lung disease or emphysema * 2. Restrictive lung disease ** 3. Lung disease with mixed restrictive/obstructive pattern 4. Hypoventilation syndromes 5. Hypoxia without lung disease (e.g., high altitude) 6. Developmental lung disorders |
| Group 4 PH associated with pulmonary artery obstructions | 1. Chronic thromboembolic PH 2. Other pulmonary artery obstructions |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryszkiewicz, P.; Schlicker, E.; Malinowska, B. Is Inducible Nitric Oxide Synthase (iNOS) Promising as a New Target Against Pulmonary Hypertension? Antioxidants 2025, 14, 377. https://doi.org/10.3390/antiox14040377
Ryszkiewicz P, Schlicker E, Malinowska B. Is Inducible Nitric Oxide Synthase (iNOS) Promising as a New Target Against Pulmonary Hypertension? Antioxidants. 2025; 14(4):377. https://doi.org/10.3390/antiox14040377
Chicago/Turabian StyleRyszkiewicz, Piotr, Eberhard Schlicker, and Barbara Malinowska. 2025. "Is Inducible Nitric Oxide Synthase (iNOS) Promising as a New Target Against Pulmonary Hypertension?" Antioxidants 14, no. 4: 377. https://doi.org/10.3390/antiox14040377
APA StyleRyszkiewicz, P., Schlicker, E., & Malinowska, B. (2025). Is Inducible Nitric Oxide Synthase (iNOS) Promising as a New Target Against Pulmonary Hypertension? Antioxidants, 14(4), 377. https://doi.org/10.3390/antiox14040377