Abstract
Heat stress (HS) is one of the most important stressors in chickens, and its adverse effects are primarily caused by disturbing the redox homeostasis. An increase in electron leakage from the mitochondrial electron transport chain is the major source of free radical production under HS, which triggers other enzymatic systems to generate more radicals. As a defense mechanism, cells have enzymatic and non-enzymatic antioxidant systems that work cooperatively against free radicals. The generation of free radicals, particularly the reactive oxygen species (ROS) and reactive nitrogen species (RNS), under HS condition outweighs the cellular antioxidant capacity, resulting in oxidative damage to macromolecules, including lipids, carbohydrates, proteins, and DNA. Understanding these detrimental oxidative processes and protective defense mechanisms is important in developing mitigation strategies against HS. This review summarizes the current understanding of major oxidative and antioxidant systems and their molecular mechanisms in generating or neutralizing the ROS/RNS. Importantly, this review explores the potential mechanisms that lead to the development of oxidative stress in heat-stressed chickens, highlighting their unique behavioral and physiological responses against thermal stress. Further, we summarize the major findings associated with these oxidative and antioxidant mechanisms in chickens.
1. Introduction
Redox homeostasis is an essential cellular mechanism present in all forms of life, responsible for the delicate balance between oxidizing and reducing reactions. Free radicals are highly reactive molecules with unpaired electrons in their outer orbital that attempt to bond with other atoms or molecules to form a stable compound [1,2]. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) together form free radicals as well as other non-radical reactive substances [3]. The ROS constitute a group of molecules originating from molecular oxygen by accepting electrons and encompass both oxygen radicals, such as superoxide (O2•−), hydroxyl (HO•), and peroxyl (ROO•), and other non-radicals like hydrogen peroxide (H2O2), ozone (O3), and singlet oxygen (1O2) [2,4,5]. The RNS consists of nitrogen-containing radicals, including nitric oxide (•NO), and nitrogen dioxide (•NO2) radicals and peroxynitrite (ONOO−) non-radicals [6]. The ROS/RNS are produced naturally as a by-product of cellular metabolisms or from exogenous sources including heavy metals, UV rays, and air pollutants. While the low level of ROS/RNS is vital in several physiological processes, particularly the signaling pathways and immune functions, vasodilation and cardiovascular functions, tissue repair, neurotransmission, and redox regulation [7,8,9,10,11,12], excessive production of ROS/RNS depletes the body antioxidant capacity, resulting in oxidative damage of polyunsaturated fatty acids (PUFAs), proteins, carbohydrates, and DNA [13,14,15]. The adaptive mechanisms involving enzymatic and non-enzymatic antioxidants allow the cells to tolerate a certain level of oxidative stress and maintain redox homeostasis. Attempts at uplifting these adaptive mechanisms to alleviate the negative effects of oxidative injury have drawn considerable attention over the last several decades.
In animals, HS has traditionally been considered the primary cause of ROS-mediated oxidative injury [16,17,18]. With the Earth’s rising temperature, HS has become a significant challenge in animal production, particularly in tropical, subtropical, and even moderate climates, affecting a wide range of animals regardless of their natural habitat. The absence of sweat glands in birds to dissipate the heat has made them more vulnerable to HS [19,20]. In particular, HS poses a serious threat to the poultry industry, which makes a significant contribution to the United States and the global economy. Commercial broiler chickens selected for high growth rates or layers selected for high egg production are especially vulnerable to HS because of their high energy requirement, resulting in increased metabolic activity and greater heat production, which is further exacerbated by their genetic predisposition for lower heat tolerance [21,22]. Some major adverse effects of HS in chickens include decreased growth, reduced egg production, and egg quality, increased susceptibility to diseases, and mortality [23,24,25].
Given the species-specific variations and their unique physiological responses, the severity of HS and the resulting oxidative stress differs in chickens compared to other animals. However, since redox regulation is fundamental for cell survival, oxidative and antioxidant mechanisms are likely to be largely conserved in all types of cells. Unfortunately, there are very limited in vitro studies comparing these mechanisms in avian cells with those in mammalian cells. Any unique features identified in chickens are highlighted in this review. There are a handful of previous reviews that have discussed the effect of HS on production parameters as well as on the oxidative and antioxidative mechanisms. However, most of them do not address potential mechanisms and the possible cross-talks between these mechanisms during HS-induced oxidative stress. Moreover, these reviews did not specifically explain the significance of those mechanisms in chickens under HS conditions. In this article, we elaborated on major oxidative and antioxidative mechanisms, their relevance, and the previous findings under HS-induced oxidative stress, particularly focusing on chickens. Additionally, we summarized various pathways in schematic diagrams aimed at aiding the readers to comprehend these mechanisms easily and coherently. A better understanding of these molecular mechanisms will help to explore the potential nutritional interventions to elevate the cellular antioxidant mechanisms during oxidative stress.
2. Heat Stress-Induced Oxidative Stress—Current Understanding
Among numerous enzymatic systems, four major systems, namely, NADPH oxidases (NOXs) [26], mitochondrial electron transport chain (ETC) [27], xanthine oxidase (XO) [28], and uncoupled endothelial nitric oxide synthase (eNOS) [29], have been identified as the major sources of ROS/RNS production in cells. These systems exhibit a significant interplay where the triggering of one source can activate the others, leading to feed-forward mechanisms that amplify ROS production and oxidative stress [30]. The concept of “ROS-induced ROS-release” has been proposed by many authors by which a small quantity of ROS stimulates a larger production of ROS from the mitochondria [31,32]. Heat stress has been shown to induce enzymatic systems, especially the mitochondrial electron transport chain (ETC), thereby increasing the production of ROS [33]. Moreover, HS-induced oxidative stress impairs the protein folding mechanism and promotes the buildup of unfolded and misfolded proteins, which in turn causes endoplasmic reticulum (ER) stress [34]. The resultant ER stress can further induce mitochondrial dysfunction and elevate mitochondrial ROS production [35,36], as summarized in Figure 1.
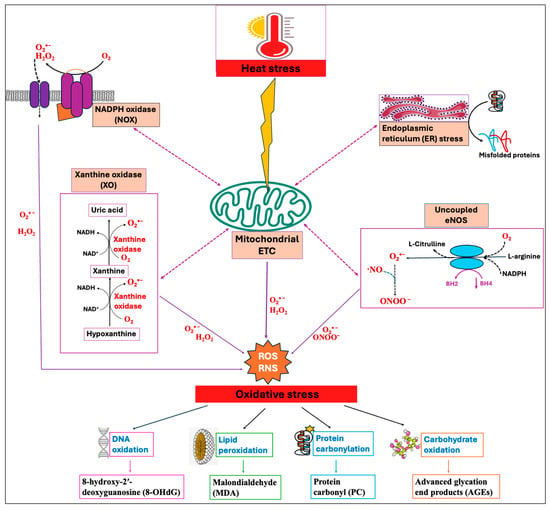
Figure 1.
Heat stress (HS)-induced oxidative stress and the indicators of oxidative stress. Primary contributors to cellular reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation include mitochondrial electron transport chain (ETC), NADPH oxidase (NOX), xanthine oxidase (XO), and uncoupled endothelial nitric oxide synthase (eNOS). Heat stress primarily triggers mitochondrial ROS production, which then activates other enzymatic systems, showing significant interplay between them. Increased levels of mitochondrial ROS disrupt the endoplasmic reticulum (ER) homeostasis, leading to the buildup of misfolded proteins, which in turn prompt mitochondria to produce more ROS. The resulting feed-forward mechanisms amplify ROS production and lead to the oxidative damage of DNA, lipids, proteins, and carbohydrates.
2.1. Mitochondrial Electron Transport Chain
During cellular metabolism, electrons transferred through the mitochondrial electron transport chain (ETC) establish a proton gradient over the mitochondrial membrane and drive ATP production. This process of ATP synthesis from ADP and Pi induced by electron transfer is called oxidative phosphorylation [37]. However, some of the electrons transferred to the ETC leak back prematurely and produce O2•− anion radicals. Mitochondrial O2•− radicals are mainly produced in two specific sites of the ETC, namely, complex-I [nicotinamide adenine dinucleotide dehydrogenase (NADH)] and complex-III (ubiquinone-cytochrome c reductase) [38]. Under normal metabolic conditions, complex-III is generally considered to be the most important site of ROS production [39]; however, conditions like HS can increase electron leakage and ROS production [40,41]. Mitochondrial uncoupling proteins (UCPs) are inner membrane carrier proteins that play a major role in the regulation of mitochondrial membrane potential and ROS levels through proton leakage [42,43]. This proton leakage across the mitochondrial membrane releases the energy of the proton gradient as heat and uncouples oxidative phosphorylation [44]. It has been suggested that increased ROS generation induces proton leakage, which in turn reduces ROS production [45,46].
The initial response following HS involves a rise in cellular energy requirements, doubling the energy expenditure during acute heat exposure [47]. Consequently, acute HS is shown to increase fatty acid transportation and mitochondrial β-oxidation to meet the cellular energy demand [48], thereby increasing the enzymatic activity of respiratory chain complexes [49]. However, this increase in mitochondrial energy production also leads to greater ROS generation as a result of elevated electron carrier reduction, increased membrane potential, and high mitochondrial oxygen levels [50]. Moreover, research has demonstrated that HS deactivates complex-I of the ETC and prompts its dissociation into smaller subcomplexes without impacting other complexes, thus slowing down the electron flow through ETC [33,51]. This causes an increase in ROS production due to the impairment of complex-I; however, it also induces manganese superoxide dismutase (Mn-SOD) that converts O2•− to hydrogen peroxide (H2O2). Furthermore, the reduction of H2O2 to water is mediated by two enzymes, catalase and glutathione peroxidase. Thus, a defect in complex-I may not lead to increased ROS production if the activity of Mn-SOD is enhanced [52].
On the other hand, the O2•− and H2O2 can react through the Haber–Weiss reaction to generate the highly reactive hydroxyl radical (HO•) [53] or the H2O2 can be reduced to HO• in the presence of transition metals, such as iron and copper through a Fenton reaction [54]. Moreover, excessive production of O2•− leads to the rapid inactivation of nitric oxide (•NO), resulting in the yield of a potent oxidant, peroxynitrite (ONOO−) [55]. Because nucleophilic CO2 is readily available, ONOO− combines quickly with CO2 to produce carbonate radical (CO3•−) and nitrogen dioxide radical (•NO2), or it may also be reduced to form HO• and •NO2 (Figure 2). The excessive production of free radicals during HS can overwhelm the body’s antioxidant defenses, causing oxidative damage to proteins, lipids, carbohydrates, and DNA [18,56]. On the other hand, free radical-induced oxidative injury is found to be more severe and persistent in mitochondrial DNA (mtDNA) compared to nuclear DNA (nDNA) since mtDNA encodes for several proteins critical for electron transport and ROS production [57,58]. Therefore, oxidative damage to mtDNA results in a vicious cycle involving ROS generation and organelle dysregulation, ultimately triggering the apoptosis process [59].
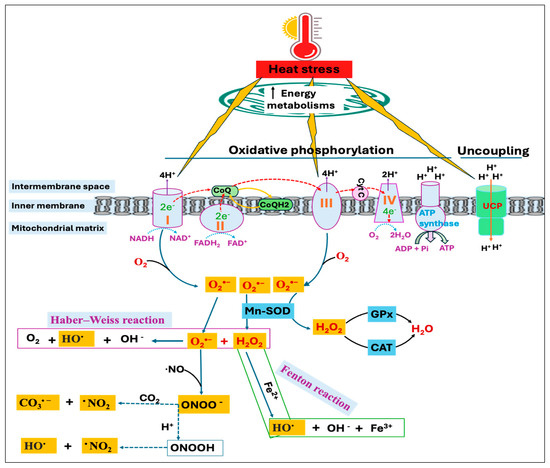
Figure 2.
Heat stress (HS)-induced reactive oxygen species (ROS) production in mitochondria. Electrons transport through complex-I, -III, and -IV drives proton transfer to intermembrane space, creating an electrochemical proton gradient that ultimately moves protons back through ATP synthase to generate ATP. Uncoupling protein (UCP), through proton leakage, uncouples oxidative phosphorylation and regulates mitochondrial ROS production. Heat stress increases energy metabolisms, affecting the enzymatic activity of respiratory chain complexes. This alters the UCP expression as well as increases electron leakage from complex-I and complex-III, resulting in superoxide (O2•−) production, which can have several fates. Mitochondrial manganese superoxide dismutase (Mn-SOD) catalyzes the conversion of O2•− to hydrogen peroxide (H2O2), which can be reduced to water by several cellular enzymes, including the glutathione peroxidase (GPx) and catalase (CAT). H2O2 can also result in the generation of hydroxyl radical (HO•) with the availability of transition metals through Fenton chemistry or by reacting with the O2•− via Haber–Weiss reaction. Rapid inactivation of nitric oxide (•NO) in the presence of O2•− resulted in the yield of peroxynitrite (ONOO−), a strong oxidant that subsequently undergoes protonation or decomposes into other reactive species, such as carbonate radical (CO3•−) and nitrogen dioxide radical (•NO2).
2.2. NADPH Oxidase (NOX)
NADPH oxidase (NOX) proteins are a group of enzymes responsible for transferring electrons across membranes to oxygen, yielding O2•− as the end product [60]. The extracellular O2•− is then dismutase to H2O2, which is commonly assumed to passively diffuse back into the cells through the plasma membrane. Emerging studies suggest that aquaporin channels might serve as a preferential route for H2O2 entry into the cells [61,62]. Currently, seven known isoforms of NOX (Nox1–5, Duox1, and Duox2) have been discovered that share a common catalytic core formed by two domains: the dehydrogenase and the transmembrane domain containing the substrates and prosthetic groups that facilitate electron transfer [63]. However, these NOX isoforms are not only regulated differently and have specific cellular distributions, but also produce different types of ROS. Nox4 regulates the basal production of H2O2 [64], and Nox1 and Nox2 produce O2•− [65], while Nox5 generates H2O2 in a Ca2+-dependent manner [66]. Nox2, also known as gp91phox, is the classic NADPH oxidase expressed predominantly in phagocytic cells. The study reported that in addition to mitochondria, NOX4 might also be involved in heat stress-induced ROS production and oxidative stress in avian muscle cells [67]. Other findings suggested that stimulation of mitochondrial O2•− could elevate the activity of NADPH oxidases, resulting in increased O2•− production in the cytosol [30,68].
2.3. Xanthine Oxidase (XO)
Xanthine oxidase (XO, type O), an isoform of xanthine oxidoreductase (XOR), is a key enzyme in purine metabolism catalyzing the oxidation of hypoxanthine to xanthine and xanthine to uric acid with consequent reduction of molecular oxygen [69]. This enzyme transfers electrons directly to O2, reducing it to generate O2•− and H2O2 via a one-electron and a two-electron reduction, respectively [70]. Another isoform of xanthine oxidoreductase, xanthine dehydrogenase (XDH, type D), however, is not involved in ROS production [71]. Under the conditions of hyperthermia, XDH (type-D) was shown to be converted into XO (type O) in rat hepatocytes [72,73]. A limited but constant presence of XO activity was reported in the liver and kidney of chickens [74,75,76]. These observations imply that avian tissues may possess some degree of XO activity under specific circumstances; nevertheless, there is no specific study to determine if HS or hyperthermia can directly activate this enzyme in avian cells.
2.4. Uncoupled Nitric Oxide Synthase (NOS)
Nitric oxide synthase (NOS) is a family of oxidoreductases that catalyzes the synthesis of nitric oxide (NO) and l-citrulline, utilizing l-arginine, molecular oxygen (O2), and NADPH-derived electrons as substrates, while tetrahydrobiopterin (BH4) is a cofactor [77]. There are three distinct subtypes of NOS, namely, inducible NOS (iNOS), neuronal NOS (nNOS), and endothelial NOS (eNOS). The expression of eNOS and nNOS is constitutive to produce their biological effects depending on their site of localization, while iNOS is induced under stress conditions [78]. Under certain conditions such as superoxide-induced oxidation of BH4, changes in the phosphorylation state of NOS, or structural change in the functional dimer, NOS will become uncoupled and lose its ability to convert l-arginine to l-citrulline, rather than accept an electron from NADPH and transfer it to molecular oxygen to yield O2•− instead of NO [79,80]. Thus, the production of O2•− instead of •NO leads to oxidative stress. On the other hand, •NO can itself modulate oxidative stress in the cell by reacting with O2•− to form peroxynitrite (ONOO−). Peroxynitrite is detrimental to the cell and exacerbates oxidative stress, even though it is less toxic compared to the O2•− [81]. Heat stress was shown to upregulate the NOS expression, thereby elevating •NO production in both in vitro and in vivo models [82,83].
2.5. Endoplasmic Reticulum (ER Stress)
The endoplasmic reticulum (ER) is the cellular organelle involved in the folding, modification, and synthesis of secretory and organelle-bound proteins. Protein synthesis and the subsequent folding steps are tightly regulated, and perturbation of ER homeostasis can be detrimental. Under normal circumstances, the cell maintains protein homeostasis within the ER; however, the homeostasis can be disrupted by a variety of physiological and pathological stimuli, leading to the buildup of unfolded and misfolded proteins, a condition known as ER stress [84]. Heat stress particularly elevates mitochondrial ROS production, which in turn induces oxidative stress, resulting in protein denaturation and the subsequent development of ER stress (Figure 1) [85,86]. The proteotoxic stress then activates a series of signaling pathways, known as the unfolded protein response (UPR), that aims at alleviating the stress and regaining ER homeostasis. Three ER transmembrane proteins, namely, inositol requiring 1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6), which are in inactive states during unstressed conditions, trigger UPR activation upon ER stress [87,88]. The UPR attempts to mitigate the buildup of unfolded proteins by lowering the secretory protein production, increasing molecular chaperones and protein processing enzymes as well as accelerating the degradation of misfolded or slowly folding proteins via the lysosomal pathway or ER-associated degradation (ERAD) [89,90]. However, the failure of UPR to reduce ER stress and to restore homeostasis during unresolvable ER stress conditions promotes cell death. In multiple in vitro and in vivo models, ER stress and oxidative stress are cooperative and synergistic in an amplification loop that impairs cell function and promotes pro-apoptotic signaling [34].
Mitochondria can serve as an efficient Ca2+ buffer in various cell types, taking up a significant portion of cytosolic Ca2+ at the expense of mitochondrial membrane potential (ΔΨm) [91]. The most important beneficial effect of Ca2+ in mitochondria is the stimulation of ATP synthesis resulting from the activation of enzymes associated with the Krebs cycle and oxidative phosphorylation [92,93]. However, several studies have reported that Ca2+ overload in mitochondria induces ROS production by activating the mitochondrial permeability transition characterized by the opening of the mitochondrial permeability transition pore (mPTP), a non-selective large conductance pore within the inner membrane. Opening of mPTP eventually leads to the uncoupling of oxidative phosphorylation, the release of cytochrome c and mitochondrial matrix, osmotic swelling as well as physical rupture of the mitochondrial outer membrane [94,95,96,97]. Moreover, Ca2+ overload has been observed to activate degradative enzymes that generate ROS capable of oxidative modifications to mitochondrial proteins and lipids [98,99,100]. During ER stress, Ca2+ released from the ER is obtained by mitochondria via mitochondria-associated ER membranes (MAMs) (Figure 3), resulting in mitochondrial ROS production [101,102].
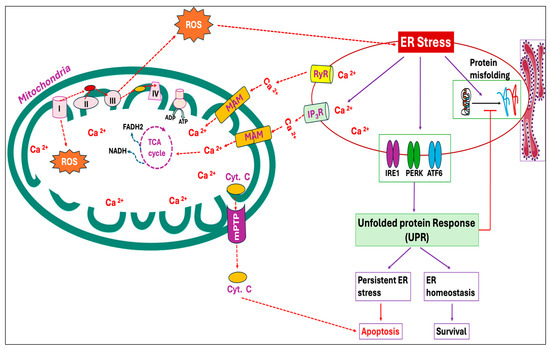
Figure 3.
Endoplasmic reticulum (ER) stress and mitochondrial reactive oxygen species (ROS) production exhibit a positive feed-forward loop. In an attempt to mitigate the accumulation of unfolded proteins and ER stress, three ER transmembrane proteins, inositol requiring 1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6), in the ER lumen are triggered and activate a set of signaling cascades called the unfolded protein response (UPR). The ER represents the primary storage site of calcium (Ca2+), and under ER stress, Ca2+ from ER cisternae flows through the calcium release channels, inositol 1,4,5-trisphosphate receptors (IP3R) and ryanodine receptors (RyR). The Ca2+ thus released from the ER is sequestered by mitochondria through mitochondria-associated ER membranes (MAMs). Calcium overload in mitochondria triggers ROS production by increasing the respiratory chain activity and opening the mitochondrial permeability transition pore (mPTP), causing the release of cytochrome C and other pro-apoptotic factors. This, in turn, exacerbates ER stress, further releasing Ca2+ and optimizing mitochondrial ROS production.
2.6. Oxylipin Pathways
Oxylipins are oxygenated metabolites generated from the oxidation of polyunsaturated fatty acids (PUFAs), particularly arachidonic acid and eicosapentaenoic acid in animals [103]. They are produced in all organisms, and involved in cellular signaling, inflammation, and oxidative stress responses [104,105]. The enzymatic pathways of various oxylipin formation are mediated by three families of enzymes, namely, cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP) [106]. The specific enzyme involved determines the classes of oxylipins produced, leading to variations between different pathways. For example, the enzyme COX metabolizes arachidonic acid into prostanoids such as prostaglandins and thromboxanes, as well as hydroxy-metabolites like 11-hydroxyeicosatetraenoic acid (11-HETE) [107]. However, CYP produces HETEs, epoxyeicosatrienoic acid (EET), and dihydroxyeicosatrienoic acids (DHETs) from arachidonic acid metabolism [108]. LOX catalyzes the synthesis of mid-chain HETEs from arachidonic acid [109], and also mediates the synthesis of hydroxy fatty acids, such as leukotrienes, lipoxins, resolvins, protectins, maresins, hepoxilins, and eoxins [110,111]. These oxylipins can have either pro-inflammatory (prostaglandins, leukotrienes, HETEs) or anti-inflammatory (lipoxins, resolvins, protectins, EETs) properties. Generally, omega-3 fatty acid-derived oxylipins have anti-inflammatory functions, while omega-6 oxylipins are largely pro-inflammatory [112]. Bioactive oxylipins, particularly prostaglandins and leukotrienes, generated from the oxidation of arachidonic acid by enzymes LOX and COX, are shown to induce NOX-dependent ROS production [113,114]. Moreover, studies have demonstrated that prostaglandins induce ROS production by dissipating the mitochondrial membrane potential and redox alteration in cells [115,116]. Heat stress is shown to increase the expression of COX-2 and prostaglandin production in endothelial cells [117]. However, details on the effect of HS on the enzymatic production of oxylipins and oxylipin-mediated ROS generation in chickens remain scarce.
The non-enzymatic pathway of oxylipin production is mainly catalyzed via free radicals produced from heat, radiation, or metal ions [118]. The hydroxyl radicals (HO•) are the most potent initiators of lipid peroxidation, with glycolipids, phospholipids, and cholesterol being highly susceptible targets of detrimental peroxidative changes [119]. Among the various products of lipid peroxidation, hydroperoxides (LOOH) are the main primary products, while MDA and 4-hydroxynonenal (HNE) are formed as secondary products [120].
2.7. Cytochrome P450 Electron Transport System
Cytochrome P450s (CYPs) are hemoproteins belonging to one of the largest enzyme families, and they possess heme-thiolate as a cofactor. They transfer electrons from pyridine nucleotides (NADPH/NADH) to catalyze diverse reactions, such as hydroxylation, dealkylation, deamination, sulfoxidation, and peroxidation [121,122]. Consequently, they can act on multiple substrates, including fatty acids, steroids, drugs, organic solvents, and carcinogens, making the CYP enzymes important in xenobiotics and drug detoxification [123]. These enzymes are external monooxygenases that mediate oxidation reactions by incorporating one oxygen atom into a substrate while reducing the other oxygen atom to water [121]. Genome-wide analyses have identified 45 CYP genes in the chicken genome, revealing the distinct features of chicken CYPs, including their rapid evolution and significant divergence [124]. It has been shown that the uncoupling of CYP reactions resulted in the production of O2•− and H2O2 [125]. Conversely, certain CYPs have been identified for their protective role against oxidative stress [126,127]. Research focusing on the effect of HS on chicken P450 enzymes is limited, particularly in understanding whether HS induces the uncoupling of P450 reactions to generate ROS or activates P450 enzymes to detoxify these ROS.
3. Filling the Gaps–Exploring the Potential Mechanisms
Heat stress, through the activation of enzymatic systems, results in oxidative stress. However, the exact mechanisms underlying the activation of these enzymatic systems under HS have not been fully established. Open mouth breathing (panting), increased peripheral circulation (wings flutter in birds), release of stress hormone (corticosterone/glucocorticoids), and reduced feed intake are some of the major immediate responses of animals, including the chickens, during HS. Discovering downstream mechanisms associated with these initial responses would potentially uncover the activation of enzymatic oxidant systems during HS.
3.1. Panting
Since birds lack sweat glands, panting is the major behavioral thermoregulatory response for evaporative cooling from the respiratory tract [128]. However, it results in hyperventilation and excessive loss of carbon dioxide (CO2), ultimately leading to respiratory alkalosis (↓ H+, ↑ HCO3−, ↑ blood pH) [129,130]. To counteract this imbalance and to maintain the normal blood pH, the kidneys increase the secretion of bicarbonate (HCO3−) ions, while decreasing the secretion of renal hydrogen (H+) ions [131]. However, this renal metabolic process develops over several days, whereas respiratory disorders have a faster impact, fluctuating CO2 levels in minutes to hours. As a result, acute respiratory alkalosis is associated with elevated HCO3− levels since there has not been enough time to lower the HCO3− levels, whereas chronic respiratory alkalosis typically involves lower-to-normal HCO3− levels [132,133]. Some of the studies reported that hyperventilation-induced respiratory alkalosis leads to the development of compensatory metabolic acidosis [134,135]. This is because the process of HCO3− excretion is followed by Cl− retention, which drives a net increase in Cl−-containing acid [136]. Moreover, the fall of pCO2 levels (hypocapnia) under hyperventilation is found to be associated with increased lactate concentration with accompanying [HCO3−] reduction in both systemic blood and cerebrospinal fluid [137,138]. Nevertheless, the current knowledge of renal acid–base balance in chickens is limited and requires further investigation. Moreover, it remains unexplored whether respiratory alkalosis has any impact on ROS production, although metabolic acidosis is shown to exhibit both direct and indirect effects on cellular ROS production (Figure 4a).
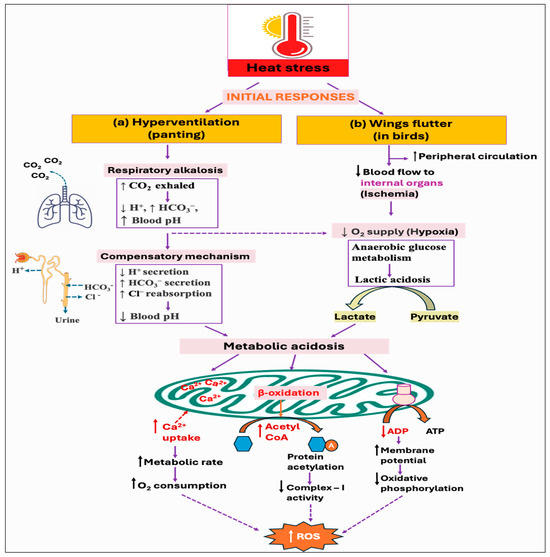
Figure 4.
Potential downstream mechanisms associated with the initial responses, hyperventilation, and wings flutter, against heat stress: (a) Panting, as a way of evaporative cooling, leads to hyperventilation. Excessive loss of carbon dioxide (CO2) develops in respiratory alkalosis, which could eventually lead to metabolic acidosis through decreasing hydrogen ion (H+) secretion while increasing bicarbonate ion (HCO3−) secretion and chloride ion (Cl−) reabsorption from the kidney. (b) On the other hand, as a result of increased peripheral circulation, the blood supply to the internal organs is reduced, creating a hypoxic environment. Cells metabolize glucose anaerobically under hypoxia, resulting in the production and accumulation of lactic acid. The resultant acidosis condition stimulates mitochondria to uptake more calcium, which increases metabolic activity and subsequent reactive oxygen species (ROS) production. Acidosis can also alter oxidative phosphorylation by reducing the signal for ATP synthesis (i.e., ADP), and by modulating the mitochondrial fatty acids oxidation that decreases complex-I activity by protein acetylation. All of these mechanisms result in increased ROS generation.
3.2. Increased Peripheral Circulation (Wings Fluttering)
Heat stress leads to increased peripheral circulation to dissipate the excess heat, thus reducing the blood flow (ischemia) to internal organs, including the liver, kidney, and gastrointestinal (GI) tract [139]. Alternatively, thermal panting-induced hypocapnia and alkalosis might elevate oxygen demands due to the added workload on the respiratory muscles [140,141], and shift the oxyhemoglobin dissociation curve to the left, reducing oxygen delivery to perfused tissues [142,143]. The resulting hypoxic condition (lack of oxygen) induces the cells to metabolize glucose anaerobically and produce lactate, thus resulting in lactic acidosis [144,145]. However, there is a contrasting finding suggesting that the blood flow distribution in chicken remained unchanged in most organs during hyperventilation-induced hypocapnic alkalosis [146].
Some studies have provided various insights into how metabolic acidosis exerts substantial stress on mitochondria. Acidosis is shown to inhibit oxidative phosphorylation in skeletal muscle and may decrease the mitochondrial oxidative capacity [147,148,149]. During acidosis, there is a reduction in the specific signal for oxidative phosphorylation (i.e., [ADP]) because of the pH-induced shift in creatine kinase (CK) equilibrium, resulting in a lower signal for ATP supply from mitochondria [150,151]. The physiological rate of mitochondrial ROS production is inversely associated with the cytosolic ADP availability [152]. Thus, a reduction in the ADP levels leads to a rise in mitochondrial membrane potential that in turn decreases the respiratory rate and stimulates ROS production due to the highly reduced state of the electron transport chain components. Moreover, mitochondria increase Ca2+ uptake in response to acid load and to counteract high cytosolic Ca2+ influx, which, in turn, stimulates ROS generation from the respiratory chain [153]. It has been suggested that Ca2+ induces ATP synthesis through the activation of enzymes involved in the mitochondrial Krebs cycle and oxidative phosphorylation. This increase in metabolic rate consumes more oxygen, thereby increasing electron leakage and free radical generation from the electron transport chain components [154]. Furthermore, Ca2+ also changes the three-dimensional conformation of the electron transport chain complexes, inducing mitochondrial ROS production. Moreover, acidosis was found to modulate fatty acid metabolism in cells and decrease the respiratory chain complex-I activity through hyperacetylation of proteins [155], as summarized in Figure 4b.
3.3. Stress Hormone
Stressors, including the HS, have been identified to activate the hypothalamus–pituitary–adrenal (HPA) axis that is accountable for the secretion and regulation of glucocorticoid (GC) hormone, also called cortisol or corticosterone, from the adrenal gland into the bloodstream [156,157]. Glucocorticoids (GCs) are well known to act through the glucocorticoid receptor (GR, NR3C1). When not bound to a ligand, the GR is mainly located in the cytosol as part of a large heterocomplex that includes chaperon proteins (HSP90, HSP70, p23) and immunophilins (FKBP51 and FKBP52) [90,158]. The binding of GC induces a conformational change in the GR, leading its associated proteins to dissociate. Subsequently, the GR translocates quickly into the nucleus, where it directly binds to glucocorticoid response elements (GREs) and mediates target gene expression, a process termed trans-activation [159]. Moreover, the GR can also modulate the target gene’s transcription by binding with other transcription factors, such as the STAT family, AP1, and NF-κB, thereby inhibiting them from interaction with their binding site on DNA, a process called trans-repression [160]. In addition, GRs are also found in mitochondria, so they can directly mediate the effect of GC on mitochondria by transcribing the genes encoded by mitochondrial DNA (mtDNA) [161,162]. It has been shown that GCs exert a biphasic response in regulating mitochondrial metabolic activities. The enzymatic activity of specific respiratory chain complex subunits and mitochondrial biogenesis was shown to be stimulated by short-term exposure to stress-level GCs, while extended GC exposure led to mitochondrial structural alterations, respiratory chain dysfunction, excessive ROS production, apoptosis, and eventual cell death [163,164]. Harmful impacts of GCs on mitochondria are exerted through downregulating the binding of GR with the cytoprotective protein, B-cell lymphoma 2 (BCL2), which has been known to prevent the formation of Bax-containing pores on the outer membrane of mitochondria, thus decreasing the leakage of calcium and cytochrome C from mitochondria [165,166]. Additionally, GCs have been found to suppress the activities of cytochrome c oxidase as well as complex-I and -V [167,168]. Beyond these mechanisms, binding of GC with the GR in the cell membrane exerts several rapid, nongenomic effects through alterations in kinase activity, including phosphatidylinositol 3-kinase (PI3K), protein kinase B (AKT), and MAP kinases (MAPKs) [160].
Studies have further demonstrated that GC enhances ROS generation through the activation of the NOX enzyme, and by inhibiting coupling to ATP formation during oxidative phosphorylation [169]. The GC-GR-induced ROS production is presumably mediated by src/syk/PI3K/AKT signaling pathways through NOX activation [169]. Glucocorticoids have also been reported to induce ROS production by limiting BH4 availability through the inhibition of inflammatory mediators, namely, GTP cyclohydrolase I (GCH1), the rate-limiting enzyme for de novo BH4 synthesis [170]. This limitation, in conjunction with the decreased availability of L-arginine due to the inhibition of cytokines affecting the CAT-1 arginine uptake transporter, leads to NOS uncoupling [170,171]. These effects are attributed to the anti-inflammatory properties of GCs, mediated either by genomic interaction of GR and NF-kB or through non-genomic anti-inflammatory pathways (Figure 5). Another mechanism whereby GCs exert a deleterious effect was found to be initiated by extracellular glucose. The stress-elicited rise in GCs subsequently increases blood glucose levels through gluconeogenesis, which has indirectly been demonstrated to decrease mitochondrial motility [167].
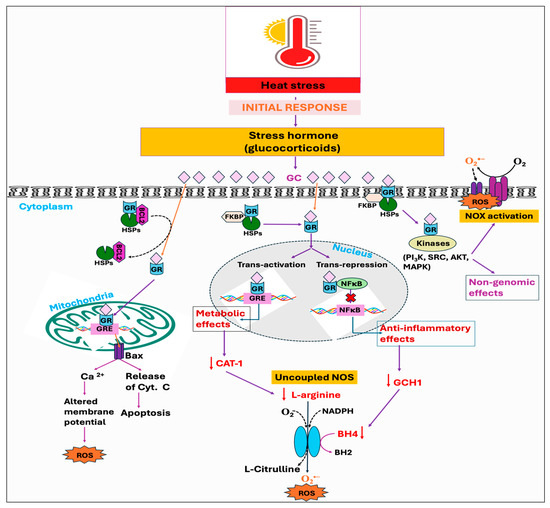
Figure 5.
Potential downstream mechanisms associated with the initial response, stress hormone, against heat stress. The conventional and well-established mode of action of glucocorticoid (GC) is through binding to its receptor, glucocorticoid receptors (GRs). Upon nuclear localization and binding to its genomic response element (GRE), the GC-GR complex exhibits various anti-inflammatory and metabolic effects, including the deficiency of GTP cyclohydrolase I (GCH1) and downregulation of cationic amino acid transporter 1 (CAT-1). This, in turn, decreases the bioavailability of tetrahydrobiopterin (BH4) and L-arginine respectively, leading to nitric oxide synthase (NOS) uncoupling. Moreover, GR associated with heat shock protein (HSP) in mitochondria forms a complex with the anti-apoptotic protein B-cell lymphoma 2 (BCL2). Chronic exposure to the GC downregulates the binding of GR with the Bcl-2, which leads to the formation of Bax pores and subsequent leakage of calcium ions and cytochrome c. The GC also exerts its rapid non-genomic effects, such as NADPH oxidase (NOX) activation, via modulation of various kinases. Therefore, the combined effects of mitochondrial dysfunction, NOS uncoupling, and NOX activation ultimately lead to a rise in reactive oxygen species (ROS) production.
3.4. Reduced Feed Intake
Reduced feed intake during HS is the adaptive strategy to minimize the generation of internal heat by decreasing metabolic activities in animals [172,173]. This is because HS increases the secretion of leptin and adiponectin, along with their receptor expression [174,175], while leptin stimulates the hypothalamic axis and reduces feed intake [176]. The energy deficit condition activates glucagon secretion from pancreatic alpha cells, resulting in lipolysis and protein breakdown in muscle [177,178,179]. Importantly, glucagon has been shown to enhance plasma lipoprotein lipase-like activity both in vivo and in vitro [180]. Moreover, the lipoprotein lipase activity is found to be elevated in plasma, muscle, and heart under stressful condition [181], thereby increasing the breakdown of triglycerides (TG) into free fatty acids (FFAs) and glycerol, and FFAs are then utilized by peripheral tissues as an energy source [182]. The elevated concentration of FFAs in circulation also increases their uptake, re-esterification into TG, and subsequent storage in adipose tissues, resulting in greater fat deposition in HS birds [183,184]. Conversely, an increased amount of free fatty acids (FFAs), particularly polyunsaturated fatty acids (PUFAs), are shown to activate the NOX-dependent ROS generation in neutrophils [185,186]. This is because the enzymatic oxidation of PUFAs produces bioactive oxylipins, such as leukotriene B4 (LTB4), which triggers NOX activation [187,188].
In addition, oxidation of FFAs in mitochondria interrupts the electron transport from complexes I and III, thereby augmenting the ROS production [189,190,191]. Because glycolysis is the central metabolic pathway, the respiratory chain is optimized for glucose oxidation, exporting cytoplasmic NADH to the mitochondria via the malate/aspartate shuttle. This means that for every two electrons that enter from FADH2, ten electrons enter through NADH. This default chain is therefore optimal at a FADH2:NADH ratio of 0.2. Using long-chain fatty acids (LCFAs), such as palmitic acid (16 carbons), results in an FADH2:NADH ratio of ~0.48 [192]. Therefore, this increased FADH2:NADH ratio resulting from the oxidation of saturated FAs in mitochondria, in turn, decreases the CoQ to CoQH2 ratio (a high CoQH2:CoQ ratio), which reduces the concentration of electron acceptors for complex-I, leading to the formation of free radicals [192]. In addition, an increased CoQH2:CoQ ratio promotes reverse electron transfer (RET)-induced mitochondrial ROS along with the elevated membrane potential [193]. The resulting ROS production from the oxidation of FFAs was demonstrated to mobilize and release the cytochrome c from mitochondria via an unknown mechanism, thereby disturbing electron transport from complex-III to complex-IV [194,195]. Moreover, palmitate-induced ROS formation impairs ER redox status and affects protein folding machinery and augments misfolded or unfolded proteins, leading to ER stress [34,196]. This ER stress can exacerbate mitochondrial dysfunction, further stimulating the generation of mitochondrial ROS [35,36]. Conversely, unsaturated fatty acids are shown to prevent ROS generation, thereby antagonizing the detrimental effects of saturated fatty acid oxidation [197,198].
In addition, it has been revealed that fatty acid oxidation enhances the acetylation of mitochondrial proteins, which are enzymes involved in energy metabolism [199]. There is direct evidence that indicated acetyl-CoA produced through β-oxidation of fatty acid drives mitochondrial protein acetylation [199,200] by both enzymatic (acetyltransferase-catalyzed acetylation) [201] and non-enzymatic [202,203] mechanisms. This increased acetylation of mitochondrial proteins reduces the activities of the ETC, which in turn inhibits the oxidation of NADH and a subsequent rise in ROS production [204]. Moreover, the resulting complex-I defect causes the NAD+:NADH ratio to decrease, suppressing the NAD+-dependent deacetylase sirtuin 3 (SIRT3) (Figure 6). SIRT3 exhibits potent deacetylase activity in mitochondria and overturns the inhibitory effects of acetylation, thereby enhancing oxidative metabolism along with the accompanying stimulation of ROS-mitigating systems, including SOD2 [205,206]. From the above-mentioned findings, it is promising that FFA oxidation increases ROS production; however, there are contrary findings indicating that an increase in β-oxidation alleviates oxidative stress, whereas limiting the mitochondrial fatty acid uptake intensifies ROS production [207,208].
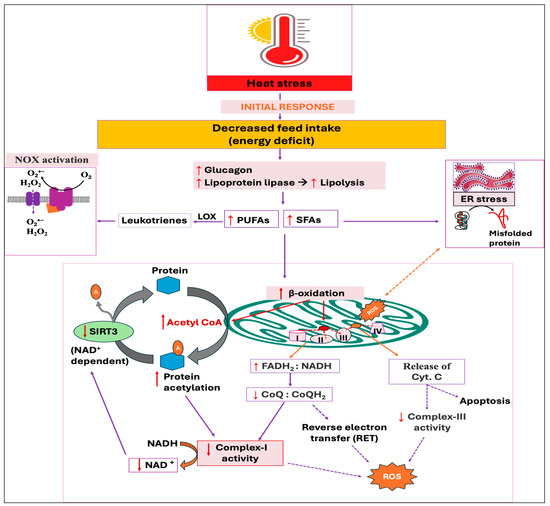
Figure 6.
Potential downstream mechanisms associated with the initial response, decreased feed intake, against heat stress. Energy deficit condition activates lipolysis and subsequent release of free fatty acids (FFAs), with polyunsaturated fatty acids (PUFAs) activating NADPH oxidase (NOX) and saturated fatty acids (SFAs) triggering endoplasmic reticulum (ER) stress. The subsequent beta-oxidation of FFAs, especially the SFAs, increases the flavin adenine dinucleotide and reduced nicotinamide adenine dinucleotide ratio (FADH2:NADH), which reduces the number of electron acceptors for complex-I, ultimately increasing electron leakage and reactive oxygen species (ROS) production. Excessive acetyl-CoA generated from beta-oxidation increases the acetylation of mitochondrial proteins via both enzymatic and non-enzymatic mechanisms, which further reduces complex-I activity. The decreased NAD+:NADH ratio resulting from complex-I defect suppresses the deacetylase sirtuin 3 (SIRT3) enzyme, aggravating protein acetylation and ROS production. The cytochrome-C that transfers electrons from complex-III to -IV is released from the mitochondria as a result of ROS accumulation, finally inducing apoptosis.
4. Indicators of Oxidative Stress
Elevated ROS production usually results in the oxidation of DNA, proteins, carbohydrates, and lipids, leading to a bioenergetic failure [209]. Biomarkers such as protein carbonyl (PC), malondialdehyde (MDA), 8-hydroxy-2′-deoxyguanosine (8-OHdG), and advanced glycation end product (AGEs) are used to assess the degree of protein, lipid, DNA, and carbohydrate oxidation, respectively [13,15].
4.1. Lipids
The primary phospholipid constituents of mitochondrial membranes are abundant in unsaturated fatty acids, making them vulnerable to free radical injury through the peroxidation of double bonds [210]. The major product of polyunsaturated fatty acid (PUFA) peroxidation is MDA, and HS is shown to increase both the mitochondrial and plasmatic MDA levels significantly [211]. This increased level of MDA then reacts with DNA bases, leading to gene mutation [212], whereas the other product of lipid peroxidation, 4-hydroxy-2-nonenal (HNE), reacts with various amino acids such as cysteine (Cys), histidine, and lysine residues, thus affecting the functions of the enzyme [213].
4.2. Proteins
ROS can oxidize amino acids (AAs), especially sulfur-containing AAs, protein backbone, and cross-link the proteins [214]. Protein oxidation can be either reversible or irreversible. It primarily involves the oxidation of cysteine thiols (-SH) into cysteine sulfenic acid (R-SOH), which then may react with nearby thiols to form disulfide bonds (S-S) or be irreversibly converted to sulfinic (R-SO2H) and sulfonic acids (R-SO3H) [215,216]. Reversible protein oxidation involves glutathionylation, nitrosylation, sulfenation, and disulfide formation of cysteine residue, while irreversible protein oxidation includes carbonylation of arginine, lysine, proline, or threonine residues and nitration of tyrosine residue [217,218]. Moreover, ROS may oxidize mitochondrial proteins directly to form disulfide bonds between cysteine residues, methionine sulphoxide from methionine residues, and carbonyl groups in the side chains of serine, arginine, lysine, proline, histidine, and threonine residues [219,220]. Aggregation and accumulation of these oxidized protein derivatives can eventually cause cell death [221]. Because protein oxidation results in the formation of carbonyl groups, PCs are easily detectable and have been employed as a quantitative indicator of protein oxidation and oxidative stress [222].
4.3. Carbohydrates
Glycation or non-enzymatic glycosylation is the process where a reducing sugar covalently attaches with amino groups in proteins, lipids, or DNA macromolecules through the Maillard reaction, leading to the formation of AGEs [223]. Although AGEs are continuously produced as a normal by-product of metabolic processes, their formation was found to be elevated under conditions of hyperglycemia and oxidative stress [224,225]. In turn, AGEs bind with their receptor RAGE, activate pro-inflammatory and pro-oxidant pathways, and further amplify oxidative stress, generating a synergistic feed-forward loop that advances their pathophysiological functions [226,227]. Moreover, aggregation of AGEs has been associated with protein misfolding, ER stress, mitochondrial dysfunction, and cellular apoptosis [228,229].
4.4. DNA
Free radical-mediated DNA oxidation can alter purine and pyrimidine bases [230], or result in double-strand DNA breaks by directly damaging the sugar backbone with high-energy impact [231]. Moreover, the accumulation of ROS can damage not only nuclear DNA, it also lead to mitochondrial DNA lesions, strand breaks, and degradation [232]. The primary products resulting from DNA base damage include thymine glycol among pyrimidines [233] and 8-OHdG among purines [234]. The enzymatic repair systems can excise the 8-OHdG residues from the DNA, allowing them to circulate in the blood and be excreted in the urine [235].
4.5. Findings in Chickens
There are extensive studies on HS-induced oxidative stress in chickens, focusing particularly on the mRNA expression and enzymatic activity of various genes, yet the knowledge regarding mitochondrial ROS production in avian cells remains limited. Avian mitochondria were found to generate significantly less ROS than mammals, with rat mitochondria producing nearly four times more hydrogen peroxide than pigeon mitochondria when using pyruvate/malate as substrates [236]. This lower ROS production has been related to the greater longevity of the birds as compared to mammals of the same body mass [237]; however, no difference in mitochondrial ROS production was observed between long-living parrots and short-living quails [238]. Moreover, liver mitochondrial membranes in pigeons exhibited more resistance to lipid peroxidation compared to rats because of the lower level of fatty acid unsaturation in the pigeon’s membrane [239]. When exposed to pro-oxidants like 95% oxygen, H2O2, and paraquat, avian cells from long-lived bird species demonstrated greater resistance to oxidative stress and DNA damage compared to mouse cells [240]. Notably, there are no supporting data directly comparing ROS production between commercial chickens and mammals. Commercial broilers or layers, which are genetically selected for their rapid growth and higher production, may exhibit distinct oxidative stress dynamics compared to their wild-type counterparts.
In the skeletal muscle mitochondria of heat-stressed broilers, there was a slight increase (2.7%) in mitochondrial membrane potential, leading to a significant rise (47%) in ROS production [241]. However, the increase in mitochondrial membrane potential and the subsequent rise in ROS production persisted only until day 9 of HS, suggesting that chickens may adapt to HS over time [242]. The avian uncoupling protein (AvUCP) that reduces ROS levels by generating a proton leak was downregulated in the muscle cells post-HS [243]. However, another study has found the upregulation of AvUCP and downregulation of NOX4 mRNA expressions during HS [244]. The variation in results among separate studies might be attributed to different experimental designs and differences in the duration of HS.
Heat stress was found to significantly increase the H2O2 and PC concentration in the liver of broilers while the MDA and 8-OHdG levels were not changed [245]. However, other studies identified that HS led to a significant increase in both the 8-OHdG content and AGE level in the chicken liver, suggesting severe oxidative damage to DNA and carbohydrates [14]. Moreover, HS significantly increased the levels of muscle carbonyl (2.39 vs. 1.91 nmol/mg), AGE (1.91 vs. 1.6 ng/mg), and MDA (222.9 vs. 181.63 nmol/g) compared to the control group [246]. Similarly, broilers exposed to chronic HS exhibited a higher MDA level and increased concentration of ROS with reduced concentration of ATP in the liver [247]. They further identified that chronic HS enhanced the mRNA expression of ER transmembrane proteins and UPR signaling components, such as PERK, ATF4, IRE1, and eukaryotic translation initiation factor 2 alpha (eIF2α). However, the mRNA expression of PERK and eIF2α in the P. major showed no significant differences between the HS and control groups, while the expression of ATF4 was significantly higher in the HS group [248]. Acute HS reduced the total nitric oxide synthase (tNOS) activity while elevating the plasma NO levels in broilers [249]. There was an increase in blood glucose levels in HS chickens compared to the control group, likely due to a stress-induced rise in glucocorticoids [250]. Finally, hyperventilation-induced respiratory alkalosis under HS conditions has been shown to reduce the activity of carbonic anhydrase, which is a critical enzyme in forming the eggshell [251]. As a result, lower concentrations of calcium and carbonate are secreted by the shell gland, resulting in thin and weak eggshells [252,253].
5. The Antioxidant Defense Mechanisms
Cells have both enzymatic and non-enzymatic antioxidant systems that function cooperatively to protect cells from oxidative injury. Antioxidants exert their effects via various mechanisms: the chain-breaking mechanism in which an electron is donated to the free radical, thereby stabilizing it and preventing the continuation of the reaction; a quenching mechanism involving deactivation of the catalyst that initiates radical formation; a co-antioxidant action; or regulation of gene expression [254,255]. The major enzymatic antioxidants consist of superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), and peroxiredoxin (PrDx), whereas the non-enzymatic antioxidants comprise glutathione (GSH), thioredoxin (Trx), lipoic acid, vitamins C and E, carotenoids, melatonin, flavonoids, and other compounds. Many authors have regarded the enzymatic antioxidants, namely, SOD, CAT, and GPx, as a first line of antioxidant defense system because of their prime importance in the overall antioxidant defense strategy, especially about O2•−. However, PrDx represents another important enzyme system, which in addition to SOD, CAT, and GPx, performs a crucial role in protecting cells against oxidative stress. Furthermore, the function of GPx and PrDx is exclusively dependent on non-enzymatic antioxidants such as GSH and Trx, and there exists a significant interplay between the enzymatic and non-enzymatic antioxidant systems (Figure 7). In this Section, we discuss various mechanisms associated with the enzymatic and non-enzymatic antioxidant systems.
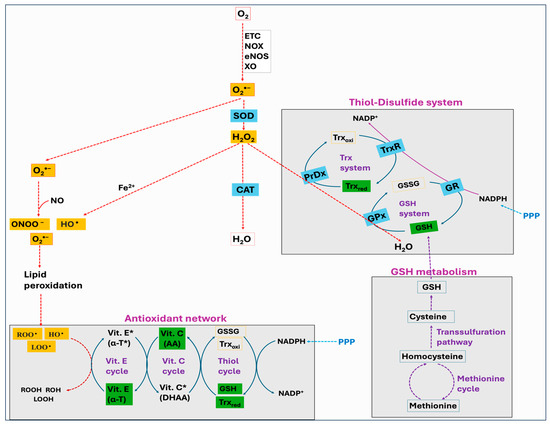
Figure 7.
Schematic representation of major enzymatic and non-enzymatic antioxidant defense systems. Superoxide dismutase (SOD) is the forefront antioxidant that neutralizes superoxide (O2•−) to hydrogen peroxide (H2O2), which is then detoxified to water by catalase (CAT) and the thiol–disulfide system, involving glutathione peroxidase (GPx) and peroxiredoxin (PrDx). The antioxidant network refers to the synergistic relationship between different antioxidants such as vitamin E, vitamin C, and glutathione (GSH), where one replenishes the original properties of another. Vitamin E scavenges lipid peroxyl radicals (LOO•), peroxyl radicals (ROO•), and hydroxyl radicals (HO•) via a chain breaking mechanism and protects the cellular membranes. Cellular GSH, synthesized endogenously from methionine and cysteine, and cellular thioredoxin (Trx) are the important components of the thiol–disulfide system and the antioxidant network. Nicotinamide adenine dinucleotide phosphate (NADPH) serves as a reducing agent by donating electrons to oxidized Trx via thioredoxin reductase (TrxR), or to oxidized glutathione (GSSG) via glutathione reductase (GR).
5.1. Superoxide Dismutase (SOD)
The SODs represent the primary and most crucial antioxidant enzymes in defending against ROS, especially O2•− radicals. They catalyze the breakdown of O2•− into H2O2, which is then further detoxified by the complementary actions of CAT and GPx [256]. Depending on the metal cofactor and cellular localization, three major types of SODs are identified, which include copper–zinc SOD (Cu/Zn-SOD or SOD1) located primarily in cytoplasm or secreted into the extracellular fluid, manganese-containing SOD (Mn-SOD or SOD2) located in the mitochondrial matrix, and an extracellular SOD (EC-SOD or SOD3) [257].
5.2. Catalase (CAT), Glutathione Peroxidase (GPx), and Peroxiredoxin (PrDx)
Three major antioxidant enzymes, namely, CAT, GPx, and PrDx, are primarily responsible for intracellular peroxide metabolism. The CAT is an iron-containing peroxidase, found primarily in peroxisomes, that catalyzes H2O2 into H2O and O2. However, CAT is activated only when the concentration of H2O2 is significantly higher than physiological thresholds, and it is expressed much lower compared to GPx and PrDx [258]. While CAT operates independently of reducing agents and catalyzes H2O2 exclusively, GPx and PrDx are dependent on intracellular electron donors to perform their peroxidase activity targeting both H2O2 and organic peroxides. Antioxidant GPx is a selenium-containing peroxidases family, and unlike CAT, GPx senses a small increase in H2O2 concentration [258]. The PrDx, distinct from heme-dependent CAT and the selenium-dependent GPx, does not require cofactors. For a long time, the role of PrDx was overshadowed by the well-studied oxidative stress defense enzymes CAT and GPx, which are considered as the major defender of cells against hydroperoxides. However, PrDxs have now been identified as ubiquitous enzymes in numerous organisms, involved in the neutralization of H2O2, alkyl hydroperoxides (ROOH), and ONOO− [259,260,261]. The GPx and PrDx are the components of the cellular thiol–disulfide redox system, the details of which will be discussed herein.
5.3. Thiol–Disulfide Redox System
Thiols are organosulfur compounds containing a constituent sulfhydryl (R-SH) group, which renders thiols highly prone to oxidation. They are present in all biological systems, and some of the physiologically significant thiols consist of amino acids such as cysteine (Cys) and homocysteine (Hcy), coenzyme A, and dihydrolipoic acid [262]. Thiols can function as reducing agents by donating electrons to ROS, thereby neutralizing ROS into less toxic byproducts while they become oxidized to form disulfides (R-S-S-R). Structural disulfide bond formation in proteins is catalyzed by different types of oxidoreductases that encompass several protein disulfide isomerase (PDI) oxidases, including PrDx and GPx [263,264,265]. In biological systems, particular reductases restore disulfides to reduced thiols by using cellular reducing agents, including NADPH or NADH. The switch between thiols (dithiol) and disulfide groups represents a redox reaction, with a reduced state of free dithiol and the oxidized state of disulfide form (Figure 8). This reaction aids in the maintenance of a balanced oxidoreductive or redox environment inside the cell [266]. The Trx and GSH systems primarily regulate the thiol–disulfide-mediated redox status of the cell [267,268]. Although these systems operate via different mechanisms, they both utilize NADPH, mainly derived from the pentose phosphate pathway (PPP), as a reducing agent (Figure 9).
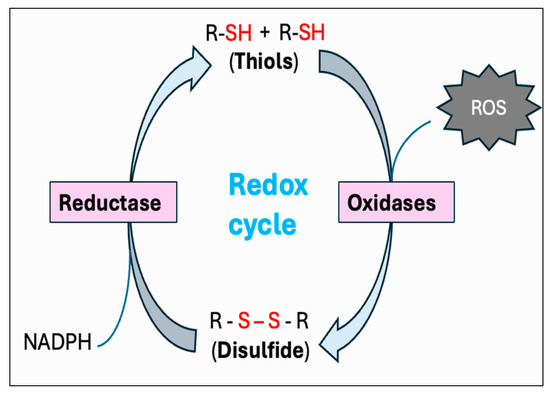
Figure 8.
Thiol–disulfide-mediated redox cycle. By donating electrons to reactive oxygen species (ROS), thiols (R-SH) function as reducing agents to neutralize ROS into less toxic byproducts, while they become oxidized to form disulfides (R-S-S-R). Conversion of thiols to disulfide is mediated by a variety of protein disulfide isomerase (PDI) oxidases, including glutathione peroxidase (GPx) and peroxiredoxin (PrDx). Specific reductase then restores disulfides to reduced thiols by utilizing the cellular reducing agents, including NADPH. This interconversion between thiols and disulfide represents the redox cycle.
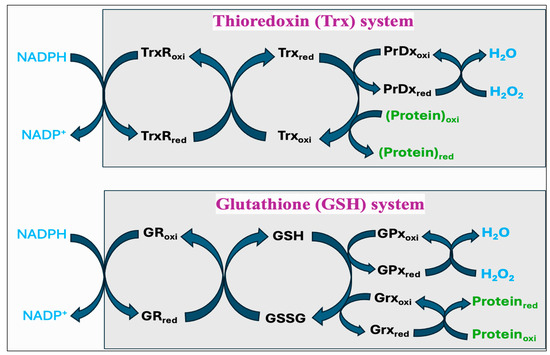
Figure 9.
The thioredoxin (Trx) system and glutathione (GSH) systems in regulating the thiol–disulfide-mediated redox state of the cell. Both systems utilize NADPH as a final electron donor; however, they operate via different components in reducing the proteins and neutralizing hydrogen peroxide (H2O2) to water. The Trx system includes thioredoxin reductase (TrxR), thioredoxin (Trx), and peroxiredoxin (PrDx) while the GSH system comprises glutathione reductase (GR), glutathione (GSH and GSSG), and glutathione peroxidase (GPx) or glutaredoxin (Grx).
The Trx system is composed of NADPH, thioredoxin reductase (TrxR), Trx, and PrDx. Reduced Trxs (Trxred) are potent reductases with broad substrate specificity that efficiently reduce disulfide bonds into thiols [269]. One of the important targets of Trxred is the antioxidant enzyme PrDx, which in turn mediates the reduction of peroxides through receiving electrons from Trxred. Moreover, Trxs are the main protein disulfide reductases in the cell to reduce and activate the proteins, while they become oxidized. The oxidized Trx (Trxoxi) is restored back to its reduced state (Trxred) by an enzyme TrxR, using NADPH as a final electron donor [270]. In mitochondria, it is reported that peroxiredoxin-3 (PrDx-3) scavenges 90% of H2O2, indicating the Trx system as the main pathway to eliminate mitochondrial H2O2 [271].
The GSH system consists of NADPH, glutathione reductase (GR), GSH, and GPx or glutaredoxin (Grx). The GSH system can protect the cells from the detrimental effects of oxidative stress through several pathways. GSH can reduce disulfide bonds in proteins through a Grx-involved reaction. This is crucial to restore the activity of several important proteins and pathways following the alterations in the cellular redox state [272,273,274]. Grx exists in two primary forms, one localized in the cytosol (Grx1) and the other present in the mitochondria and nuclei of mammalian cells (Grx2) [275]. While Grx1 obtains electrons from GSH, Grx2 can accept electrons from both GSH and TrxR2 [276]. The GSH-Grx system can therefore function as an alternative system to decrease the levels of Trx when the electrons flow from TrxR1 is inhibited [277]. Moreover, GSH transfers an electron to GPx that alleviates oxidative stress by neutralizing different types of ROS [267,278]. The GR provides electrons to GSSG by utilizing NADPH as the final electron donor, reconverting GSSG back to GSH to maintain a normal GSH:GSSG ratio [279].
5.4. Significance of Thiol–Disulfide Redox Status
The cellular thiol–disulfide redox balance is shaped by the distributions of thiols and disulfides across various subcellular compartments. The reduced forms of Trx and GSH serve as vital cellular thiol reservoirs, primarily associated with the neutralization of ROS. Reduction in the GSH:GSSG or Trxred:Trxoxi ratios is often linked to the detrimental impacts of oxidative species, eliciting death or survival signals [280]. Consequently, the thiol:disulfide ratio serves as an important indicator of oxidative stress, since maintaining reduced states of GSH and Trx is critical for numerous cellular processes, including the stability and activity of various proteins such as enzymes GPx and PrDx, chaperones, and transcription factors [281]. The ratio of GSH:GSSG in a resting cell is typically higher than 100:1; however, in different oxidative stress models, this ratio was shown to drop as low as 10:1, even to 1:1 [282].
5.5. Methionine Cycle and Transsulfuration Pathway
Methionine is an essential sulfur-containing amino acid that undergoes breakdown and regeneration through a series of metabolic reactions, known as the methionine cycle. In this process, methionine is first transformed into S-adenosyl-methionine (SAM), a universal methyl donor, which subsequently converts to S-adenosyl-homocysteine (SAH) after donating its methyl group. Hydrolysis of SAH then results in homocysteine (Hcy), which either undergoes a transsulfuration pathway to generate cysteine or, is converted back to methionine through a remethylation pathway with a methyl donation from the folate cycle [283]. Another enzyme that catalyzes the remethylation of Hcy to methionine is betaine-homocysteine-S-methyltransferase (BHMT) with betaine as the methyl donor. Additionally, the methionine salvage pathway or 5′-methylthioadenosine (MTA) cycle is involved in the regeneration of methionine from SAM as well as in the synthesis of polyamines [284,285].
The transsulfuration pathway, alternatively known as the cystathionine pathway, enables the cell to use methionine for the synthesis of GSH [286]. This pathway is particularly active in hepatocytes, and either absent or present in small quantities in other tissues outside the liver [287]. It represents a two-step process, converting Hcy into cysteine. In the first step, vitamin B6-dependent enzyme cystathionine β synthase (CBS) facilitates the synthesis of cystathionine by combining Hcy and serine. The second step involves the breakdown of cystathionine by a separate vitamin B6-dependent enzyme γ-cystathionase (also called cystathionine-γ-lyase, CGL), generating free cysteine for the synthesis of GSH [287].
5.6. Cysteine Metabolism and GSH Biosynthesis
The primary pathways by which cysteine is catabolized in the body include its utilization for GSH synthesis or its degradation through cysteine sulfinate-dependent pathways. Since the transsulfuration pathway converts nearly all methionine sulfur into cysteine sulfur before it is oxidized and eliminated, consuming a specific molar amount of methionine generates an almost equivalent molar amount of cysteine [288,289]. The enzyme cysteine dioxygenase (CDO) plays an important role in cysteine catabolism by oxidizing the sulfhydryl group of cysteine to cysteinesulfinate. This precursor is then used in the production of taurine and can be further transaminated into pyruvate and inorganic sulfur [290]. Taurine is an additional antioxidant that reduces oxidative stress by directly detoxifying ROS, enhancing GSH oxidation activity, and maintaining membrane permeability against ROS-induced disruptions [291]. The rate-limiting enzyme in GSH synthesis, glutamate-cysteine ligase (GCL), known as γ-glutamylcysteine synthetase (GCS), also utilizes cysteine as a substrate and hence competes with the enzyme CDO [292]. Thus, the activities of CDO and GCL are critical in regulating the partitioning of cysteine to meet diverse metabolic requirements and also to maintain its concentrations in the body [293]. These two enzymes, CDO and GCL, respond reciprocally with GCL activity decreasing and CDO activity increasing when the dietary protein or sulfur amino acid level is increased [294,295,296].
Glutathione (GSH), also known as γ-L-glutamyl-L-cysteinylglycine, is a tripeptide and the most abundant non-protein thiol that protects cells from oxidative injury, and is found in all mammalian tissues in the ranges of 1–10 mM with the highest concentrations in liver [297]. Synthesis of GSH occurs via a two-step ATP-dependent enzymatic reaction with the initial step driven by the rate-limiting enzyme GCL, catalyzing the formation of γ-glutamyl-L-cysteine from cysteine and glutamate. In the second step of GSH synthesis, the enzyme glutathione synthetase (GSS) catalyzes the addition of glycine to γ-glutamyl-L-cysteine, yielding GSH [297]. Despite its overexpression, GSS failed to elevate the GSH level, whereas the overexpression of GCL did result in a higher GSH level, indicating GCL as the rate-limiting enzyme in the process [298]. However, there is growing evidence that GSS is crucial in regulating the overall GSH synthesis in particular tissues or during stressful periods [299]. Glutathione is present as the thiol-reduced (GSH) and disulfide-oxidized (GSSG) forms, with the predominant thiol-reduced GSH accounting for nearly 98% of total GSH [300,301]. The antioxidant function of GSH is carried through GPx-catalyzed reactions, reducing peroxides with the oxidation of GSH to GSSG. The enzyme GR, in turn, restores GSH from GSSG by using NADPH as a reducing agent, therefore creating a continuous redox cycle [299]. All the major pathways, including the methionine cycle, transsulfuration pathway, cysteine catabolism, and GSH synthesis are summarized in Figure 10.
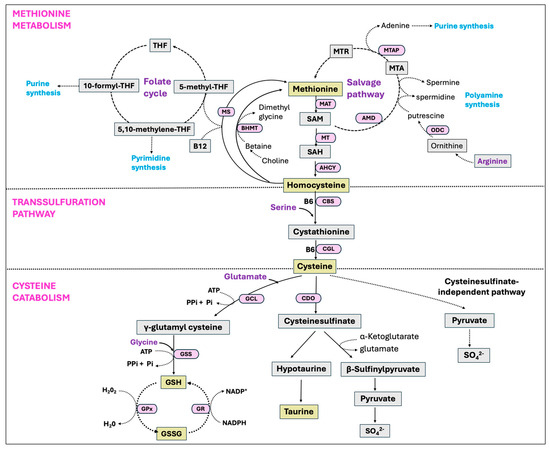
Figure 10.
Methionine metabolism, transsulfuration pathway, and the cysteine catabolic pathways. Methionine is an essential amino acid that is converted to homocysteine through the transmethylation pathway. Homocysteine can then be reconverted back into methionine via remethylation pathways, involving a methyl donation from the folate cycle or the betaine. Methionine can also be regenerated via the salvage pathway involving the recycling of methylthioadenosine (MTA), a by-product of polyamine biosynthesis. Alternatively, homocysteine can be converted into cysteine via the transsulfuration pathway. Cysteine has multiple fates and is catabolized to produce various nonprotein compounds, such as glutathione (GSH), taurine, sulfate, and coenzyme A depending on the physiological and nutritional situations.
5.7. Findings in Chickens
Antioxidant mechanisms in chickens share fundamental similarities with those in mammals, as both rely on enzymatic and non-enzymatic antioxidant systems to combat oxidative stress [302]. A study demonstrated that pigeons have approximately double the activity of SOD and peroxidase compared to rats, while their CAT activity was considerably lower [303]. Early researchers had predicted that species with longer lifespans would possess a stronger antioxidant defense [304,305]. More recent studies between rats and pigeons, however, yielded inconsistent results. Instead of directly correlating with longevity, elevated antioxidant enzyme activity is now widely believed to be an adaptation to greater oxidative stress, suggesting the greater antioxidant defense as a possible indicator of increased oxidative stress rather than enhanced protection [306,307]. In a separate study, rats and pigeons showed similar plasma total antioxidant capacity (0.8 vs. 1.2 mM Trolox equivalents), but pigeons had higher antioxidant capacity (0.9 vs. 2.1 μmol Fe(II)/g tissue) in the skeletal muscle [308]. This study also showed that non-enzymatic antioxidants, including the plasma GSH, were higher in pigeons (11.8 vs. 19.9 mM), while enzymatic antioxidants such as SOD (94.1 vs. 16.8 U/g tissue) and GPx (34.7 vs. 4.6 U/g tissue) were generally higher in rats, especially in heart mitochondria. Moreover, rats showed significantly greater CAT levels in plasma (124.6 vs. 18.5 U/mL), heart (5469 vs. 1778 U/g tissue), and liver (218.6 vs. 34.1 U/g tissue) compared to pigeons. In contrast, pigeons had higher total skeletal muscle SOD (18.3 vs. 28.2 U/mg tissue) and heart SOD (136.8 vs. 271.5 U/g tissue). Despite such variations, total antioxidant status in plasma and liver were similar in both species due to counterbalancing contributions of non-enzymatic and enzymatic antioxidants [308].
Various studies have shown the elevated mRNA expression and enzymatic activity of SOD, CAT, and GPx2 in broilers reared under HS conditions [17,18,309,310]. There was a significantly higher enzymatic activity of SOD (1.30 vs. 1.72 U SOD/mg protein) and CAT (223.45 vs. 292.77 µmol H2O2/min/mg protein) without affecting the GPx activity (299 vs. 293 nmol NADPH oxidized/min × mg protein), while the GSH content (4.56 vs. 2.05 µg GSH/mg protein) was reduced in the liver of heat-stressed broilers [309]. However, a separate study found a decreased activity of GPx (9.62 vs. 5.6 U/mg protein) in the liver of heat-stressed chickens compared to control birds without significantly affecting the GR activity (0.27 vs. 0.17 U/mg protein) [311]. Higher Trx expression in the ovarian follicles of chicken has been linked with improved egg production rates [312]. However, the in vitro study revealed that chickens have extremely low TrxR activities compared to mammalian TrxR activity [313]. Moreover, the expression of PrDx is not tissue-specific in chickens, highlighting its crucial function as a ubiquitous housekeeping gene protective against oxidative injury [314]. In growing chickens, HS negatively affected the mitochondrial Trx system, including Trx2, TrxR2, and PrDx3 [315]. Acute HS was shown to upregulate the protein levels of PrDx1, PrDx3, and PrDx4 in the small yellow follicles of layer chickens [316], whereas chronic HS significantly downregulated the relative mRNA expression of NADPH and GPx genes in broiler chickens [17]. Further, separate studies found the enzymatic activity of GR, GSH, GSSG, and GSH:GSSG ratio to be adversely affected during HS [14,245].
The first limiting amino acid in a typical corn–soya-based poultry feed is methionine, which participates in an important biosynthesis pathway of other essential molecules. Multiple studies have reported the dose-dependent beneficial effects of different isoforms of methionine supplementation in poultry diets during the time of oxidative stress [317,318,319]. Supplementing broilers under acute HS conditions with adequate and high levels of DL-methionine increased the expression of GSS and GPx-7 genes [320]. Cysteine can be produced endogenously from the transsulfuration pathway [321,322]; therefore, it is not considered an essential amino acid in poultry feed. However, for GSH synthesis, the availability of cysteine is regarded as the rate-limiting factor among the three amino acids-cysteine, glutamate, and glycine [323,324]. During periods of oxidative stress, the need for GSH is greater, and cysteine may be limiting when attempting to produce adequate levels of GSH. Among many amino acids, cysteine showed the highest incorporation into the tissues of chickens during HS [325]. Furthermore, there was an increased expression of the hepatic CBS gene in chickens in response to a cysteine-deficient diet [326], suggesting the active intracellular compensatory mechanism to produce sufficient levels of cysteine through the transsulfuration pathway. Numerous studies have identified the beneficial effects of different isoforms of cysteine, such as N-acetylcysteine [327,328] and S-allyl-cysteine [329], under HS conditions. Arginine is also a limiting amino acid in poultry diet because birds lack a functional urea cycle, and are thus unable to synthesize endogenous arginine [330]. Arginine was shown to increase the antioxidant potential of birds by slowing down the cellular nitric oxide breakdown [331], and by increasing the levels of GSH [332]. Various amino acids, phytochemicals, and natural compounds that enhance antioxidant defense mechanisms when supplemented to chickens are summarized in Table 1.

Table 1.
Overview of nutritional supplements that enhance antioxidant capacity in chickens under heat stress conditions.
6. The Antioxidant Network and Energy Metabolism
The principal roles of antioxidants include the prevention of ROS production, free radicals scavenging before they react with biological molecules, and repairing the damage. To achieve these functions, different antioxidants work together within the defense network cooperatively and synergistically. The interaction between different antioxidants helps them to work efficiently against oxidative stress, while also restoring their original properties. This process is generally referred to as the antioxidant network [345,346]. Vitamin E (α-tocopherol) works as a lipophilic chain-breaking antioxidant and prevents peroxidative damage to cell membranes by neutralizing free radicals via hydrogen atom transfer [347]. In doing this, vitamin E produces a nonradical molecule and an α-tocopherol radical, which can interact with another radical to form stable compounds, attack a lipid molecule, or regenerate vitamin E by interacting with reducing agents like vitamin C or ubiquinol [348]. Reports suggest that vitamin E and vitamin C operate together cyclically to achieve the antioxidant function [349,350,351]. Additionally, this interaction between vitamin E and vitamin C is important to recycle and replenish the antioxidant potential of vitamin E.
Vitamin C (ascorbate/ascorbic acid) is a water-soluble antioxidant that contains radical scavenging activity. In addition to directly deactivating the peroxyl radicals formed in the aqueous phase, ascorbate can also recycle α-tocopherol from its radical form (α-tocopherol radical) [352,353]. Ascorbate, upon donating an electron to α-tocopherol, becomes oxidized to a less reactive compound called semidehydroascorbate (SDA) radical. This SDA radical has two potential routes; it can either be regenerated back to ascorbic acid or converted to dehydroascorbate (DHAA). The enzyme DHAA reductase can then catalyze the regeneration of ascorbic acid from DHAA by utilizing GSH as a reducing agent [354,355,356]. The series of oxidation–reduction reactions enabling ascorbic acid and GSH to interact with each other has been well elucidated [357,358,359]. In addition, NADPH, mainly derived from the PPP, plays a key role in the antioxidant network by providing reducing equivalents to restore the reduced forms of GSH or Trx. This series of interconnected reactions regenerates the reduced compounds, thereby enhancing their antioxidant properties (Figure 7). Oxidative stress has been shown to deplete both the NADPH pool and the NADPH:NADP+ ratio, while also altering NADP-dependent metabolic pathways [360,361,362]. Moreover, to regenerate NADPH during the oxidative stress, there was a shift in glycolytic flux toward the oxidative PPP [362,363,364].
Findings in Chickens
Birds can endogenously synthesize vitamin C, although its level depletes quickly during the HS condition [365]. In contrast, chickens cannot synthesize vitamin E, rather they rely on dietary intake to fulfill their requirements [366,367]. Numerous studies revealed the protective role of vitamin E and vitamin C during the period of oxidative stress in chickens [368,369,370]. Vitamin E has been reported to enhance growth rates, egg production, and hatchability [371,372,373]. Dietary supplementation of vitamin E in poultry feed increased α-tocopherol levels, while the MDA concentration was decreased in tissues and serum [373,374,375]. In another study, vitamin E has been found to reduce oxidative stress and histopathological alterations in the duodenum and jejunum induced by the Newcastle disease virus [376]. Vitamin C, on the other hand, has been shown to lower high plasma corticosterone levels, and to maintain normal leukocytic count in chickens [377,378]. The combined supplementation of vitamin C (200 mg/kg) and vitamin E (250 mg/kg) was more effective than individual supplementation in enhancing erythrocytic antioxidant status in colored broiler breeders subjected to hot and humid conditions [370]. Moreover, as compared to supplementation with vitamin E (125 IU/Kg) alone, a combination of vitamin C (200 mg/kg) and vitamin E (125 IU/kg) synergistically reduced the activity of lipid peroxidase (1.7 vs 1.54 nmol/MDA/mg protein) while increasing GR activity (71.08 vs. 79.82 U/mL) in layers under tropical summer conditions [379]. Enzymes and metabolites associated with energy metabolism, such as glucose, glucose-6-phosphate (G6P), and the member of the pentose phosphate pathway were found to be elevated during HS in chickens [248,380]. Moreover, a separate study demonstrated that glucose supplementation during chronic HS improved the production parameters possibly by shifting the excess glucose into protein synthesis [23].
7. Ubiquitin–Proteasome System (UPS) and the Molecular Chaperones
Protein oxidation from free radicals typically results in structural damage and functional alterations, thereby affecting protein homeostasis (proteostasis). This disturbance can lead to numerous detrimental effects on cells, including the functional loss of essential proteins or the toxic gain-of-function caused by misfolded protein conformations [381,382]. However, cells have protective mechanisms, known as protein quality control systems, which can prevent proteins from misfolding and toxic aggregations induced by stressful stimuli, including oxidative stress. This quality control system depends on three main strategies: the proteolytic system-mediated degradation of misfolded proteins, the ATP-dependent chaperone-mediated refolding of non-native proteins, and their sequestration into inert inclusions [383,384]. One of the primary pathways in the clearance of misfolded proteins is the ubiquitin–proteasome system (UPS), which depends on the ubiquitination of misfolded proteins followed by 26S proteasomal degradation [385]. Studies showed that the degradation of misfolded protein through the proteasome accelerated over ten-fold upon exposure to H2O2 or O2•− [386].
Molecular chaperones are proteins that assist with the folding and/or assembling of other macromolecules into more orderly structures, without themselves being a part of these final structures [387]. Chaperons can be broadly divided into two groups based on their dependency on metabolic energy: those depending on the metabolic energy, e.g., all ATP-dependent chaperones, and those independent of metabolic energy, e.g., small heat shock proteins (sHSPs) and protein disulfide isomerase [381,388,389]. ATP-dependent chaperones can either be constitutively expressed to perform the essential housekeeping roles or induced by brief exposure to increased temperatures or other stressors that lead to protein denaturation. Chaperones that are expressed constitutively are termed heat shock cognates (HSCs), whereas stress-inducible chaperones are referred to as heat shock proteins (HSPs) [390].
Among different HSPs, HSP70 and HSP90 are the most important chaperones, which are highly conserved and ubiquitous in all organisms for the maintenance of life [391,392]. These chaperones work cooperatively and execute a wide range of functions, such as folding and assembly of newly made proteins, refolding of misfolded or aggregated proteins, facilitating the translocation of organelle and secretory proteins across membranes, and regulating specific protein activities (Figure 11) [393,394,395,396,397]. Although the major function of HSPs is to help improperly folded proteins recover their normal folded structure, they also cooperate with the UPS system to enhance the removal of terminally misfolded proteins [398,399]. Besides their importance in protein folding, HSP70 and HSP90 also function as a negative regulator for heat stress transcription factors (HSFs) by maintaining the inactive state of HSFs (Figure 12) [400,401]. Furthermore, experimental findings have indicated an association between HSP70 and redox status, where the HSP70 significantly enhanced the enzymatic activities of GPx and GR [402].
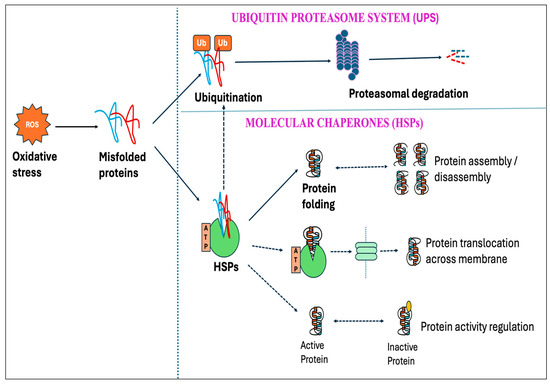
Figure 11.
Schematic representation of the ubiquitin–proteasome system (UPS) and the molecular chaperones in regulating proteostasis. Misfolded protein resulting from oxidative injury activates the UPS, leading to the ubiquitination of misfolded proteins and 26S proteasomal degradation. The primary role of molecular chaperones, including the heat shock protein 70 (HSP70) and heat shock protein 90 (HSP90), is to refold the misfolded proteins; however, they have also been demonstrated to assist in the degradation of misfolded proteins through the UPS. In addition, HSPs play roles in protein assembly/disassembly, their translocation across membranes, and activity regulation of secretory proteins.
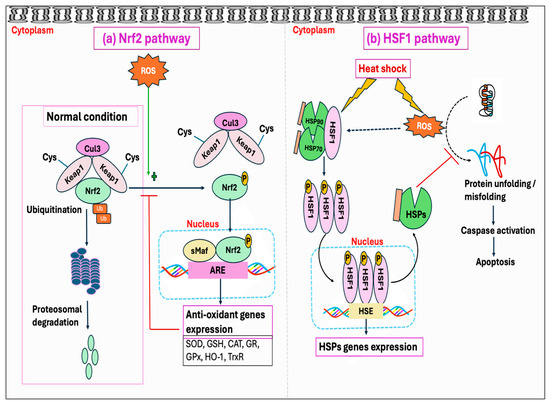
Figure 12.
Schematic representation of the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway and heat shock factor 1 (HSF1) pathway in regulating oxidative stress and proteotoxic stress, respectively: (a) Under normal conditions, kelch-like ECH-associated protein 1 (Keap1) binds with Nrf2, which will be subsequently ubiquitinated by Cullin 3 (Cul3) ligase for proteasomal degradation. During oxidative stress, reactive oxygen species (ROS) modifies cysteine residues in Keap1, leading to Nrf2 stabilization, nuclear translocation, and target gene binding to trigger the expression of multiple antioxidant genes. (b) HSF, particularly HSF1, is activated by both proteotoxic stimuli such as heat shock and oxidative stress. Upon activation, it trimerizes and interacts with the heat shock element (HSE) of its target genes in the nucleus, thereby regulating the expression of heat shock proteins (HSPs). As a molecular chaperone, HSPs assist in proper protein folding and mitigate proteotoxic stress.
Findings in Chickens
The skeletal muscle of long-lived pigeons demonstrated significantly lower proteasome activity than short-lived rats, suggesting the higher protein oxidation levels in pigeons result from reduced proteasome activity rather than reduced ROS production [403]. However, the ubiquitin and HSP70 expression increased upon heat shock in chicken testicular cells but not in mouse testicular cells, contributing to greater thermotolerance in avian spermatogenesis [404]. While there are numerous studies demonstrating the role and expression of HSPs, there is limited information on the role of the UPS system in degrading the misfolded protein during HS conditions in chickens. One of the studies showed that mitochondrial O2•− production promotes the expression of atrogin-1 mRNA in HS-cultured muscle cells, leading to protein degradation via the UPS pathway [405]. Moreover, cortisol-induced mitochondrial O2•− production also resulted in UPS activation and subsequent degradation of muscle proteins in broiler chickens [406]. Moreover, an increased level of HSP70 was found to protect the intestinal mucosa from HS damage by enhancing the antioxidant enzyme activity and inhibiting the progression of lipid peroxidation [407]. Researchers also examined HSP70 expression in avian erythroid cells during their maturation, demonstrating that definite red blood cells react to heat shock by increasing the synthesis of HSP70 protein ten-to-twenty-fold without significantly changing the HSP70 mRNA levels [408]. However, a separate study found that HSP70 members were among the most downregulated genes within the ileal transcriptome of HS birds compared to the control group [409]. Multiple studies have agreed that the long-term upregulation of HSP70 is associated with improved thermotolerance acquisition in chickens [410,411,412].
8. Transcriptional Regulation of Oxidant and Antioxidant Mechanisms
Several transcription factors are involved in regulating the oxidant and antioxidant mechanisms during oxidative stress. The major transcription factors/pathways include nuclear factor erythroid 2-related factor 2 (Nrf2), nuclear factor kappa B (NF-κB), activator protein-1 (AP-1), mitogen-activated protein kinase (MAPK), and forkhead box O (FoxO) [413,414,415]. Moreover, a family of heat shock factors (HSFs) drives proteotoxic stress-inducible transcription [416]. Once activated, these transcription factors induce the expression of multiple genes related to redox homeostasis, inflammation, immunity, cell survival, and apoptosis [413].
8.1. Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Pathway
The Nrf2 is widely recognized as the redox-sensitive master regulator of cellular responses against oxidative stress [417]. During normal physiological conditions, kelch-like ECH-associated protein 1 (Keap1) attaches to Nrf2 in the cytoplasm and prevents it from nuclear translocation, leading to the ubiquitination and proteasomal degradation of Nrf2 [418,419]. During oxidative stress, the modification of specific stress-sensing cysteine residues results in conformational changes in Keap1 [420,421], thereby inhibiting Keap1 from facilitating the Nrf2 ubiquitination by Cullin 3 (Cul3) [422]. As a result, Nrf2 stabilizes and translocates into the nucleus where it heterodimerizes with small Maf proteins (sMaf) and binds to the antioxidant response element (ARE) within the promoter region of its target gene, resulting in the rapid activation of antioxidant genes, including SOD, CAT, GPx, and GSH, associated with ROS detoxification (Figure 12a) [311,423].
8.2. Heat Shock Factors (HSFs) Pathway
Proteotoxic stress disrupts proper protein folding, leading the misfolded proteins to accumulate and aggregate. Heat shock factors (HSFs) are the master trans-activators that are triggered by multiple proteotoxic stimuli, including heat shock [424]. In response, HSFs quickly activate the transcription of genes that encode HSPs, which subsequently operate as molecular chaperones [416]. In mammalian systems, three HSFs, namely, HSF1, HSF2, and HSF4, have been identified [425]; however, chickens have an additional HSF3, which is co-expressed with HSF1 in chicken cells and tissues [426]. This suggests that both HSF1 and HSF3 are the primary genes of the HSF family in chickens involved in heat shock response [426,427]. Among these various HSFs, HSF1 is considered the principal regulator of HSP expression, while HSF2 heterodimerizes with HSF1 and regulates specific HSP gene expression [428,429]. Moreover, HSF1 is also activated in response to oxidative stress besides heat shock response and other proteotoxic stresses [430]. These stressful stimuli trigger HSF1 to dissociate from HSP90 and HSP70, thereby activating HSF1. HSF1 then trimerizes, followed by several post-translational modifications and binding to the heat shock elements (HSEs) in the promoter region of its target genes (Figure 12b). This results in the transcriptional activation of target genes by HSF1 [416,431,432].
8.3. Nuclear Factor Kappa B (NF-κB) Pathway
The NF-κB proteins represent a group of transcription factors that have a key role in regulating both inflammation and immunity. Generally, the activity of NF-κB is regulated through IκB proteins, which typically prevents NF-κB from its DNA binding. However, the activation of NF-κB through a canonical pathway causes IκB proteins to phosphorylate, ubiquitinate, and degrade subsequently in the proteasome. Consequently, activated NF-κB moves into the nucleus and triggers the activation of target genes [433,434]. Free radicals have a dual role in NF-κB activity, either stimulating or suppressing it in a phase- and context-dependent manner. Particularly, the initial phase of oxidative stress triggers the activation of the NF-κB pathway while prolonged oxidative stress could inhibit proteasomal activity, thereby blocking the degradation of IκB and consequently preventing NF-κB activation [435]. Moreover, the NF-κB pathway has the potential to exhibit both antioxidant and pro-oxidant roles during the time of oxidative stress [436]. Antioxidant roles of NF-κB include anti-apoptotic function via downregulation of the JNK pathway and reduction in ROS accumulation via antioxidant targets, including the Mn-SOD, heme oxygenase-1 (HO-1), and GPx [437,438]. Conversely, NF-κB signaling may exert a pro-oxidant role by activating the expression of certain genes like NOX2, XO, and iNOS (Figure 13a) [439,440,441].
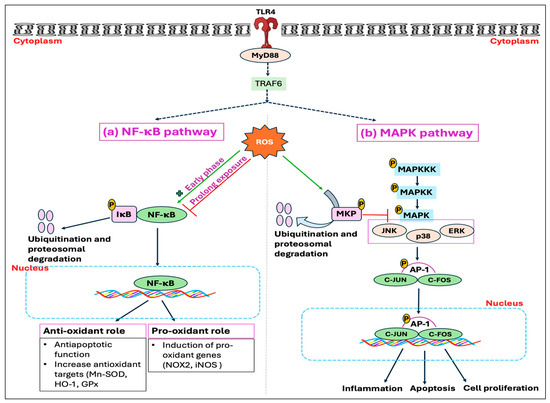
Figure 13.
Schematic representation of the nuclear factor kappa B (NF-κB) pathway and mitogen-activated protein kinase (MAPK) pathway in regulating oxidative stress: (a) Reactive oxygen species (ROS)-induced oxidative stress has a dual role in NF-κB activity; the early phase activates the NF-κB pathway while prolonged exposure can inhibit the degradation of the inhibitor of nuclear factor kappa B (IκB), thus preventing NF-κB activation. Additionally, the NF-κB pathway can exert both antioxidant as well as pro-oxidant functions during the time of oxidative stress by regulating the expression of various antioxidant genes and pro-oxidant genes, respectively. (b) Activation of another transcription factor, activator protein-1 (AP-1), is linked with cellular proliferation, inflammation, and death. AP-1 is the primary target of MAPK, which has three subfamilies, namely, extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38. One mechanism by which oxidative stress triggers MAPK and AP-1 is the inactivation and degradation of mitogen-activated protein kinase phosphatase (MKP). Under normal conditions, MKP negatively regulates MAPK by dephosphorylation, thereby rendering its activity. However, ROS can promote the degradation of MKP, resulting in the activation of MAPK signaling.
8.4. Mitogen-Activated Protein Kinase (MAPK) Pathway
The ubiquitous protein complex AP-1 is activated by numerous signals and mediates various cellular responses. It comprises a group of related dimeric complexes from the FOS and JUN protein families rather than a singular transcription factor [442]. Enhanced activity of AP-1 has been linked to cellular proliferation, differentiation, stress, and death [443,444]. Various growth factors and proinflammatory cytokines, as well as UV radiation, have been shown to activate AP-1 activity through a range of distinct mechanisms. This AP-1 represents one of the major targets of the mitogen-activated protein kinases (MAPKs), a group of serine/threonine protein kinases that are closely engaged in regulating various cellular processes [445]. Three main subfamilies of MAPK, namely, extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38, are identified. The activation of each MAPK subgroup occurs through a series of phosphorylation steps, starting from the activation of MAPK kinase kinases (MAPKKK). Therefore, the dephosphorylation of MAPKs by phosphatases could be the most effective mechanism for its negative regulation. Evidence from studies suggested that ROS can trigger or facilitate the induction of MAPK signaling pathways [446,447]. One of the potential mechanisms for ROS-mediated MAPK activation involves inactivation and degradation of the mitogen-activated protein kinase phosphatase (MKP), resulting in the long-term activation of the JNK, p38, and ERK pathways (Figure 13b) [448,449].
8.5. Findings in Chickens
The protective effects of different polyphenols and carotenoids in poultry have been shown to mediate through the Nrf2 pathway. Elevating dietary lycopene levels mitigated the adverse effects of HS on the Nrf2 system, and had a positive impact on chicken performance [337,450]. Moreover, dietary resveratrol has been shown to alleviate heat-induced liver injury through the Nrf2–Keap1 pathway [336]. On the other hand, cadmium, an environmental pollutant, induced oxidative stress and led to the upregulation of Nrf2 and downstream target genes in the chicken’s kidney [451]. However, Nrf2 showed a slight downregulation on day 1 post-HS, while no significant difference was observed in the mRNA expression of Nrf2 at 12 days post-HS in broiler chickens [17]. Under HS conditions, these genes were over-expressed in backyard chicken breeds that proved to be more resistant to high temperatures compared with commercial broilers [412]. Furthermore, thermal manipulation with cold stress in broiler chickens decreased the hepatic HSF3 expression, while the splenic HSF3 remained unaffected [452].
The expression of chicken myeloid differentiation factor 88 (MyD88-2) was observed in various tissues, and the overexpression of MyD88-2 was shown to activate NF-κB significantly [453]. Both the mRNA expressions and protein levels of NF-κB and other inflammatory genes were significantly higher in the hypothalamus of the HS group than in the control group [454]. Similarly, at 35 and 42 days of age, the HS group exhibited a substantial increase in the relative expression of MyD88 and NF-κB genes compared to the control group [455]. A separate study demonstrated increased mRNA levels of toll-like receptor 4 (TLR4), C-JUN, C-FOS, caspase-3, and p38 of MAPK in the jejunal mucosa of black-boned chicken under HS condition [456]. Moreover, the mRNA expressions of NF-κB subunits (p50, p65, p52, and RelB), AP-1 (C-JUN and C-FOS), and IRF3 were significantly elevated in the spleen of chickens under HS when compared with the control group [457].
9. Conclusions
Heat stress is becoming a major threat for both humans and animals. In livestock production, HS is directly related to higher mortality rates and decreased production, imposing not only economic burdens but also increasing food insecurity on a global scale. Mitigating the adverse effects of HS requires an in-depth understanding of major pathways related to ROS production and detoxification. Heat stress triggers various cellular enzymatic systems, leading to the excessive production of ROS and subsequent oxidative injury to lipids, proteins, carbohydrates, and DNA. However, the exact mechanisms of how HS stimulates the enzymatic systems to generate excessive amounts of ROS, ultimately developing into oxidative stress, have not been fully uncovered. Animals, including chickens, fight against HS through different physiological and hormonal responses, such as panting, increased peripheral circulation, reduced feed intake, and secretion of stress hormones. However, these adaptive responses have been directly or indirectly linked to the development of oxidative stress. A detailed investigation into these initial responses against HS and an exploration of their downstream mechanisms may establish the connection between HS and oxidative stress in chickens. Conversely, cells also have enzymatic and non-enzymatic antioxidant systems that detoxify ROS/RNS, thereby protecting cells from oxidative injury. The interconnected series of reactions between various antioxidants, such as the antioxidant network, enhance the potency of each antioxidant in combating oxidative stress. The process of ROS/RNS generation and elimination is tightly regulated through multiple transcriptional factors, and there exists a significant interplay between different systems in maintaining the redox homeostasis of cells. Extensive studies in chickens have reported the mRNA expression and protein activity of different oxidant and antioxidant genes. Although the primary mechanisms of oxidative stress and antioxidant defense mechanisms between chickens and mammals have fundamental similarities, further molecular studies are required to identify any unique pathways in chickens and to develop effective nutritional interventions against HS.
Author Contributions
Writing—original draft preparation, B.A.; writing—review and editing, B.A., J.K., O.W.A., A.F.A.G., M.C.M., A.L.F., S.K., R.R. and S.E.A.; supervision, S.E.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ADP | Adenosine diphosphate |
| AGEs | Advanced glycation end products |
| AHCY | Adenosylhomocysteinase |
| AMD | Adenosylmethionine decarboxylase |
| AP-1 | Activator protein-1 |
| ARE | Antioxidant response element |
| ATF6 | Activating transcription factor 6 |
| ATP | Adenosine triphosphate |
| BCL2 | B-cell lymphoma 2 |
| BH4 | Tetrahydrobiopterin |
| BHMT | Betaine-homocysteine methyltransferase |
| CAT | Catalase |
| CAT-1 | Cationic amino acid transporter 1 |
| CBS | Cystathionine-beta-synthase |
| CDO | Cysteine dioxygenase |
| C-FOS | c-Fos proto-oncogene |
| CGL | Cystathionine-gamma-lyase |
| C-JUN | c-Jun proto-oncogene |
| CO3•− | Carbonate radical |
| CoQ | Ubiquinone |
| CoQH2 | Reduced coenzyme Q (ubiquinol) |
| COX | Cyclooxygenase |
| Cul3 | Cullin 3 |
| Cyt.C | Cytochrome C |
| DHAA | Dehydroascorbate |
| DNA | Deoxyribonucleic acid |
| eNOS | Endothelial nitric oxide synthase |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| ETC | Electron transport chain |
| FADH2 | Flavin adenine dinucleotide |
| FFAs | Free fatty acids |
| FKBP | FK506 binding proteins |
| FoxO | Forkhead box O |
| GC | Glucocorticoids |
| GCH1 | GTP cyclohydrolase I |
| GCL | Glutamine cysteine ligase |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GREs | Glucocorticoid response elements |
| Grx | Glutaredoxin |
| GSH | Reduced glutathione |
| GS | Glutamine synthetase |
| GSS | Glutathione synthetase |
| GSSG | Oxidized glutathione |
| GST | Glutathione S-transferase |
| H+ | Hydrogen ion |
| HCO3− | Bicarbonate ion |
| HO• | Hydroxyl radical |
| H2O2 | Hydrogen peroxide |
| HS | Heat stress |
| HSC | Heat shock cognate |
| HSE | Heat shock element |
| HSF | Heat shock factor |
| HSP | Heat shock protein |
| IκB | Inhibitor of nuclear factor kappa B |
| IL-8 | Interleukin-8 |
| IL-18 | Interleukin-18 |
| iNOS | Inducible nitric oxide synthase |
| IP3R | Inositol 1,4,5-trisphosphate receptors |
| IRE1 | Inositol requiring 1 protein |
| JNK | c-Jun N-terminal kinase |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LOO• | Lipid peroxyl radical |
| LOX | Lipoxygenase |
| MAMs | Mitochondria-associated ER membranes |
| MAPK | Mitogen-activated protein kinase |
| MAPKK | MAPK kinase |
| MAPKKK | MAPK kinase kinase |
| MAT | Methionine adenosyl transferase |
| MDA | Malondialdehyde |
| MKP | Mitogen-activated protein kinase phosphatase |
| mPTP | Mitochondrial permeability transition pore |
| MT | Methyl transferase |
| MTAP | Methylthioadenosine phosphorylase |
| MTR | Methylthioribose |
| NADH | Nicotinamide-adenine dinucleotide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| •NO | Nitric oxide radical |
| •NO2 | Nitrogen dioxide radical |
| NOS | Nitric oxide synthase |
| nNOS | Neuronal nitric oxide synthase |
| NOX | NADPH oxidase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| O2•− | Superoxide radical |
| ODC | Ornithine decarboxylase |
| 8-OHdG | 8-hydroxy-2′-deoxyguanosine |
| ONOO− | Peroxynitrite |
| p62 | Sequestosome-1 |
| PC | Protein carbonyl |
| PDI | Protein disulfide isomerase |
| PERK | PKR-like ER kinase |
| PI3K | Phosphatidylinositol 3-kinase |
| PPP | Pentose phosphate pathway |
| PrDx | Peroxiredoxin |
| ROO• | Peroxyl radical |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptors |
| SAH | S-adenosyl-homocysteine |
| SAM | S-adenosyl-methionine |
| SDA | Semidehydroascorbate |
| SIRT3 | Sirtuin 3 |
| sMaf | small Maf protein |
| SOD | Superoxide dismutase |
| TAC | Total antioxidant capacity |
| THF | Tetra-hydrofolate |
| TNF-α | Tumor necrosis factor-alpha |
| Trx | Thioredoxin |
| TrxR | Thioredoxin reductase |
| Ub | Ubiquitin |
| UCP | Uncoupling proteins |
| UPR | Unfolded protein response |
| UPS | Ubiquitin–proteasome system |
| XO | Xanthine oxidase |
References
- Sisein, E.A. Biochemistry of free radicals and antioxidants. Sch. Acad. J. Biosci. 2014, 2, 110–118. [Google Scholar]
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free radical properties, source and targets, antioxidant consumption and health. Oxygen 2022, 2, 48–78. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals and other reactive species in disease. In eLS; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Do Yoo, Y. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, D.-Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009, 20, 223–230. [Google Scholar] [CrossRef]
- Naseem, K.M. The role of nitric oxide in cardiovascular diseases. Mol. Asp. Med. 2005, 26, 33–65. [Google Scholar] [CrossRef] [PubMed]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and methods to measure oxidative stress in clinical samples: Research applications in the cancer field. Oxidative Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef]
- Habashy, W.S.; Milfort, M.C.; Rekaya, R.; Aggrey, S.E. Cellular antioxidant enzyme activity and biomarkers for oxidative stress are affected by heat stress. Int. J. Biometeorol. 2019, 63, 1569–1584. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxidative Med. Cell. Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef]
- Bernabucci, U.; Ronchi, B.; Lacetera, N.; Nardone, A. Markers of oxidative status in plasma and erythrocytes of transition dairy cows during hot season. J. Dairy Sci. 2002, 85, 2173–2179. [Google Scholar] [CrossRef]
- Habashy, W.S.; Milfort, M.C.; Rekaya, R.; Aggrey, S.E. Expression of genes that encode cellular oxidant/antioxidant systems are affected by heat stress. Mol. Biol. Rep. 2018, 45, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Decuypere, E.; Buyse, J. Acute heat stress induces oxidative stress in broiler chickens. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2006, 144, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Nichelmann, M.; Burmeister, A.; Janke, O.; Höchel, J.; Tzschentke, B. Avian embryonic thermoregulation: Role of Q10 in interpretation of endothermic reactions. J. Therm. Biol. 1998, 23, 369–376. [Google Scholar] [CrossRef]
- Renaudeau, D.; Collin, A.; Yahav, S.; De Basilio, V.; Gourdine, J.-L.; Collier, R. Adaptation to hot climate and strategies to alleviate heat stress in livestock production. Animal 2012, 6, 707–728. [Google Scholar] [CrossRef]
- Settar, P.; Yalcin, S.; Turkmut, L.; Ozkan, S.; Cahanar, A. Season by genotype interaction related to broiler growth rate and heat tolerance. Poult. Sci. 1999, 78, 1353–1358. [Google Scholar] [CrossRef]
- Deeb, N.; Cahaner, A. Genotype-by-environment interaction with broiler genotypes differing in growth rate. 3. Growth rate and water consumption of broiler progeny from weight-selected versus nonselected parents under normal and high ambient temperatures. Poult. Sci. 2002, 81, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Ariyo, O.W.; Kwakye, J.; Sovi, S.; Aryal, B.; Ghareeb, A.F.; Hartono, E.; Milfort, M.C.; Fuller, A.L.; Rekaya, R.; Aggrey, S.E. Glucose Supplementation Improves Performance and Alters Glucose Transporters’ Expression in Pectoralis major of Heat-Stressed Chickens. Animals 2023, 13, 2911. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-R.; Ryu, C.; Lee, S.-D.; Cho, J.-H.; Kang, H. Effects of Heat Stress on the Laying Performance, Egg Quality, and Physiological Response of Laying Hens. Animals 2024, 14, 1076. [Google Scholar] [CrossRef]
- Tang, L.-P.; Li, W.-H.; Liu, Y.-L.; Lun, J.-C.; He, Y.-M. Heat stress aggravates intestinal inflammation through TLR4-NF-κB signaling pathway in Ma chickens infected with Escherichia coli O157: H7. Poult. Sci. 2021, 100, 101030. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD (P) H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef]
- Fridovich, I. Mitochondria: Are they the seat of senescence? Aging Cell 2004, 3, 13–16. [Google Scholar] [CrossRef]
- Spiekermann, S.; Landmesser, U.; Dikalov, S.; Bredt, M.; Gamez, G.; Tatge, H.; Reepschläger, N.; Hornig, B.; Drexler, H.; Harrison, D.G. Electron spin resonance characterization of vascular xanthine and NAD (P) H oxidase activity in patients with coronary artery disease: Relation to endothelium-dependent vasodilation. Circulation 2003, 107, 1383–1389. [Google Scholar] [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.-O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (Ros-Induced) Ros release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef]
- Kimura, S.; Zhang, G.-X.; Nishiyama, A.; Shokoji, T.; Yao, L.; Fan, Y.-Y.; Rahman, M.; Abe, Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: Comparison of angiotensin II and diazoxide. Hypertension 2005, 45, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Downs, C.A.; Heckathorn, S.A. The mitochondrial small heat-shock protein protects NADH: Ubiquinone oxidoreductase of the electron transport chain during heat stress in plants. FEBS Lett. 1998, 430, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef]
- Senior, A.E. ATP synthesis by oxidative phosphorylation. Physiol. Rev. 1988, 68, 177–231. [Google Scholar] [CrossRef]
- Selivanov, V.A.; Votyakova, T.V.; Pivtoraiko, V.N.; Zeak, J.; Sukhomlin, T.; Trucco, M.; Roca, J.; Cascante, M. Reactive oxygen species production by forward and reverse electron fluxes in the mitochondrial respiratory chain. PLoS Comput. Biol. 2011, 7, e1001115. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- White, M.G.; Saleh, O.; Nonner, D.; Barrett, E.F.; Moraes, C.T.; Barrett, J.N. Mitochondrial dysfunction induced by heat stress in cultured rat CNS neurons. J. Neurophysiol. 2012, 108, 2203–2214. [Google Scholar] [CrossRef]
- Wang, Z.; Cai, F.; Chen, X.; Luo, M.; Hu, L.; Lu, Y. The role of mitochondria-derived reactive oxygen species in hyperthermia-induced platelet apoptosis. PLoS ONE 2013, 8, e75044. [Google Scholar] [CrossRef]
- Ricquier, D.; Bouillaud, F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem. J. 2000, 345, 161–179. [Google Scholar] [CrossRef]
- Abe, T.; Mujahid, A.; Sato, K.; Akiba, Y.; Toyomizu, M. Possible role of avian uncoupling protein in down-regulating mitochondrial superoxide production in skeletal muscle of fasted chickens. FEBS Lett. 2006, 580, 4815–4822. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S. Mitochondrial H+ leak and ROS generation: An odd couple. Free Radic. Biol. Med. 2005, 38, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Yang, L.; Tan, G.-Y.; Fu, Y.-Q.; Feng, J.-H.; Zhang, M.-H. Effects of acute heat stress and subsequent stress removal on function of hepatic mitochondrial respiration, ROS production and lipid peroxidation in broiler chickens. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2010, 151, 204–208. [Google Scholar] [CrossRef]
- Mujahid, A.; Akiba, Y.; Warden, C.H.; Toyomizu, M. Sequential changes in superoxide production, anion carriers and substrate oxidation in skeletal muscle mitochondria of heat-stressed chickens. FEBS Lett. 2007, 581, 3461–3467. [Google Scholar] [CrossRef]
- Akbarian, A.; Michiels, J.; Degroote, J.; Majdeddin, M.; Golian, A.; De Smet, S. Association between heat stress and oxidative stress in poultry; mitochondrial dysfunction and dietary interventions with phytochemicals. J. Anim. Sci. Biotechnol. 2016, 7, 37. [Google Scholar] [CrossRef]
- Green, K.; Brand, M.D.; Murphy, M.P. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 2004, 53, S110–S118. [Google Scholar] [CrossRef]
- Pobezhimova, T.; Voinikov, V.; Varakina, N. Inactivation of complex I of the respiratory chain of maize mitochondria incubated in vitro by elevated temperature. J. Therm. Biol. 1996, 21, 283–288. [Google Scholar] [CrossRef]
- Pitkanen, S.; Robinson, B.H. Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J. Clin. Investig. 1996, 98, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Loeb, L.A. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2001, 477, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. LXXIII.—Oxidation of tartaric acid in presence of iron. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Gryglewski, R.; Palmer, R.; Moncada, S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986, 320, 454–456. [Google Scholar] [CrossRef]
- Altan, Ö.; Pabuçcuoğlu, A.; Altan, A.; Konyalioğlu, S.; Bayraktar, H. Effect of heat stress on oxidative stress, lipid peroxidation and some stress parameters in broilers. Br. Poult. Sci. 2003, 44, 545–550. [Google Scholar] [CrossRef]
- Melegh, B.; Bock, I.; Gáti, I.; Méhes, K. Multiple mitochondrial DNA deletions and persistent hyperthermia in a patient with Brachmann-de Lange phenotype. Am. J. Med. Genet. 1996, 65, 82–88. [Google Scholar] [CrossRef]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Bienert, G.P.; Møller, A.L.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef]
- Magnani, F.; Nenci, S.; Millana Fananas, E.; Ceccon, M.; Romero, E.; Fraaije, M.W.; Mattevi, A. Crystal structures and atomic model of NADPH oxidase. Proc. Natl. Acad. Sci. USA 2017, 114, 6764–6769. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Dikalova, A.E.; Bikineyeva, A.T.; Schmidt, H.H.; Harrison, D.G.; Griendling, K.K. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Kikusato, M.; Yoshida, H.; Furukawa, K.; Toyomizu, M. Effect of heat stress-induced production of mitochondrial reactive oxygen species on NADPH oxidase and heme oxygenase-1 mRNA levels in avian muscle cells. J. Therm. Biol. 2015, 52, 8–13. [Google Scholar] [CrossRef]
- Rathore, R.; Zheng, Y.-M.; Niu, C.-F.; Liu, Q.-H.; Korde, A.; Ho, Y.-S.; Wang, Y.-X. Hypoxia activates NADPH oxidase to increase [ROS] i and [Ca2+] i through the mitochondrial ROS-PKCε signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef]
- Parks, D.A.; Granger, D.N. Xanthine oxidase: Biochemistry, distribution and physiology. Acta Physiol. Scand. Suppl. 1986, 548, 87–99. [Google Scholar]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase-derived reactive species: Physiological and pathological effects. Oxidative Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef]
- Waud, W.R.; Rajagopalan, K. Purification and properties of the NAD+-dependent (type D) and O2-dependent (type O) forms of rat liver xanthine dehydrogenase. Arch. Biochem. Biophys. 1976, 172, 354–364. [Google Scholar] [CrossRef]
- Stirpe, F.; Della Corte, E.; Lorenzoni, E. The regulation of rat liver xanthine oxidase: Conversion in vitro of the enzyme activity from dehydrogenase (type D) to oxidase (type O). J. Biol. Chem. 1969, 244, 3855–3863. [Google Scholar] [CrossRef]
- Skibba, J.L.; Powers, R.H.; Stadnicka, A.; Kalbfleisch, J.H. The effect of hyperthermia on conversion of rat hepatic xanthine dehydrogenase to xanthine oxidase. Biochem. Pharmacol. 1988, 37, 4592–4595. [Google Scholar] [CrossRef]
- Nishino, T.; Schopfer, L.; Massey, V. The reactivity of chicken liver xanthine dehydrogenase with molecular oxygen. J. Biol. Chem. 1989, 264, 2518–2527. [Google Scholar] [CrossRef]
- Strittmatter, C.F. Studies on avian xanthine dehydrogenases: Properties and patterns of appearance during development. J. Biol. Chem. 1965, 240, 2557–2564. [Google Scholar] [CrossRef]
- Remy, C.; Richert, D.A.; Westerfeld, W. The determination of xanthine dehydrogenase in chicken tissues. J. Biol. Chem. 1951, 193, 649–657. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994, 23, 1121–1131. [Google Scholar] [CrossRef]
- Xia, Y.; Tsai, A.-L.; Berka, V.; Zweier, J.L. Superoxide generation from endothelial nitric-oxide synthase: A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J. Biol. Chem. 1998, 273, 25804–25808. [Google Scholar] [CrossRef]
- Vásquez-Vivar, J.; Kalyanaraman, B.; MARTAsek, P.; Hogg, N.; Masters, B.S.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef]
- Wink, D.A.; Miranda, K.M.; Espey, M.G.; Pluta, R.M.; Hewett, S.J.; Colton, C.; Vitek, M.; Feelisch, M.; Grisham, M.B. Mechanisms of the antioxidant effects of nitric oxide. Antioxid. Redox Signal. 2001, 3, 203–213. [Google Scholar] [CrossRef]
- Le Greves, P.; Sharma, H.S.; Westman, J.; Alm, P.; Nyberg, F. Acute heat stress induces edema and nitric oxide synthase upregulation and down-regulates mRNA levels of the NMDAR1, NMDAR2A and NMDAR2B subunits in the rat hippocampus. In Brain Edema X, Proceedings of the Tenth International Symposium San Diego, San Diego, CA, USA, 20–23 October 1996; Springer: Vienna, Austria, 1997; pp. 275–278. [Google Scholar]
- Harris, M.B.; Blackstone, M.A.; Ju, H.; Venema, V.J.; Venema, R.C. Heat-induced increases in endothelial NO synthase expression and activity and endothelial NO release. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H333–H340. [Google Scholar] [CrossRef]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef]
- Lepock, J.R. How do cells respond to their thermal environment? Int. J. Hyperth. 2005, 21, 681–687. [Google Scholar] [CrossRef]
- Sharma, S.; Chaudhary, P.; Sandhir, R.; Bharadwaj, A.; Gupta, R.K.; Khatri, R.; Bajaj, A.C.; Baburaj, T.; Kumar, S.; Pal, M. Heat-induced endoplasmic reticulum stress in soleus and gastrocnemius muscles and differential response to UPR pathway in rats. Cell Stress Chaperones 2021, 26, 323–339. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Unfolded protein response. Curr. Biol. 2012, 22, R622–R626. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Scheuner, D.; Schröder, M.; Shen, X.; Lee, K.; Liu, C.Y.; Arnold, S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 2002, 3, 411–421. [Google Scholar] [CrossRef]
- Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039. [Google Scholar] [CrossRef]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Moraes, A.A.; França, L.P.; Tucci-Viegas, V.M.; Lasakosvitsch, F.; Gaiba, S.; Azevedo, F.L.; Nogueira, A.P.; Segreto, H.R.; Ferreira, A.T.; Franca, J.P. Gamma Radiation Induces p53-Mediated Cell Cycle Arrest in Bone Marrow Cells. In Flow Cytometry-Recent Perspectives; IntechOpen: London, UK, 2012. [Google Scholar]
- Das, A.M.; Harris, D.A. Control of mitochondrial ATP synthase in heart cells: Inactive to active transitions caused by beating or positive inotropic agents. Cardiovasc. Res. 1990, 24, 411–417. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979, 180, 533–544. [Google Scholar] [CrossRef]
- Friberg, H.; Wieloch, T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie 2002, 84, 241–250. [Google Scholar] [CrossRef]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef]
- Siesjö, B.K.; Elmer, E.; Janelidze, S.; Keep, M.; Kristian, T.; Ouyang, Y.-B.; Uchino, H. Role and mechanisms of secondary mitochondrial failure. In Current Progress in the Understanding of Secondary Brain Damage from Trauma and Ischemia, Proceedings of the 6th International Symposium: Mechanisms of Secondary Brain Damage-Novel Developments; Mauls/Sterzing, Italy, February 1998; Springer: Vienna, Austria, 1999; pp. 7–13. [Google Scholar]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar] [CrossRef]
- Wingrave, J.M.; Schaecher, K.E.; Sribnick, E.A.; Wilford, G.G.; Ray, S.K.; Hazen-Martin, D.J.; Hogan, E.L.; Banik, N.L. Early induction of secondary injury factors causing activation of calpain and mitochondria-mediated neuronal apoptosis following spinal cord injury in rats. J. Neurosci. Res. 2003, 73, 95–104. [Google Scholar] [CrossRef]
- Almeida, A.; Allen, K.L.; Bates, T.E.; Clark, J.B. Effect of reperfusion following cerebral ischaemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J. Neurochem. 1995, 65, 1698–1703. [Google Scholar] [CrossRef]
- Smaili, S.; Hirata, H.; Ureshino, R.; Monteforte, P.T.; Morales, A.P.; Muler, M.L.; Terashima, J.; Oseki, K.; Rosenstock, T.R.; Lopes, G.S. Calcium and cell death signaling in neurodegeneration and aging. Anais Acad. Bras. Cienc. 2009, 81, 467–475. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 213–224. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Botta, E.; Holinstat, M. Eicosanoids in Inflammation in the Blood and the Vessel. Front. Pharmacol. 2022, 13, 997403. [Google Scholar] [CrossRef]
- Misheva, M.; Johnson, J.; McCullagh, J. Role of oxylipins in the inflammatory-related diseases NAFLD, obesity, and type 2 diabetes. Metabolites 2022, 12, 1238. [Google Scholar] [CrossRef]
- Reinbothe, C.; Springer, A.; Samol, I.; Reinbothe, S. Plant oxylipins: Role of jasmonic acid during programmed cell death, defence and leaf senescence. FEBS J. 2009, 276, 4666–4681. [Google Scholar] [CrossRef] [PubMed]
- Massey, K.A.; Nicolaou, A. Lipidomics of oxidized polyunsaturated fatty acids. Free Radic. Biol. Med. 2013, 59, 45–55. [Google Scholar] [CrossRef]
- Thuresson, E.D.; Lakkides, K.M.; Smith, W.L. Different catalytically competent arrangements of arachidonic acid within the cyclooxygenase active site of prostaglandin endoperoxide H synthase-1 lead to the formation of different oxygenated products. J. Biol. Chem. 2000, 275, 8501–8507. [Google Scholar] [CrossRef]
- Tourdot, B.E.; Ahmed, I.; Holinstat, M. The emerging role of oxylipins in thrombosis and diabetes. Front. Pharmacol. 2014, 4, 176. [Google Scholar] [CrossRef]
- Askari, A.A.; Thomson, S.; Edin, M.L.; Lih, F.B.; Zeldin, D.C.; Bishop-Bailey, D. Basal and inducible anti-inflammatory epoxygenase activity in endothelial cells. Biochem. Biophys. Res. Commun. 2014, 446, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in our understanding of oxylipins derived from dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef] [PubMed]
- Nayeem, M.A. Role of oxylipins in cardiovascular diseases. Acta Pharmacol. Sin. 2018, 39, 1142–1154. [Google Scholar] [CrossRef]
- Schmitz, G.; Ecker, J. The opposing effects of n−3 and n−6 fatty acids. Prog. Lipid Res. 2008, 47, 147–155. [Google Scholar] [CrossRef]
- Shin, S.-W.; Seo, C.-Y.; Han, H.; Han, J.-Y.; Jeong, J.-S.; Kwak, J.-Y.; Park, J.-I. 15d-PGJ2 induces apoptosis by reactive oxygen species–mediated inactivation of Akt in leukemia and colorectal cancer cells and shows in vivo antitumor activity. Clin. Cancer Res. 2009, 15, 5414–5425. [Google Scholar] [CrossRef]
- Kumar, K.A.; Arunasree, K.M.; Roy, K.R.; Reddy, N.P.; Aparna, A.; Reddy, G.V.; Reddanna, P. Effects of (15S)-hydroperoxyeicosatetraenoic acid and (15S)-hydroxyeicosatetraenoic acid on the acute-lymphoblastic-leukaemia cell line Jurkat: Activation of the Fas-mediated death pathway. Biotechnol. Appl. Biochem. 2009, 52, 121–133. [Google Scholar] [CrossRef]
- Kondo, M.; Oya-Ito, T.; Kumagai, T.; Osawa, T.; Uchida, K. Cyclopentenone prostaglandins as potential inducers of intracellular oxidative stress. J. Biol. Chem. 2001, 276, 12076–12083. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, M.; Sánchez-Rodríguez, J.; Santos, A.; Perez-Castillo, A. 15-Deoxy-Δ-12, 14-prostaglandin J 2 induces programmed cell death of breast cancer cells by a pleiotropic mechanism. Carcinogenesis 2005, 26, 81–92. [Google Scholar] [CrossRef]
- Rossi, A.; Coccia, M.; Trotta, E.; Angelini, M.; Santoro, M.G. Regulation of cyclooxygenase-2 expression by heat: A novel aspect of heat shock factor 1 function in human cells. PLoS ONE 2012, 7, e31304. [Google Scholar] [CrossRef] [PubMed]
- Durand, T.; Bultel-Poncé, V.; Guy, A.; Berger, S.; Mueller, M.J.; Galano, J.-M. New bioactive oxylipins formed by non-enzymatic free-radical-catalyzed pathways: The phytoprostanes. Lipids 2009, 44, 875–888. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, R. Cytochromes P450 as versatile biocatalysts. J. Biotechnol. 2006, 124, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef]
- Ren, J.; Yang, L.; Li, Q.; Zhang, Q.; Sun, C.; Liu, X.; Yang, N. Global investigation of cytochrome P450 genes in the chicken genome. Genes 2019, 10, 617. [Google Scholar] [CrossRef]
- White, R.E.; Coon, M.J. Oxygen activation by cytochrome P-4501. Annu. Rev. Biochem. 1980, 49, 315–356. [Google Scholar] [CrossRef]
- Shi, W.; Sun, J.; Xu, B.; Li, H. Molecular characterization and oxidative stress response of a cytochrome P450 gene (CYP4G11) from Apis cerana cerana. Z. Naturforsch. C 2013, 68, 509–521. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, W.; Li, Z.; Ma, L.; Yu, J.; Wang, H.; Liu, Z.; Xu, B. Identification and characterization of three new cytochrome P450 genes and the use of RNA interference to evaluate their roles in antioxidant defense in Apis cerana cerana Fabricius. Front. Physiol. 2018, 9, 1608. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.-A. Physiology of thermal panting in birds. In Annales de Biologie Animale Biochimie Biophysique; EDP Sciences: Les Ulis, France, 1970; pp. 151–168. [Google Scholar]
- Teeter, R.; Smith, M.; Owens, F.; Arp, S.; Sangiah, S.; Breazile, J. Chronic heat stress and respiratory alkalosis: Occurrence and treatment in broiler chicks. Poult. Sci. 1985, 64, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Calder, W., Jr.; Schmidt-Nielsen, K. Evaporative cooling and respiratory alkalosis in the pigeon. Proc. Natl. Acad. Sci. USA 1966, 55, 750–756. [Google Scholar] [CrossRef]
- Sur, M.; Hashmi, M.F. Alkalosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Castro, D.; Patil, S.; Zubair, M.; Keenaghan, M. Arterial blood gas. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Hopkins, E.; Sanvictores, T.; Sharma, S. Physiology, Acid Base Balance. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Sanchez, W.; McGuire, M.; Beede, D. Macromineral nutrition by heat stress interactions in dairy cattle: Review and original research. J. Dairy Sci. 1994, 77, 2051–2079. [Google Scholar] [CrossRef] [PubMed]
- Kadzere, C.T.; Murphy, M.R.; Silanikove, N.; Maltz, E. Heat stress in lactating dairy cows: A review. Livest. Prod. Sci. 2002, 77, 59–91. [Google Scholar] [CrossRef]
- Burhans, W.; Burhans, C.R.; Baumgard, L. Invited review: Lethal heat stress: The putative pathophysiology of a deadly disorder in dairy cattle. J. Dairy Sci. 2022, 105, 3716–3735. [Google Scholar] [CrossRef]
- Kazemi, H.; Johnson, D. Regulation of cerebrospinal fluid acid-base balance. Physiol. Rev. 1986, 66, 953–1037. [Google Scholar] [CrossRef]
- Cain, S.M. Increased oxygen uptake with passive hyperventilation of dogs. J. Appl. Physiol. 1970, 28, 4–7. [Google Scholar] [CrossRef]
- Lian, P.; Braber, S.; Varasteh, S.; Wichers, H.J.; Folkerts, G. Hypoxia and heat stress affect epithelial integrity in a Caco-2/HT-29 co-culture. Sci. Rep. 2021, 11, 13186. [Google Scholar] [CrossRef] [PubMed]
- Theye, R.A.; Gronert, G.A.; Heffron, J. Oxygen uptake of canine whole body and hind limb with hypocapnic alkalosis. Anesthesiology 1977, 47, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Rastegar, H.; Woods, M.; Harken, A.H. Respiratory alkalosis increases tissue oxygen demand. J. Surg. Res. 1979, 26, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Harken, A.H.; Woods, M. The influence of oxyhemoglobin affinity on tissue oxygen consumption. Ann. Surg. 1976, 183, 130–135. [Google Scholar] [CrossRef]
- Oakes, G.K.; Walker, A.M.; Ehrenkranz, R.A.; Cefalo, R.C.; Chez, R.A. Uteroplacental blood flow during hyperthermia with and without respiratory alkalosis. J. Appl. Physiol. 1976, 41, 197–201. [Google Scholar] [CrossRef]
- Robbins, N.M.; Swanson, R.A. Opposing effects of glucose on stroke and reperfusion injury: Acidosis, oxidative stress, and energy metabolism. Stroke 2014, 45, 1881–1886. [Google Scholar] [CrossRef]
- Marder, J.; Arad, Z. Panting and acid-base regulation in heat stressed birds. Comp. Biochem. Physiol. A 1989, 94, 395–400. [Google Scholar] [CrossRef]
- Wolfenson, D.; Frei, Y.F.; Berman, A. Blood flow distribution during artificially induced respiratory hypocapnic alkalosis in the fowl. Resp. Physiol. 1982, 50, 87–92. [Google Scholar] [CrossRef]
- Walsh, B.; Tiivel, T.; Tonkonogi, M.; Sahlin, K. Increased concentrations of Pi and lactic acid reduce creatine-stimulated respiration in muscle fibers. J. Appl. Physiol. 2002, 92, 2273–2276. [Google Scholar] [CrossRef]
- Harkema, S.; Meyer, R.A. Effect of acidosis on control of respiration in skeletal muscle. Am. J. Physiol. Cell Physiol. 1997, 272, C491–C500. [Google Scholar] [CrossRef]
- Jubrias, S.A.; Crowther, G.J.; Shankland, E.G.; Gronka, R.K.; Conley, K.E. Acidosis inhibits oxidative phosphorylation in contracting human skeletal muscle in vivo. J. Physiol. 2003, 553, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Tonkonogi, M.; Sahlin, K. Actively phosphorylating mitochondria are more resistant to lactic acidosis than inactive mitochondria. Am. J. Physiol. Cell Physiol. 1999, 277, C288–C293. [Google Scholar] [CrossRef]
- Conley, K.E.; Kemper, W.F.; Crowther, G.J. Limits to sustainable muscle performance: Interaction between glycolysis and oxidative phosphorylation. J. Exp. Biol. 2001, 204, 3189–3194. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Bento, L.M.A.; Fagian, M.M.; Vercesi, A.E.; Gontijo, J.A.R. Effects of NH4Cl-induced systemic metabolic acidosis on kidney mitochondrial coupling and calcium transport in rats. Nephrol. Dial. Transplant. 2007, 22, 2817–2823. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Corbet, C.; Pinto, A.; Martherus, R.; de Jesus, J.P.S.; Polet, F.; Feron, O. Acidosis drives the reprogramming of fatty acid metabolism in cancer cells through changes in mitochondrial and histone acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Smulders, T.V. Telencephalic regulation of the HPA axis in birds. Neurobiol. Stress 2021, 15, 100351. [Google Scholar] [CrossRef] [PubMed]
- Quinteiro-Filho, W.M.; Rodrigues, M.; Ribeiro, A.; Ferraz-de-Paula, V.; Pinheiro, M.; Sá, L.R.M.D.; Ferreira, A.J.P.; Palermo-Neto, J. Acute heat stress impairs performance parameters and induces mild intestinal enteritis in broiler chickens: Role of acute hypothalamic-pituitary-adrenal axis activation. J. Anim. Sci. 2012, 90, 1986–1994. [Google Scholar] [CrossRef]
- Pratt, W.B.; Toft, D.O. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997, 18, 306–360. [Google Scholar]
- Beato, M. Gene regulation by steroid hormones. Cell 1989, 56, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Psarra, A.-M.G.; Sekeris, C.E. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: Role of the mitochondrial glucocorticoid receptor. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Demonacos, C.; Tsawdaroglou, N.; Djordjevic-Markovic, R.; Papalopoulou, M.; Galanopoulos, V.; Papadogeorgaki, S.; Sekeris, C.E. Import of the glucocorticoid receptor into rat liver mitochondria in vivo and in vitro. J. Steroid Biochem. Mol. Biol. 1993, 46, 401–413. [Google Scholar] [CrossRef]
- Duclos, M.; Gouarne, C.; Martin, C.; Rocher, C.; Mormede, P.; Letellier, T. Effects of corticosterone on muscle mitochondria identifying different sensitivity to glucocorticoids in Lewis and Fischer rats. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E159–E167. [Google Scholar] [CrossRef]
- Munck, A.; Náray-Fejes-Tóth, A. The ups and downs of glucocorticoid physiology permissive and suppressive effects revisited. Mol. Cell. Endocrinol. 1992, 90, C1–C4. [Google Scholar] [CrossRef]
- Armstrong, J.S. Mitochondria: A target for cancer therapy. Br. J. Pharmacol. 2006, 147, 239–248. [Google Scholar] [CrossRef]
- Du, J.; McEwen, B.; Manji, H.K. Glucocorticoid receptors modulate mitochondrial function: A novel mechanism for neuroprotection. Commun. Integr. Biol. 2009, 2, 350–352. [Google Scholar] [CrossRef]
- Simon, N.; Jolliet, P.; Morin, C.; Zini, R.; Urien, S.K.; Tillement, J.-P. Glucocorticoids decrease cytochrome c oxidase activity of isolated rat kidney mitochondria. FEBS Lett. 1998, 435, 25–28. [Google Scholar] [CrossRef]
- Morin, C.; Zini, R.; Simon, N.; Charbonnier, P.; Tillement, J.P.; Louet, H.L. Low glucocorticoid concentrations decrease oxidative phosphorylation of isolated rat brain mitochondria: An additional effect of dexamethasone. Fundam. Clin. Pharmacol. 2000, 14, 493–500. [Google Scholar] [CrossRef]
- Signorello, M.G.; Ravera, S.; Leoncini, G. Oxidative Stress Induced by Cortisol in Human Platelets. Int. J. Mol. Sci. 2024, 25, 3776. [Google Scholar] [CrossRef]
- Simmons, W.W.; Ungureanu-Longrois, D.; Smith, G.K.; Smith, T.W.; Kelly, R.A. Glucocorticoids regulate inducible nitric oxide synthase by inhibiting tetrahydrobiopterin synthesis and L-arginine transport. J. Biol. Chem. 1996, 271, 23928–23937. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; Wallerath, T.; Closs, E.; Schmidt, C.; Schwarz, P.; Forstermann, U.; Lehr, H.-A. Dexamethasone suppresses eNOS and CAT-1 and induces oxidative stress in mouse resistance arterioles. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H436–H444. [Google Scholar] [CrossRef] [PubMed]
- de Souza, L.F.A.; Espinha, L.P.; de Almeida, E.A.; Lunedo, R.; Furlan, R.L.; Macari, M. How heat stress (continuous or cyclical) interferes with nutrient digestibility, energy and nitrogen balances and performance in broilers. Livest. Sci. 2016, 192, 39–43. [Google Scholar] [CrossRef]
- Renaudeau, D.; Gourdine, J.-L.; St-Pierre, N. A meta-analysis of the effects of high ambient temperature on growth performance of growing-finishing pigs. J. Anim. Sci. 2011, 89, 2220–2230. [Google Scholar] [CrossRef]
- Bernabucci, U.; Basirico, L.; Morera, P.; Lacetera, N.; Ronchi, B.; Nardone, A. Heat shock modulates adipokines expression in 3T3-L1 adipocytes. J. Mol. Endocrinol. 2009, 42, 139–147. [Google Scholar] [CrossRef]
- Morera, P.; Basiricò, L.; Hosoda, K.; Bernabucci, U. Chronic heat stress up-regulates leptin and adiponectin secretion and expression and improves leptin, adiponectin and insulin sensitivity in mice. J. Mol. Endocrinol. 2012, 48, 129. [Google Scholar] [CrossRef]
- Rabe, K.; Lehrke, M.; Parhofer, K.G.; Broedl, U.C. Adipokines and insulin resistance. Mol. Med. 2008, 14, 741–751. [Google Scholar] [CrossRef]
- Grabner, G.F.; Xie, H.; Schweiger, M.; Zechner, R. Lipolysis: Cellular mechanisms for lipid mobilization from fat stores. Nat. Metab. 2021, 3, 1445–1465. [Google Scholar] [CrossRef]
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 2007, 27, 79–101. [Google Scholar] [CrossRef]
- Hayashi, Y. Glucagon regulates lipolysis and fatty acid oxidation through inositol triphosphate receptor 1 in the liver. J. Diabetes Investig. 2020, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Caren, R.; Corbo, L. Glucagon and plasma lipoprotein lipase. Proc. Soc. Exp. Biol. Med. 1974, 146, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Ricart-Jané, D.; Cejudo-Martín, P.; Peinado-Onsurbe, J.; López-Tejero, M.D.; Llobera, M. Changes in lipoprotein lipase modulate tissue energy supply during stress. J. Appl. Physiol. 2005, 99, 1343–1351. [Google Scholar] [CrossRef]
- Emami, N.K.; Jung, U.; Voy, B.; Dridi, S. Radical response: Effects of heat stress-induced oxidative stress on lipid metabolism in the avian liver. Antioxidants 2020, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wen, J.; Zhang, H. Effect of chronic heat exposure on fat deposition and meat quality in two genetic types of chicken. Poult. Sci. 2007, 86, 1059–1064. [Google Scholar] [CrossRef]
- Geraert, P.; Padilha, J.; Guillaumin, S. Metabolic and endocrine changes induced by chronic heatexposure in broiler chickens: Growth performance, body composition and energy retention. Br. J. Nutr. 1996, 75, 195–204. [Google Scholar] [CrossRef]
- Curnutte, J.; Badwey, J.; Robinson, J.M.; Karnovsky, M.; Karnovsky, M. Studies on the mechanism of superoxide release from human neutrophils stimulated with arachidonate. J. Biol. Chem. 1984, 259, 11851–11857. [Google Scholar] [CrossRef]
- Corey, S.; Rosoff, P. Unsaturated fatty acids and lipoxygenase products regulate phagocytic NADPH oxidase activity by a nondetergent mechanism. J. Lab. Clin. Med. 1991, 118, 343–351. [Google Scholar]
- Woo, C.-H.; You, H.-J.; Cho, S.-H.; Eom, Y.-W.; Chun, J.-S.; Yoo, Y.-J.; Kim, J.-H. Leukotriene B4 stimulates Rac-ERK cascade to generate reactive oxygen species that mediates chemotaxis. J. Biol. Chem. 2002, 277, 8572–8578. [Google Scholar] [CrossRef]
- Serezani, C.H.; Aronoff, D.M.; Jancar, S.; Mancuso, P.; Peters-Golden, M. Leukotrienes enhance the bactericidal activity of alveolar macrophages against Klebsiella pneumoniae through the activation of NADPH oxidase. Blood 2005, 106, 1067–1075. [Google Scholar] [CrossRef]
- Schönfeld, P.; Wojtczak, L. Fatty acids decrease mitochondrial generation of reactive oxygen species at the reverse electron transport but increase it at the forward transport. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Cocco, T.; Di, M.; Papa, P.; Lorusso, M. Arachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generation. Free Radic. Biol. Med. 1999, 27, 51–59. [Google Scholar] [CrossRef]
- Schonfeld, P.; Reiser, G. Rotenone-like action of the branched-chain phytanic acid induces oxidative stress in mitochondria. J. Biol. Chem. 2006, 281, 7136–7142. [Google Scholar] [CrossRef] [PubMed]
- Speijer, D. Oxygen radicals shaping evolution: Why fatty acid catabolism leads to peroxisomes while neurons do without it: FADH2/NADH flux ratios determining mitochondrial radical formation were crucial for the eukaryotic invention of peroxisomes and catabolic tissue differentiation. BioEssays 2011, 33, 88–94. [Google Scholar]
- Cogliati, S.; Cabrera-Alarcón, J.L.; Enriquez, J.A. Regulation and functional role of the electron transport chain supercomplexes. Biochem. Soc. Trans. 2021, 49, 2655–2668. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, M.; Cocco, T.; Lorusso, M. Arachidonic acid causes cytochrome c release from heart mitochondria. Biochem. Biophys. Res. Commun. 2000, 277, 128–133. [Google Scholar] [CrossRef]
- Korge, P.; Honda, H.M.; Weiss, J.N. Effects of fatty acids in isolated mitochondria: Implications for ischemic injury and cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H259–H269. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Gehrmann, W.; Würdemann, W.; Plötz, T.; Jörns, A.; Lenzen, S.; Elsner, M. Antagonism between saturated and unsaturated fatty acids in ROS mediated lipotoxicity in rat insulin-producing cells. Cell. Physiol. Biochem. 2015, 36, 852–865. [Google Scholar] [CrossRef]
- Han, C.Y.; Kargi, A.Y.; Omer, M.; Chan, C.K.; Wabitsch, M.; O’Brien, K.D.; Wight, T.N.; Chait, A. Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes: Dissociation of adipocyte hypertrophy from inflammation. Diabetes 2010, 59, 386–396. [Google Scholar] [CrossRef]
- Pougovkina, O.; Te Brinke, H.; Ofman, R.; Van Cruchten, A.G.; Kulik, W.; Wanders, R.J.; Houten, S.M.; De Boer, V.C. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum. Mol. Genet. 2014, 23, 3513–3522. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Thompson, C.B. A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 270–276. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Paik, W.K.; Pearson, D.; Lee, H.W.; Kim, S. Nonenzymatic acetylation of histones with acetyl-CoA. Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1970, 213, 513–522. [Google Scholar] [CrossRef]
- Vazquez, E.J.; Berthiaume, J.M.; Kamath, V.; Achike, O.; Buchanan, E.; Montano, M.M.; Chandler, M.P.; Miyagi, M.; Rosca, M.G. Mitochondrial complex I defect and increased fatty acid oxidation enhance protein lysine acetylation in the diabetic heart. Cardiovasc. Res. 2015, 107, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Baeza, J.; Smallegan, M.J.; Denu, J.M. Mechanisms and dynamics of protein acetylation in mitochondria. Trends Biochem. Sci. 2016, 41, 231–244. [Google Scholar] [CrossRef]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.-H.; Jiang, H.; Kim, H.-S.; Flynn, C.R.; Hill, S.; McDonald, W.H. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef]
- Choi, S.-E.; Jung, I.-R.; Lee, Y.-J.; Lee, S.-J.; Lee, J.-H.; Kim, Y.; Jun, H.-S.; Lee, K.-W.; Park, C.B.; Kang, Y. Stimulation of lipogenesis as well as fatty acid oxidation protects against palmitate-induced INS-1 β-cell death. Endocrinology 2011, 152, 816–827. [Google Scholar] [CrossRef]
- Namgaladze, D.; Lips, S.; Leiker, T.J.; Murphy, R.C.; Ekroos, K.; Ferreiros, N.; Geisslinger, G.; Brüne, B. Inhibition of macrophage fatty acid β-oxidation exacerbates palmitate-induced inflammatory and endoplasmic reticulum stress responses. Diabetologia 2014, 57, 1067–1077. [Google Scholar] [CrossRef]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Slimen, I.B.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 2014, 30, 513–523. [Google Scholar] [CrossRef]
- Mujahid, A.; Pumford, N.R.; Bottje, W.; Nakagawa, K.; Miyazawa, T.; Akiba, Y.; Toyomizu, M. Mitochondrial oxidative damage in chicken skeletal muscle induced by acute heat stress. J. Poult. Sci. 2007, 44, 439–445. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Valle, N.R.-D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef]
- Schaur, R. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Asp. Med. 2003, 24, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Scaloni, A.; Giustarini, D.; Cavarra, E.; Tell, G.; Lungarella, G.; Colombo, R.; Rossi, R.; Milzani, A. Proteins as biomarkers of oxidative/nitrosative stress in diseases: The contribution of redox proteomics. Mass Spectrom. Rev. 2005, 24, 55–99. [Google Scholar] [CrossRef]
- Yuan, K.; Liu, Y.; Chen, H.N.; Zhang, L.; Lan, J.; Gao, W.; Dou, Q.; Nice, E.C.; Huang, C. Thiol-based redox proteomics in cancer research. Proteomics 2015, 15, 287–299. [Google Scholar] [CrossRef]
- Peluffo, G.; Radi, R. Biochemistry of protein tyrosine nitration in cardiovascular pathology. Cardiovasc. Res. 2007, 75, 291–302. [Google Scholar] [CrossRef]
- Cai, Z.; Yan, L.-J. Protein oxidative modifications: Beneficial roles in disease and health. J. Biochem. Pharmacol. Res. 2013, 1, 15. [Google Scholar]
- Agarwal, A.; Prabakaran, S.A. Mechanism, measurement, and prevention of oxidative stress in male reproductive physiology. Indian J. Exp. Biol. 2005, 43, 963–974. [Google Scholar] [PubMed]
- Yokoyama, Y.; Beckman, J.; Beckman, T.; Wheat, J.; Cash, T.; Freeman, B.; Parks, D. Circulating xanthine oxidase: Potential mediator of ischemic injury. Am. J. Physiol. Gastrointest. Liver Physiol. 1990, 258, G564–G570. [Google Scholar] [CrossRef]
- Poppek, D.; Grune, T. Proteasomal defense of oxidative protein modifications. Antioxid. Redox Signal. 2006, 8, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.T.; Fu, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997, 324, 1–18. [Google Scholar] [CrossRef]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox signaling and advanced glycation endproducts (AGEs) in diet-related diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef]
- Baynes, J.W. Role of oxidative stress in development of complications in diabetes. Diabetes 1991, 40, 405–412. [Google Scholar] [CrossRef]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.-B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating glycotoxins and dietary advanced glycation endproducts: Two links to inflammatory response, oxidative stress, and aging. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.-J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188. [Google Scholar] [CrossRef]
- Chan, C.M.; Huang, D.Y.; Huang, Y.P.; Hsu, S.H.; Kang, L.Y.; Shen, C.M.; Lin, W.W. Methylglyoxal induces cell death through endoplasmic reticulum stress-associated ROS production and mitochondrial dysfunction. J. Cell. Mol. Med. 2016, 20, 1749–1760. [Google Scholar] [CrossRef]
- Alikhani, M.; Alikhani, Z.; Boyd, C.; MacLellan, C.M.; Raptis, M.; Liu, R.; Pischon, N.; Trackman, P.C.; Gerstenfeld, L.; Graves, D.T. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone 2007, 40, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA damage induced by ROS-modulating agents with the ability to target DNA: A comparison of the biological characteristics of citrus pectin and apple pectin. Sci. Rep. 2018, 8, 13902. [Google Scholar] [CrossRef]
- Griffin, R.J. Radiobiology for the Radiologist, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006; Volume 66, p. 656. [Google Scholar]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Kreutzer, D.A.; Essigmann, J.M. Mutagenicity and repair of oxidative DNA damage: Insights from studies using defined lesions. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1998, 400, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Tomicic, M.T.; Roos, W.P.; Kaina, B. Mechanisms of human DNA repair: An update. Toxicology 2003, 193, 3–34. [Google Scholar] [CrossRef]
- Ock, C.-Y.; Kim, E.-H.; Choi, D.J.; Lee, H.J.; Hahm, K.-B.; Chung, M.H. 8-Hydroxydeoxyguanosine: Not mere biomarker for oxidative stress, but remedy for oxidative stress-implicated gastrointestinal diseases. World J. Gastroenterol. 2012, 18, 302. [Google Scholar] [CrossRef]
- Herrero, A.; Barja, G. Sites and mechanisms responsible for the low rate of free radical production of heart mitochondria in the long-lived pigeon. Mech. Ageing Dev. 1997, 98, 95–111. [Google Scholar] [CrossRef]
- Holmes, D.; Flückiger, R.; Austad, S. Comparative biology of aging in birds: An update. Exp. Gerontol. 2001, 36, 869–883. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Hulbert, A.J.; Buttemer, W.A. Does the oxidative stress theory of aging explain longevity differences in birds? I. Mitochondrial ROS production. Exp. Gerontol. 2012, 47, 203–210. [Google Scholar] [CrossRef]
- Pamplona, R.; Prat, J.; Cadenas, S.; Rojas, C.; Perez-Campo, R.; Torres, M.L.; Barja, G. Low fatty acid unsaturation protects against lipid peroxidation in liver mitochondria from long-lived species: The pigeon and human case. Mech. Ageing Dev. 1996, 86, 53–66. [Google Scholar] [CrossRef]
- Ogburn, C.E.; Austad, S.N.; Holmes, D.J.; Kiklevich, J.V.; Gollahon, K.; Rabinovitch, P.S.; Martin, G.M. Cultured renal epithelial cells from birds and mice: Enhanced resistance of avian cells to oxidative stress and DNA damage. J. Gerontol. A Biol. Sci. Med. Sci. 1998, 53, B287–B292. [Google Scholar] [CrossRef] [PubMed]
- Kikusato, M.; Toyomizu, M. Crucial role of membrane potential in heat stress-induced overproduction of reactive oxygen species in avian skeletal muscle mitochondria. PLoS ONE 2013, 8, e64412. [Google Scholar] [CrossRef]
- Azad, M.; Kikusato, M.; Sudo, S.; Amo, T.; Toyomizu, M. Time course of ROS production in skeletal muscle mitochondria from chronic heat-exposed broiler chicken. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2010, 157, 266–271. [Google Scholar] [CrossRef]
- Mujahid, A.; Sato, K.; Akiba, Y.; Toyomizu, M. Acute heat stress stimulates mitochondrial superoxide production in broiler skeletal muscle, possibly via downregulation of uncoupling protein content. Poult. Sci. 2006, 85, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Al-Zghoul, M.; Sukker, H.; Ababneh, M. Effect of thermal manipulation of broilers embryos on the response to heat-induced oxidative stress. Poult. Sci. 2019, 98, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Miao, Q.; Si, X.; Xie, Y.; Chen, L.; Liu, Z.; Liu, L.; Tang, X.; Zhang, H. Effects of acute heat stress at different ambient temperature on hepatic redox status in broilers. Poult. Sci. 2020, 99, 4113–4122. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Z.; Xing, T.; Li, J.; Zhang, L.; Zhao, L.; Gao, F. Effect of chronic heat stress on the carbonylation of glycolytic enzymes in breast muscle and its correlation with the growth performance of broilers. Poult. Sci. 2023, 102, 103103. [Google Scholar] [CrossRef]
- Jing, J.; Zeng, H.; Shao, Q.; Tang, J.; Wang, L.; Jia, G.; Liu, G.; Chen, X.; Tian, G.; Cai, J. Selenomethionine alleviates environmental heat stress induced hepatic lipid accumulation and glycogen infiltration of broilers via maintaining mitochondrial and endoplasmic reticulum homeostasis. Redox Biol. 2023, 67, 102912. [Google Scholar] [CrossRef]
- Kwakye, J.; Ariyo, O.W.; Ghareeb, A.F.; Hartono, E.; Sovi, S.; Aryal, B.; Milfort, M.C.; Fuller, A.L.; Rekaya, R.; Aggrey, S.E. Effect of Glucose Supplementation on Apoptosis in the Pectoralis major of Chickens Raised under Thermoneutral or Heat Stress Environment. Genes 2023, 14, 1922. [Google Scholar] [CrossRef]
- Uyanga, V.A.; Wang, M.; Tong, T.; Zhao, J.; Wang, X.; Jiao, H.; Onagbesan, O.M.; Lin, H. L-citrulline influences the body temperature, heat shock response and nitric oxide regeneration of broilers under thermoneutral and heat stress condition. Front. Physiol. 2021, 12, 671691. [Google Scholar] [CrossRef]
- Ariyo, O.W.; Kwakye, J.; Sovi, S.; Aryal, B.; Hartono, E.; Ghareeb, A.F.; Milfort, M.C.; Fuller, A.L.; Rekaya, R.; Aggrey, S.E. mRNA expression of kidney aquaporins and blood composition of meat-type chickens raised with or without glucose supplementation under cyclic heat or thermoneutral condition. J. Therm. Biol. 2024, 126, 104003. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Sarwar, M. Dietary electrolyte balance: Implications in heat stressed broilers. World Poult. Sci. J. 2006, 62, 638–653. [Google Scholar]
- Mongin, P. Acid-base balance during eggshell formation. In Proceedings of the Respiratory Function in Birds, Adult and Embryonic: Satellite Symposium of the 27th International Congress of Physiological Sciences, Paris, France, 28–30 July 1977; Springer: Berlin/Heidelberg, Germany, 1978; pp. 247–259. [Google Scholar]
- Hyline. Understanding Heat Stress in Layers: Management Tips to Improve Hot Weather Flock Performance. Available online: https://www.hyline.com/ViewFile?id=ff054c39-aa45-43ae-adbe-1488017266f1 (accessed on 19 October 2024).
- Krinsky, N.I. Mechanism of action of biological antioxidants. Proc. Soc. Exp. Biol. Med. 1992, 200, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Rice-Evans, C.A.; Diplock, A.T. Current status of antioxidant therapy. Free Radic. Biol. Med. 1993, 15, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Dunning, S.; ur Rehman, A.; Tiebosch, M.H.; Hannivoort, R.A.; Haijer, F.W.; Woudenberg, J.; van den Heuvel, F.A.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Glutathione and antioxidant enzymes serve complementary roles in protecting activated hepatic stellate cells against hydrogen peroxide-induced cell death. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 2027–2034. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef]
- Bryk, R.; Griffin, P.; Nathan, C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature 2000, 407, 211–215. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Karplus, P.A. A primer on peroxiredoxin biochemistry. Free Radic. Biol. Med. 2015, 80, 183–190. [Google Scholar] [CrossRef]
- Alvarez, B.; Salinas, G. Basic concepts of thiol chemistry and biology. In Redox Chemistry and Biology of Thiols; Elsevier: Amsterdam, The Netherlands, 2022; pp. 1–18. [Google Scholar]
- Hatahet, F.; Ruddock, L.W. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.-R.; Lappi, A.-K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.; Wang, C.-C.; Ruddock, L.W. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef]
- Sen, C.K.; Packer, L.; Hänninen, O. Biological thiols and redox regulation of cellular signal transduction pathways. In Handbook of Oxidants and Antioxidants in Exercise Amsterdam; Elsevier: Amsterdam, The Netherlands, 2000; pp. 375–401. [Google Scholar]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Nagy, P. Kinetics and mechanisms of thiol–disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid. Redox Signal. 2013, 18, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Thioredoxin. Annu. Rev. Biochem. 1985, 54, 237–271. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid. Redox Signal. 2011, 15, 781–794. [Google Scholar] [CrossRef]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2010, 425, 313–325. [Google Scholar] [CrossRef]
- Lind, C.; Gerdes, R.; Schuppe-Koistinen, I.; Cotgreave, I.A. Studies on the mechanism of oxidative modification of human glyceraldehyde-3-phosphate dehydrogenase by glutathione: Catalysis by glutaredoxin. Biochem. Biophys. Res. Commun. 1998, 247, 481–486. [Google Scholar] [CrossRef]
- Davis, D.A.; Newcomb, F.M.; Starke, D.W.; Ott, D.E.; Mieyal, J.J.; Yarchoan, R. Thioltransferase (glutaredoxin) is detected within HIV-1 and can regulate the activity of glutathionylated HIV-1 protease in vitro. J. Biol. Chem. 1997, 272, 25935–25940. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 1304–1317. [Google Scholar] [CrossRef]
- Lundberg, M.; Johansson, C.; Chandra, J.; Enoksson, M.; Jacobsson, G.; Ljung, J.; Johansson, M.; Holmgren, A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J. Biol. Chem. 2001, 276, 26269–26275. [Google Scholar] [CrossRef]
- Johansson, C.; Lillig, C.H.; Holmgren, A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J. Biol. Chem. 2004, 279, 7537–7543. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R. Tissue-specific functions of individual glutathione peroxidases. Free Radic. Biol. Med. 1999, 27, 951–965. [Google Scholar] [CrossRef]
- Bindoli, A.; Fukuto, J.M.; Forman, H.J. Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid. Redox Signal. 2008, 10, 1549–1564. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Toledano, M.B.; Delaunay-Moisan, A.; Outten, C.E.; Igbaria, A. Functions and cellular compartmentation of the thioredoxin and glutathione pathways in yeast. Antioxid. Redox Signal. 2013, 18, 1699–1711. [Google Scholar] [CrossRef]
- Chai, Y.-C.; Ashraf, S.S.; Rokutan, K.; Johnston, R.B.; Thomas, J.A. S-thiolation of individual human neutrophil proteins including actin by stimulation of the respiratory burst: Evidence against a role for glutathione disulfide. Arch. Biochem. Biophys. 1994, 310, 273–281. [Google Scholar] [CrossRef]
- Sanderson, S.M.; Gao, X.; Dai, Z.; Locasale, J.W. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat. Rev. Cancer 2019, 19, 625–637. [Google Scholar] [CrossRef]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in aging and disease. Aging 2011, 3, 716. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Functions of polyamines in mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef] [PubMed]
- Tarver, H.; Schmidt, C. The conversion of methionine to cystine: Experiments with radioactive sulfur. J. Biol. Chem. 1939, 130, 67–80. [Google Scholar] [CrossRef]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Stipanuk, M.H. Metabolism of sulfur-containing amino acids. Annu. Rev. Nutr. 1986, 6, 179–209. [Google Scholar] [CrossRef]
- Shils, M.E.; Olson, J.A.; Shike, M. Modern Nutrition in Health and Disease; Lea & Febiger: Philadelphia, PA, USA, 1994; Volume 2. [Google Scholar]
- Stipanuk, M.; Ueki, I.; Dominy, J.; Simmons, C.; Hirschberger, L. Cysteine dioxygenase: A robust system for regulation of cellular cysteine levels. Amino Acids 2009, 37, 55–63. [Google Scholar] [CrossRef]
- Colovic, M.B.; Vasic, V.M.; Djuric, D.M.; Krstic, D.Z. Sulphur-containing amino acids: Protective role against free radicals and heavy metals. Curr. Med. Chem. 2018, 25, 324–335. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Londono, M.; Lee, J.-I.; Hu, M.; Anthony, F.Y. Enzymes and metabolites of cysteine metabolism in nonhepatic tissues of rats show little response to changes in dietary protein or sulfur amino acid levels. J. Nutr. 2002, 132, 3369–3378. [Google Scholar] [CrossRef]
- Cresenzi, C.L.; Lee, J.I.; Stipanuk, M.H. Cysteine is the metabolic signal responsible for dietary regulation of hepatic cysteine dioxygenase and glutamate cysteine ligase in intact rats. J. Nutr. 2003, 133, 2697–2702. [Google Scholar] [CrossRef]
- Bagley, P.J.; Stipanuk, M.H. The activities of rat hepatic cysteine dioxygenase and cysteinesulfinate decarboxylase are regulated in a reciprocal manner in response to dietary casein level. J. Nutr. 1994, 124, 2410–2421. [Google Scholar] [CrossRef]
- Bagley, P.J.; Stipanuk, M.H. Rats fed a low protein diet supplemented with sulfur amino acids have increased cysteine dioxygenase activity and increased taurine production in hepatocytes. J. Nutr. 1995, 125, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Bella, D.L.; Stipanuk, M.H. High levels of dietary protein or methionine have different effects on cysteine metabolism in rat hepatocytes. In Taurine 2 Basic and Clinical Aspects; Springer: Boston, MA, USA, 1996; pp. 73–84. [Google Scholar]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.M.; MacIver, F.H.; Dawes, I.W. Glutathione synthetase is dispensable for growth under both normal and oxidative stress conditions in the yeast Saccharomyces cerevisiae due to an accumulation of the dipeptide gamma-glutamylcysteine. Mol. Biol. Cell 1997, 8, 1699–1707. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Akerboom, T.; Bilzer, M.; Sies, H. The relationship of biliary glutathione disulfide efflux and intracellular glutathione disulfide content in perfused rat liver. J. Biol. Chem. 1982, 257, 4248–4252. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Surai, P.F.; Kochish, I.I.; Fisinin, V.I.; Kidd, M.T. Antioxidant defence systems and oxidative stress in poultry biology: An update. Antioxidants 2019, 8, 235. [Google Scholar] [CrossRef]
- Matkovics, B.; Novak, R.; Hanh, H.D.; Szabo, L.; Varga, S.I. A comparative study of some more important experimental animal peroxide metabolism enzymes. Comp. Biochem. Physiology. B Comp. Biochem. 1977, 56, 397–402. [Google Scholar] [CrossRef]
- Ku, H.-H.; Sohal, R. Comparison of mitochondrial pro-oxidant generation and anti-oxidant defenses between rat and pigeon: Possible basis of variation in longevity and metabolic potential. Mech. Ageing Dev. 1993, 72, 67–76. [Google Scholar] [CrossRef]
- Barja, G.; Cadenas, S.; Rojas, C.; Lopez-Torres, M.; Perez-Campo, R. A decrease of free radical production near critical targets as a cause of maximum longevity in animals. Comp. Biochem. Physiol. Part B Comp. Biochem. 1994, 108, 501–512. [Google Scholar] [CrossRef]
- Cutler, R.G. Peroxide-producing potential of tissues: Inverse correlation with longevity of mammalian species. Proc. Natl. Acad. Sci. USA 1985, 82, 4798–4802. [Google Scholar] [CrossRef] [PubMed]
- Perez-Campo, R.; Lopez-Torres, M.; Rojas, C.; Cadenas, S.; Barja, G. Longevity and antioxidant enzymes, non-enzymatic antioxidants and oxidative stress in the vertebrate lung: A comparative study. J. Comp. Physiol. B 1994, 163, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Hulbert, A.J.; Buttemer, W.A. The long life of birds: The rat-pigeon comparison revisited. PLoS ONE 2011, 6, e24138. [Google Scholar] [CrossRef]
- Del Vesco, A.; Khatlab, A.; Goes, E.; Utsunomiya, K.; Vieira, J.; Neto, A.O.; Gasparino, E. Age-related oxidative stress and antioxidant capacity in heat-stressed broilers. Animal 2017, 11, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.-Y.; Yang, L.; Fu, Y.-Q.; Feng, J.-H.; Zhang, M.-H. Effects of different acute high ambient temperatures on function of hepatic mitochondrial respiration, antioxidative enzymes, and oxidative injury in broiler chickens. Poult. Sci. 2010, 89, 115–122. [Google Scholar] [CrossRef]
- Zhang, J.; Bai, K.; Su, W.; Wang, A.; Zhang, L.; Huang, K.; Wang, T. Curcumin attenuates heat-stress-induced oxidant damage by simultaneous activation of GSH-related antioxidant enzymes and Nrf2-mediated phase II detoxifying enzyme systems in broiler chickens. Poult. Sci. 2018, 97, 1209–1219. [Google Scholar] [CrossRef]
- Xiao, R.; Power, R.; Mallonee, D.; Routt, K.; Spangler, L.; Pescatore, A.; Cantor, A.; Ao, T.; Pierce, J.; Dawson, K. Effects of yeast cell wall-derived mannan-oligosaccharides on jejunal gene expression in young broiler chickens. Poult. Sci. 2012, 91, 1660–1669. [Google Scholar] [CrossRef]
- Smith, A.D.; Morris, V.C.; Levander, O.A. Rapid determination of glutathione peroxidase and thioredoxin reductase activities using a 96-well microplate format: Comparison to standard cuvette-based assays. Int. J. Vitam. Nutr. Res. 2001, 71, 87–92. [Google Scholar] [CrossRef]
- Han, J.Y.; Song, K.; Shin, J.; Han, B.; Park, T.; Park, H.; Kim, J.; Lillehoj, H.; Lim, J.; Kim, H. Identification and characterization of the peroxiredoxin gene family in chickens. Poult. Sci. 2005, 84, 1432–1438. [Google Scholar] [CrossRef]
- Zhang, J.; Bai, K.W.; He, J.; Niu, Y.; Lu, Y.; Zhang, L.; Wang, T. Curcumin attenuates hepatic mitochondrial dysfunction through the maintenance of thiol pool, inhibition of mtDNA damage, and stimulation of the mitochondrial thioredoxin system in heat-stressed broilers. J. Anim. Sci. 2018, 96, 867–879. [Google Scholar] [CrossRef]
- Cheng, C.-Y.; Tu, W.-L.; Chen, C.-J.; Chan, H.-L.; Chen, C.-F.; Chen, H.-H.; Tang, P.-C.; Lee, Y.-P.; Chen, S.-E.; Huang, S.-Y. Functional genomics study of acute heat stress response in the small yellow follicles of layer-type chickens. Sci. Rep. 2018, 8, 1320. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, A.D.; Liu, G.; Sun, T.; Tolba, S.A.; Xi, L.; Whelan, R.; Lei, X.G. Supplemental methionine and stocking density affect antioxidant status, fatty acid profiles, and growth performance of broiler chickens. J. Anim. Sci. 2020, 98, skaa092. [Google Scholar] [CrossRef] [PubMed]
- Zduńczyk, Z.; Jankowski, J.; Kubińska, M.; Ognik, K.; Czech, A.; Juśkiewicz, J. The effect of different dietary levels of dl-methionine and dl-methionine hydroxy analogue on the antioxidant and immune status of young turkeys. Arch. Anim. Nutr. 2017, 71, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Khatlab, A.d.S.; Del Vesco, A.P.; de Oliveira Neto, A.R.; Fernandes, R.P.M.; Gasparino, E. Dietary supplementation with free methionine or methionine dipeptide mitigates intestinal oxidative stress induced by Eimeria spp. challenge in broiler chickens. J. Anim. Sci. Biotechnol. 2019, 10, 58. [Google Scholar] [CrossRef]
- Del Vesco, A.P.; Gasparino, E.; de Oliveira Grieser, D.; Zancanela, V.; Soares, M.A.M.; de Oliveira Neto, A.R. Effects of methionine supplementation on the expression of oxidative stress-related genes in acute heat stress-exposed broilers. Br. J. Nutr. 2015, 113, 549–559. [Google Scholar] [CrossRef]
- Aggrey, S.; González-Cerón, F.; Rekaya, R.; Mercier, Y. Gene expression differences in the methionine remethylation and transsulphuration pathways under methionine restriction and recovery with D, L-methionine or D, L-HMTBA in meat-type chickens. J. Anim. Physiol. Anim. Nutr. 2018, 102, e468–e475. [Google Scholar] [CrossRef]
- D’mello, J. Amino acids as multifunctional molecules. In Amino Acids in Animal Nutrition; Cabi Publishing: Wallingford, Oxfordshire, UK, 2003; pp. 1–14. [Google Scholar]
- Bannai, S.; Tateishi, N. Role of membrane transport in metabolism and function of glutathione in mammals. J. Membr. Biol. 1986, 89, 1–8. [Google Scholar] [CrossRef]
- Ishii, T.; Sugita, Y.; Bannai, S. Regulation of glutathione levels in mouse spleen lymphocytes by transport of cysteine. J. Cell. Physiol. 1987, 133, 330–336. [Google Scholar] [CrossRef]
- Habashy, W.; Milfort, M.; Attia, Y.; Rekaya, R.; Aggrey, S. Effect of heat stress on amino acid digestibility and transporters in meat-type chickens. Poult. Sci. 2017, 96, 2312–2319. [Google Scholar] [CrossRef]
- Vilar da Silva, J.H.; González-Cerón, F.; Howerth, E.W.; Rekaya, R.; Aggrey, S.E. Alteration of dietary cysteine affects activities of genes of the transsulfuration and glutathione pathways, and development of skin tissues and feather follicles in chickens. Anim. Biotechnol. 2020, 31, 203–208. [Google Scholar] [CrossRef]
- Cao, X.; Guo, L.; Zhou, C.; Huang, C.; Li, G.; Zhuang, Y.; Yang, F.; Liu, P.; Hu, G.; Gao, X. Effects of N-acetyl-l-cysteine on chronic heat stress-induced oxidative stress and inflammation in the ovaries of growing pullets. Poult. Sci. 2023, 102, 102274. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Hou, Y.; Tan, L.; Liao, M.; Xie, J.; Wang, L.; Ding, B.; Yang, Y.; Gong, J. N-acetylcysteine improves the growth performance and intestinal function in the heat-stressed broilers. Anim. Feed Sci. Technol. 2016, 220, 83–92. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.-L.; Xing, G.-D.; Qian, Y.; Zhong, J.-F.; Chen, K.-L. S-allyl cysteine ameliorates heat stress-induced oxidative stress by activating Nrf2/HO-1 signaling pathway in BMECs. Toxicol. Appl. Pharmacol. 2021, 416, 115469. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.I.M.; Murakami, A.E. Arginine metabolism in uricotelic species. Acta Sci. Anim. Sci. 2010, 32, 357–366. [Google Scholar] [CrossRef]
- Subramaniyan, S.A.; Kang, D.R.; Park, J.R.; Siddiqui, S.H.; Ravichandiran, P.; Yoo, D.J.; Na, C.S.; Shim, K.S. Effect of in ovo injection of l-arginine in different chicken embryonic development stages on post-hatchability, immune response, and Myo-D and myogenin proteins. Animals 2019, 9, 357. [Google Scholar] [CrossRef]
- Flynn, N.; Meininger, C.; Haynes, T.; Wu, G. The metabolic basis of arginine nutrition and pharmacotherapy. Biomed. Pharmacother. 2002, 56, 427–438. [Google Scholar] [CrossRef]
- Elnesr, S.; Elwan, H.; Xu, Q.; Xie, C.; Dong, X.; Zou, X. Effects of in ovo injection of sulfur-containing amino acids on heat shock protein 70, corticosterone hormone, antioxidant indices, and lipid profile of newly hatched broiler chicks exposed to heat stress during incubation. Poult. Sci. 2019, 98, 2290–2298. [Google Scholar] [CrossRef]
- El-Hadad, E.; Madian, H.; Hassan, M.; Saad, M.; El-Shhat, A.; Eliraqy, E. Impact of dietary arginine supplementation on hemato-biochmical, histological, antioxidant, and immunity variables of geese under heat stress condition in Egypt. Adv. Anim. Vet. Sci. 2024, 12, 411–421. [Google Scholar] [CrossRef]
- Sifa, D.; Bai, X.; Zhang, D.; Hu, H.; Wu, X.; Wen, A.; He, S.; Zhao, L. Dietary glutamine improves meat quality, skeletal muscle antioxidant capacity and glutamine metabolism in broilers under acute heat stress. J. Appl. Anim. Res. 2018, 46, 1412–1417. [Google Scholar] [CrossRef]
- Ding, K.-N.; Lu, M.-H.; Guo, Y.-N.; Liang, S.-S.; Mou, R.-W.; He, Y.-M.; Tang, L.-P. Resveratrol relieves chronic heat stress-induced liver oxidative damage in broilers by activating the Nrf2-Keap1 signaling pathway. Ecotoxicol. Environ. Saf. 2023, 249, 114411. [Google Scholar] [CrossRef]
- Sahin, K.; Orhan, C.; Tuzcu, M.; Sahin, N.; Hayirli, A.; Bilgili, S.; Kucuk, O. Lycopene activates antioxidant enzymes and nuclear transcription factor systems in heat-stressed broilers. Poult. Sci. 2016, 95, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Onderci, M.; Sahin, K.; Sahin, N.; Gursu, M.; Doerge, D.; Sarkar, F.; Kucuk, O. The effect of genistein supplementation on performance and antioxidant status of Japanese quail under heat stress. Arch. Anim. Nutr. 2004, 58, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Shan, A.; Chen, Z.; Du, J.; Song, K.; Li, J.; Xu, Q. Effect of Ligustrum lucidum and Schisandra chinensis on the egg production, antioxidant status and immunity of laying hens during heat stress. Arch. Anim. Nutr. 2005, 59, 439–447. [Google Scholar] [CrossRef]
- Hosseindoust, A.; Oh, S.M.; Ko, H.S.; Jeon, S.M.; Ha, S.H.; Jang, A.; Son, J.S.; Kim, G.Y.; Kang, H.K.; Kim, J.S. Muscle antioxidant activity and meat quality are altered by supplementation of astaxanthin in broilers exposed to high temperature. Antioxidants 2020, 9, 1032. [Google Scholar] [CrossRef]
- Pirgozliev, V.R.; Mansbridge, S.C.; Westbrook, C.A.; Woods, S.L.; Rose, S.P.; Whiting, I.M.; Yovchev, D.G.; Atanasov, A.G.; Kljak, K.; Staykova, G.P. Feeding dihydroquercetin and vitamin E to broiler chickens reared at standard and high ambient temperatures. Arch. Anim. Nutr. 2020, 74, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, C.; Wang, Z.; Li, W.; Sun, W.; Zhang, F.; Hong, Y.; Liu, X.; Liu, X.; Lyu, Q. Antioxidant effect of taurine on chronic heat-stressed broilers. In Taurine 12 A Conditionally Essential Amino Acid; Springer: Berlin/Heidelberg, Germany, 2022; pp. 161–169. [Google Scholar]
- Attia, Y.A.; El-Naggar, A.S.; Abou-Shehema, B.M.; Abdella, A.A. Effect of supplementation with trimethylglycine (betaine) and/or vitamins on semen quality, fertility, antioxidant status, DNA repair and welfare of roosters exposed to chronic heat stress. Animals 2019, 9, 547. [Google Scholar] [CrossRef]
- Del Vesco, A.; Gasparino, E.; Zancanela, V.; Grieser, D.; Stanquevis, C.; Pozza, P.; Oliveira Neto, A. Effects of selenium supplementation on the oxidative state of acute heat stress-exposed quails. J. Anim. Physiol. Anim. Nutr. 2017, 101, 170–179. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2015, 15, 71. [Google Scholar] [CrossRef]
- Packer, L.; Obermüller-Jevic, U. Vitamin E: An introduction. In The Antioxidant Vitamins C and E, Proceedings of the a Symposium Held at the 2002 World Congress of the Oxygen Club of California, Santa Barbara, CA, USA, 6–9 March 2002; AOCS Press: Champaign, IL, USA, 2002. [Google Scholar]
- Diplock, A. The role of vitamin E in biological membranes. In Proceedings of the Ciba Foundation Symposium 101-Biology of Vitamin E, London, UK, 8–10 March 1983; pp. 45–55. [Google Scholar]
- Niki, E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: In vitro and in vivo evidence. Free Radic. Biol. Med. 2014, 66, 3–12. [Google Scholar] [CrossRef]
- Wang, X.; Quinn, P.J. The location and function of vitamin E in membranes. Mol. Membr. Biol. 2000, 17, 143–156. [Google Scholar]
- Packer, J.E.; Slater, T.; Willson, R. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature 1979, 278, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Rigo, A.; Maiorino, M.; Ursini, F.; Gregolin, C. Formation of α-tocopherol radical and recycling of α-tocopherol by ascorbate during peroxidation of phosphatidylcholine liposomes: An electron paramagnetic resonance study. Biochim. Biophys. Acta Gen. Subj. 1984, 801, 215–219. [Google Scholar] [CrossRef]
- Doba, T.; Burton, G.W.; Ingold, K.U. Antioxidant and co-antioxidant activity of vitamin C. The effect of vitamin C, either alone or in the presence of vitamin E or a water-soluble vitamin E analogue, upon the peroxidation of aqueous multilamellar phospholipid liposomes. Biochim. Biophys. Acta Lipids Lipid Metab. 1985, 835, 298–303. [Google Scholar] [CrossRef]
- Niki, E.; Kawakami, A.; Yamamoto, Y.; Kamiya, Y. Oxidation of lipids. VIII. Synergistic inhibition of oxidation of phosphatidylcholine liposome in aqueous dispersion by vitamin E and vitamin C. Bull. Chem. Soc. Jpn. 1985, 58, 1971–1975. [Google Scholar] [CrossRef]
- Linster, C.L.; Van Schaftingen, E. Vitamin C: Biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef]
- Biswas, P.; Dellanoce, C.; Vezzoli, A.; Mrakic-Sposta, S.; Malnati, M.; Beretta, A.; Accinni, R. Antioxidant activity with increased endogenous levels of vitamin C, E and A following dietary supplementation with a combination of glutathione and resveratrol precursors. Nutrients 2020, 12, 3224. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Several lines of antioxidant defense against oxidative stress: Antioxidant enzymes, nanomaterials with multiple enzyme-mimicking activities, and low-molecular-weight antioxidants. Arch. Toxicol. 2024, 98, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Glutathione-ascorbic acid antioxidant system in animals. J. Biol. Chem. 1994, 269, 9397–9400. [Google Scholar] [CrossRef]
- Lee, E.; Park, H.-Y.; Kim, S.-W.; Kim, J.; Lim, K. Vitamin C and glutathione supplementation: A review of their additive effects on exercise performance. Phys. Act. Nutr. 2023, 27, 36. [Google Scholar] [CrossRef]
- Mårtensson, J.; Meister, A. Glutathione deficiency increases hepatic ascorbic acid synthesis in adult mice. Proc. Natl. Acad. Sci. USA 1992, 89, 11566–11568. [Google Scholar] [CrossRef]
- Pollak, N.; Dölle, C.; Ziegler, M. The power to reduce: Pyridine nucleotides–small molecules with a multitude of functions. Biochem. J. 2007, 402, 205–218. [Google Scholar] [CrossRef]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef]
- Kuehne, A.; Emmert, H.; Soehle, J.; Winnefeld, M.; Fischer, F.; Wenck, H.; Gallinat, S.; Terstegen, L.; Lucius, R.; Hildebrand, J. Acute activation of oxidative pentose phosphate pathway as first-line response to oxidative stress in human skin cells. Mol. Cell 2015, 59, 359–371. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Moon, S.J.; Dong, W.; Stephanopoulos, G.N.; Sikes, H.D. Oxidative pentose phosphate pathway and glucose anaplerosis support maintenance of mitochondrial NADPH pool under mitochondrial oxidative stress. Bioeng. Transl. Med. 2020, 5, e10184. [Google Scholar] [CrossRef]
- Thornton, P. Increased environmental temperature influences on ascorbic acid activity in the domestic fowl. Fed. Proc. 1961, 20, 210A. [Google Scholar]
- Da Rocha, C.; Maiorka, A.; de Paula Valle, F.; Schramm, V.G.; Angeli, A.; da Silva, A.F. The effect of soybean oil quality and vitamin E supplementation on turkey diet nutrition. J. Appl. Poult. Res. 2012, 21, 318–324. [Google Scholar] [CrossRef]
- Chan, K.M.; Decker, E.A.; Feustman, C. Endogenous skeletal muscle antioxidants. Crit. Rev. Food Sci. Nutr. 1994, 34, 403–426. [Google Scholar] [CrossRef]
- Cinar, M.; Yildirim, E.; Yigit, A.A.; Yalcinkaya, I.; Duru, O.; Kisa, U.; Atmaca, N. Effects of dietary supplementation with vitamin C and vitamin E and their combination on growth performance, some biochemical parameters, and oxidative stress induced by copper toxicity in broilers. Biol. Trace Elem. Res. 2014, 158, 186–196. [Google Scholar] [CrossRef]
- Hashem, M.A.; Abd El Hamied, S.S.; Ahmed, E.M.; Amer, S.A.; Hassan, A.M. Alleviating effects of vitamins C and E supplementation on oxidative stress, hematobiochemical, and histopathological alterations caused by copper toxicity in broiler chickens. Animals 2021, 11, 1739. [Google Scholar] [CrossRef]
- Jena, B.; Panda, N.; Patra, R.; Mishra, P.; Behura, N.; Panigrahi, B. Supplementation of vitamin E and C reduces oxidative stress in broiler breeder hens during summer. Food Nutr. Sci. 2013, 4, 33–37. [Google Scholar] [CrossRef]
- Rengaraj, D.; Hong, Y.H. Effects of dietary vitamin E on fertility functions in poultry species. Int. J. Mol. Sci. 2015, 16, 9910–9921. [Google Scholar] [CrossRef]
- Surai, P.F.; Fisinin, V.I.; Karadas, F. Antioxidant systems in chick embryo development. Part 1. Vitamin E, carotenoids and selenium. Anim. Nutr. 2016, 2, 1–11. [Google Scholar] [CrossRef]
- Sahin, K.; Sahin, N.; Onderci, M. Vitamin E supplementation can alleviate negative effects of heat stress on egg production, egg quality, digestibility of nutrients and egg yolk mineral concentrations of Japanese quails. Res. Vet. Sci. 2002, 73, 307–312. [Google Scholar] [CrossRef]
- Vakili, R.; Rashidi, A.A.; Sobhanirad, S. Effects of dietary fat, vitamin E and zinc supplementation on tibia breaking strength in female broilers under heat stress. Afr. J. Agric. Res. 2010, 5, 3151–3156. [Google Scholar]
- Morrissey, P.; Brandon, S.; Buckley, D.; Sheehy, P.; Frigg, M. Tissue content of α-tocopherol and oxidative stability of broilers receiving dietary α-tocopheryl acetate supplement for various periods pre-slaughter. Br. Poult. Sci. 1997, 38, 84–88. [Google Scholar] [CrossRef]
- Rehman, Z.U.; Meng, C.; Sun, Y.; Mahrose, K.M.; Umar, S.; Ding, C.; Munir, M. Pathobiology of Avian avulavirus 1: Special focus on waterfowl. Vet. Res. 2018, 49, 94. [Google Scholar] [CrossRef]
- Schmeling, S.K.; Nockels, C.F. Effects of age, sex, and ascorbic acid ingestion on chicken plasma corticosterone levels. Poult. Sci. 1978, 57, 527–533. [Google Scholar] [CrossRef]
- Julian, R.J. Physiological Management & Environmental Triggers of The Ascites Syndrome. Poult. Int. 1998, 37, 28–33. [Google Scholar]
- Panda, A.; Ramarao, S.; Raju, M.; Chatterjee, R. Effect of dietary supplementation with vitamins E and C on production performance, immune responses and antioxidant status of White Leghorn layers under tropical summer conditions. Br. Poult. Sci. 2008, 49, 592–599. [Google Scholar] [CrossRef]
- Jastrebski, S.F.; Lamont, S.J.; Schmidt, C.J. Chicken hepatic response to chronic heat stress using integrated transcriptome and metabolome analysis. PLoS ONE 2017, 12, e0181900. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef]
- Martin, T.G.; Kirk, J.A. Under construction: The dynamic assembly, maintenance, and degradation of the cardiac sarcomere. J. Mol. Cell. Cardiol. 2020, 148, 89–102. [Google Scholar] [CrossRef]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem. 2017, 86, 97–122. [Google Scholar] [CrossRef]
- Finley, D.; Prado, M.A. The proteasome and its network: Engineering for adaptability. Cold Spring Harb. Perspect. Biol. 2020, 12, a033985. [Google Scholar] [CrossRef]
- Davies, K.; Goldberg, A. Oxygen radicals stimulate intracellular proteolysis and lipid peroxidation by independent mechanisms in erythrocytes. J. Biol. Chem. 1987, 262, 8220–8226. [Google Scholar] [CrossRef]
- Quinlan, R.A.; Ellis, R.J. Chaperones: Needed for both the good times and the bad times. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20130091. [Google Scholar] [CrossRef]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef]
- Haslbeck, M.; Vierling, E. A first line of stress defense: Small heat shock proteins and their function in protein homeostasis. J. Mol. Biol. 2015, 427, 1537–1548. [Google Scholar] [CrossRef]
- Jeng, W.; Lee, S.; Sung, N.; Lee, J.; Tsai, F.T. Molecular chaperones: Guardians of the proteome in normal and disease states. F1000Research 2015, 4, 1448. [Google Scholar] [CrossRef]
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Deuerling, E.; Pfund, C.; Craig, E.A. Getting newly synthesized proteins into shape. Cell 2000, 101, 119–122. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef]
- Young, J.C.; Barral, J.M.; Hartl, F.U. More than folding: Localized functions of cytosolic chaperones. Trends Biochem. Sci. 2003, 28, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Neupert, W.; Brunner, M. The protein import motor of mitochondria. Nat. Rev. Mol. Cell Biol. 2002, 3, 555–565. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Park, S.-H.; Bolender, N.; Eisele, F.; Kostova, Z.; Takeuchi, J.; Coffino, P.; Wolf, D.H. The cytoplasmic Hsp70 chaperone machinery subjects misfolded and endoplasmic reticulum import-incompetent proteins to degradation via the ubiquitin–proteasome system. Mol. Biol. Cell 2007, 18, 153–165. [Google Scholar] [CrossRef]
- Hoffmann, J.H.; Linke, K.; Graf, P.C.; Lilie, H.; Jakob, U. Identification of a redox-regulated chaperone network. EMBO J. 2004, 23, 160–168. [Google Scholar] [CrossRef]
- Shi, Y.; Mosser, D.D.; Morimoto, R.I. Molecularchaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998, 12, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Guettouche, T.; Fenna, M.; Boellmann, F.; Pratt, W.B.; Toft, D.O.; Smith, D.F.; Voellmy, R. Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J. Biol. Chem. 2001, 276, 45791–45799. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wharton, W.; Moseley, P.; Shi, H. Heat shock protein 70 regulates cellular redox status by modulating glutathione-related enzyme activities. Cell Stress Chaperones 2007, 12, 245. [Google Scholar] [CrossRef]
- Portero-Otín, M.; Requena, J.R.; Bellmunt, M.J.; Ayala, V.; Pamplona, R. Protein nonenzymatic modifications and proteasome activity in skeletal muscle from the short-lived rat and long-lived pigeon. Exp. Gerontol. 2004, 39, 1527–1535. [Google Scholar] [CrossRef]
- Mezquita, B.; Mezquita, C.; Mezquita, J. Marked differences between avian and mammalian testicular cells in the heat shock induction and polyadenylation of Hsp70 and ubiquitin transcripts. FEBS Lett. 1998, 436, 382–386. [Google Scholar] [CrossRef]
- Furukawa, K.; Kikusato, M.; Kamizono, T.; Yoshida, H.; Toyomizu, M. Possible involvement of mitochondrial reactive oxygen species production in protein degradation induced by heat stress in avian muscle cells. J. Poult. Sci. 2015, 52, 260–267. [Google Scholar] [CrossRef]
- Lin, H.; Decuypere, E.; Buyse, J. Oxidative stress induced by corticosterone administration in broiler chickens (Gallus gallus domesticus): 1. Chronic exposure. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2004, 139, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Hao, Y.; Wang, X. Overexpression of heat shock protein 70 and its relationship to intestine under acute heat stress in broilers: 2. Intestinal oxidative stress. Poult. Sci. 2012, 91, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, J.E.; da Mota, A.F.; Boleli, I.C.; Macari, M.; Coutinho, L.L. Effect of moderate and severe heat stress on avian embryonic hsp70 gene expression. Growth Dev. Aging 2002, 66, 27–33. [Google Scholar]
- Ghareeb, A.F.; Foutz, J.C.; Schneiders, G.H.; Richter, J.N.; Milfort, M.C.; Fuller, A.L.; Rekaya, R.; Aggrey, S.E. Host transcriptome response to heat stress and Eimeria maxima infection in meat-type chickens. PLoS ONE 2024, 19, e0296350. [Google Scholar] [CrossRef]
- Wang, S.; Edens, F. Heat conditioning induces heat shock proteins in broiler chickens and turkey poults. Poult. Sci. 1998, 77, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Al-Zghoul, M. Thermal manipulation during broiler chicken embryogenesis increases basal mRNA levels and alters production dynamics of heat shock proteins 70 and 60 and heat shock factors 3 and 4 during thermal stress. Poult. Sci. 2018, 97, 3661–3670. [Google Scholar] [CrossRef] [PubMed]
- Cedraz, H.; Gromboni, J.G.G.; Garcia, A.A.P.; Farias Filho, R.V.; Souza, T.M.; Oliveira, E.R.d.; Oliveira, E.B.D.; Nascimento, C.S.D.; Meneghetti, C.; Wenceslau, A.A. Heat stress induces expression of HSP genes in genetically divergent chickens. PLoS ONE 2017, 12, e0186083. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis. 2019, 10, 637. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef]
- Wang, X.; Hai, C. Novel insights into redox system and the mechanism of redox regulation. Mol. Biol. Rep. 2016, 43, 607–628. [Google Scholar] [CrossRef]
- Himanen, S.V.; Sistonen, L. New insights into transcriptional reprogramming during cellular stress. J. Cell Sci. 2019, 132, jcs238402. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox Signal. 2010, 13, 1665–1678. [Google Scholar] [CrossRef]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.-I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.-I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef]
- Nakai, A.; Morimoto, R.I. Characterization of a novel chicken heat shock transcription factor, heat shock factor 3, suggests a new regulatory pathway. Mol. Cell. Biol. 1993, 13, 1983–1997. [Google Scholar]
- Xie, J.; Tang, L.; Lu, L.; Zhang, L.; Xi, L.; Liu, H.-C.; Odle, J.; Luo, X. Differential expression of heat shock transcription factors and heat shock proteins after acute and chronic heat stress in laying chickens (Gallus gallus). PLoS ONE 2014, 9, e102204. [Google Scholar] [CrossRef]
- Östling, P.; Björk, J.K.; Roos-Mattjus, P.; Mezger, V.; Sistonen, L. Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J. Biol. Chem. 2007, 282, 7077–7086. [Google Scholar] [CrossRef]
- Jaeger, A.M.; Pemble IV, C.W.; Sistonen, L.; Thiele, D.J. Structures of HSF2 reveal mechanisms for differential regulation of human heat-shock factors. Nat. Struct. Mol. Biol. 2016, 23, 147–154. [Google Scholar] [CrossRef]
- Ahn, S.-G.; Thiele, D.J. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003, 17, 516–528. [Google Scholar] [CrossRef]
- Rabindran, S.K.; Haroun, R.I.; Clos, J.; Wisniewski, J.; Wu, C. Regulation of heat shock factor trimer formation: Role of a conserved leucine zipper. Science 1993, 259, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, A.M.; Makley, L.N.; Gestwicki, J.E.; Thiele, D.J. Genomic heat shock element sequences drive cooperative human heat shock factor 1 DNA binding and selectivity. J. Biol. Chem. 2014, 289, 30459–30469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Wu, M.; Bian, Q.; Liu, Y.; Fernandes, A.F.; Taylor, A.; Pereira, P.; Shang, F. Sustained oxidative stress inhibits NF-κB activation partially via inactivating the proteasome. Free Radic. Biol. Med. 2009, 46, 62–69. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besançon, F. NF-κB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFα-treated Ewing sarcoma cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-g. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Anrather, J.; Racchumi, G.; Iadecola, C. NF-κB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 2006, 281, 5657–5667. [Google Scholar] [CrossRef]
- Xu, P.; Huecksteadt, T.P.; Hoidal, J.R. Molecular cloning and characterization of the human xanthine dehydrogenase gene (XDH). Genomics 1996, 34, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.R.; Lutz, R.D.; Choi, H.-S.; Kamitani, T.; Chmura, K.; Chan, E.D. Role of the NF-κB signaling pathway and κB cis-regulatory elements on the IRF-1 and iNOS promoter regions in mycobacterial lipoarabinomannan induction of nitric oxide. Infect. Immun. 2003, 71, 1442–1452. [Google Scholar] [CrossRef]
- Zenz, R.; Eferl, R.; Scheinecker, C.; Redlich, K.; Smolen, J.; Schonthaler, H.B.; Kenner, L.; Tschachler, E.; Wagner, E.F. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res. Ther. 2008, 10, 201. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef]
- Wisdom, R. AP-1: One switch for many signals. Exp. Cell Res. 1999, 253, 180–185. [Google Scholar] [CrossRef]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 1995, 270, 16483–16486. [Google Scholar] [CrossRef]
- McCubrey, J.A.; LaHair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Forman, H.J. Redox signaling and the MAP kinase pathways. Biofactors 2003, 17, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-H.; Hur, E.-M.; Lee, J.-H.; Jun, D.-J.; Kim, K.-T. Protein kinase Cδ-mediated proteasomal degradation of MAP kinase phosphatase-1 contributes to glutamate-induced neuronal cell death. J. Cell Sci. 2006, 119, 1329–1340. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.-i.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef]
- Liu, X.; Lin, X.; Zhang, S.; Guo, C.; Li, J.; Mi, Y.; Zhang, C. Lycopene ameliorates oxidative stress in the aging chicken ovary via activation of Nrf2/HO-1 pathway. Aging 2018, 10, 2016. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Zhang, C.; Sun, Y.-C.; Zhang, Q.; Lv, M.-W.; Guo, K.; Li, J.-L. Cadmium exposure triggers mitochondrial dysfunction and oxidative stress in chicken (Gallus gallus) kidney via mitochondrial UPR inhibition and Nrf2-mediated antioxidant defense activation. Sci. Total Environ. 2019, 689, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Tarkhan, A.H.; Saleh, K.M.; Al-Zghoul, M.B. HSF3 and Hsp70 expression during post-hatch cold stress in broiler chickens subjected to embryonic thermal manipulation. Vet. Sci. 2020, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Shen, Y.; Li, X.; Ding, C.; Ma, Z. Molecular cloning and functional characterization of a novel isoform of chicken myeloid differentiation factor 88 (MyD88). Dev. Comp. Immunol. 2008, 32, 1522–1530. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhuang, Y.; Shi, Y.; Xu, Z.; Zhou, C.; Guo, L.; Liu, P.; Wu, C.; Hu, R.; Hu, G.; et al. Effects of N-acetyl-l-cysteine on heat stress-induced oxidative stress and inflammation in the hypothalamus of hens. J. Therm. Biol. 2021, 98, 102927. [Google Scholar] [CrossRef]
- Chen, H.; Wang, F.; Wu, X.; Yuan, S.; Dong, H.; Zhou, C.; Feng, S.; Zhao, Z.; Si, L. Chronic Heat Stress Induces Oxidative Stress and Induces Inflammatory Injury in Broiler Spleen via TLRs/MyD88/NF-κB Signaling Pathway in Broilers. Vet. Sci. 2024, 11, 293. [Google Scholar] [CrossRef]
- Liu, L.; Gong, X.; Zhang, X.; Zhang, D.; Tang, Y.; Liu, J.; Li, Y.; Pan, D. Resveratrol alleviates heat-stress-induced impairment of the jejunal mucosa through TLR4/MAPK signaling pathway in black-boned chicken. Poult. Sci. 2024, 103, 103242. [Google Scholar] [CrossRef]
- Ma, D.; Zhang, M.; Feng, J. Dietary Peppermint Extract Inhibits Chronic Heat Stress-Induced Activation of Innate Immunity and Inflammatory Response in the Spleen of Broiler Chickens. Animals 2024, 14, 1157. [Google Scholar] [CrossRef]
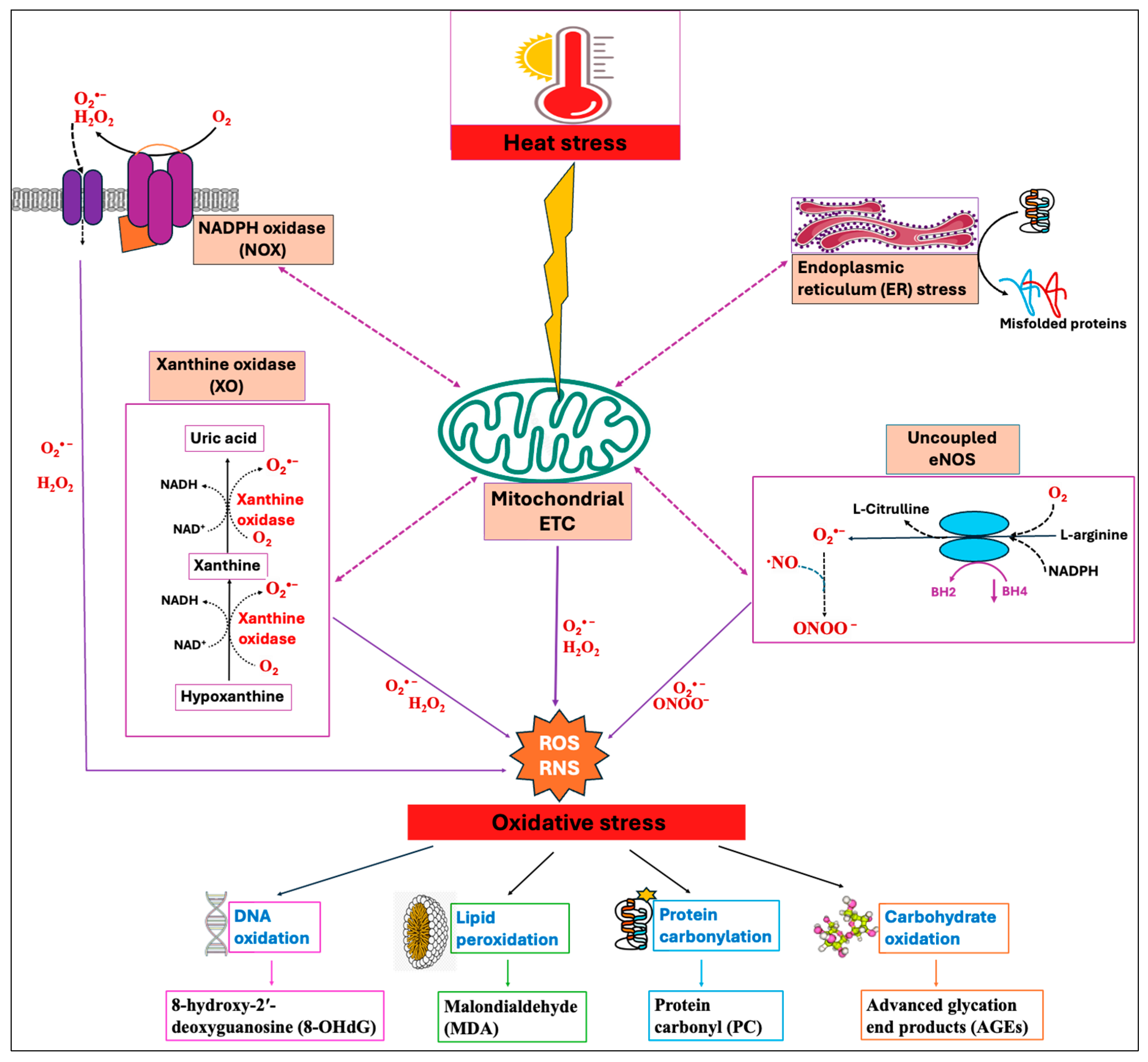
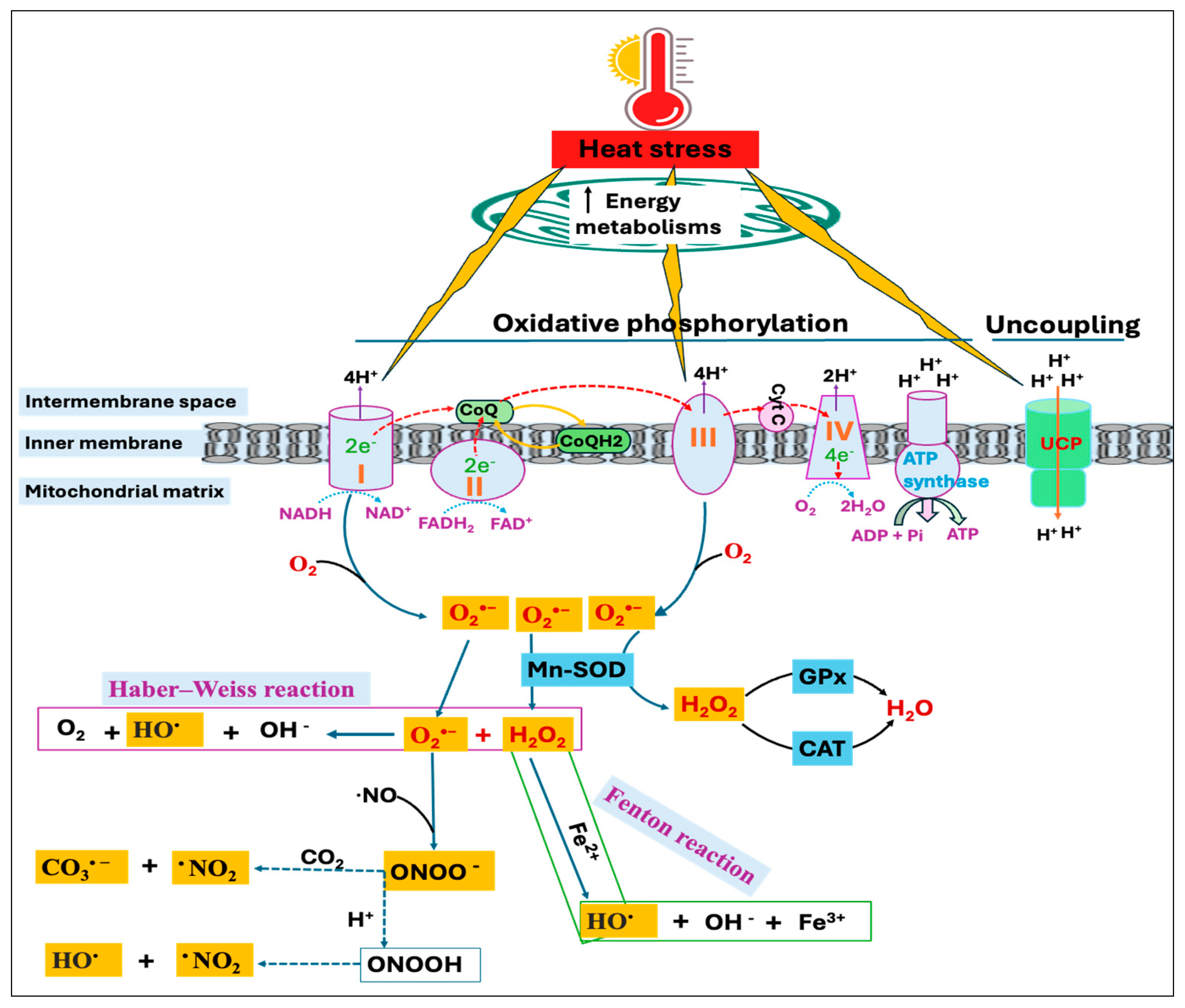
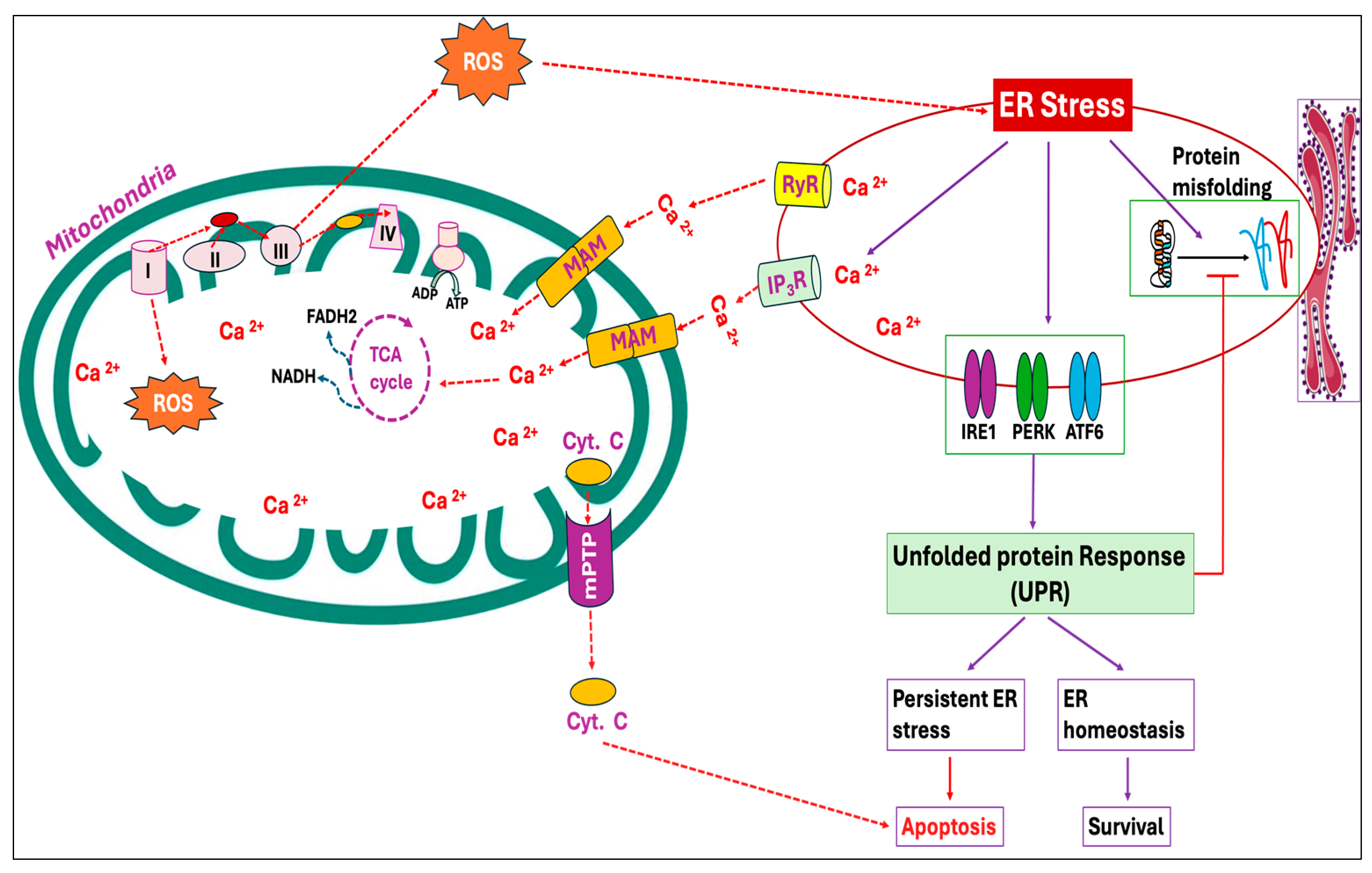
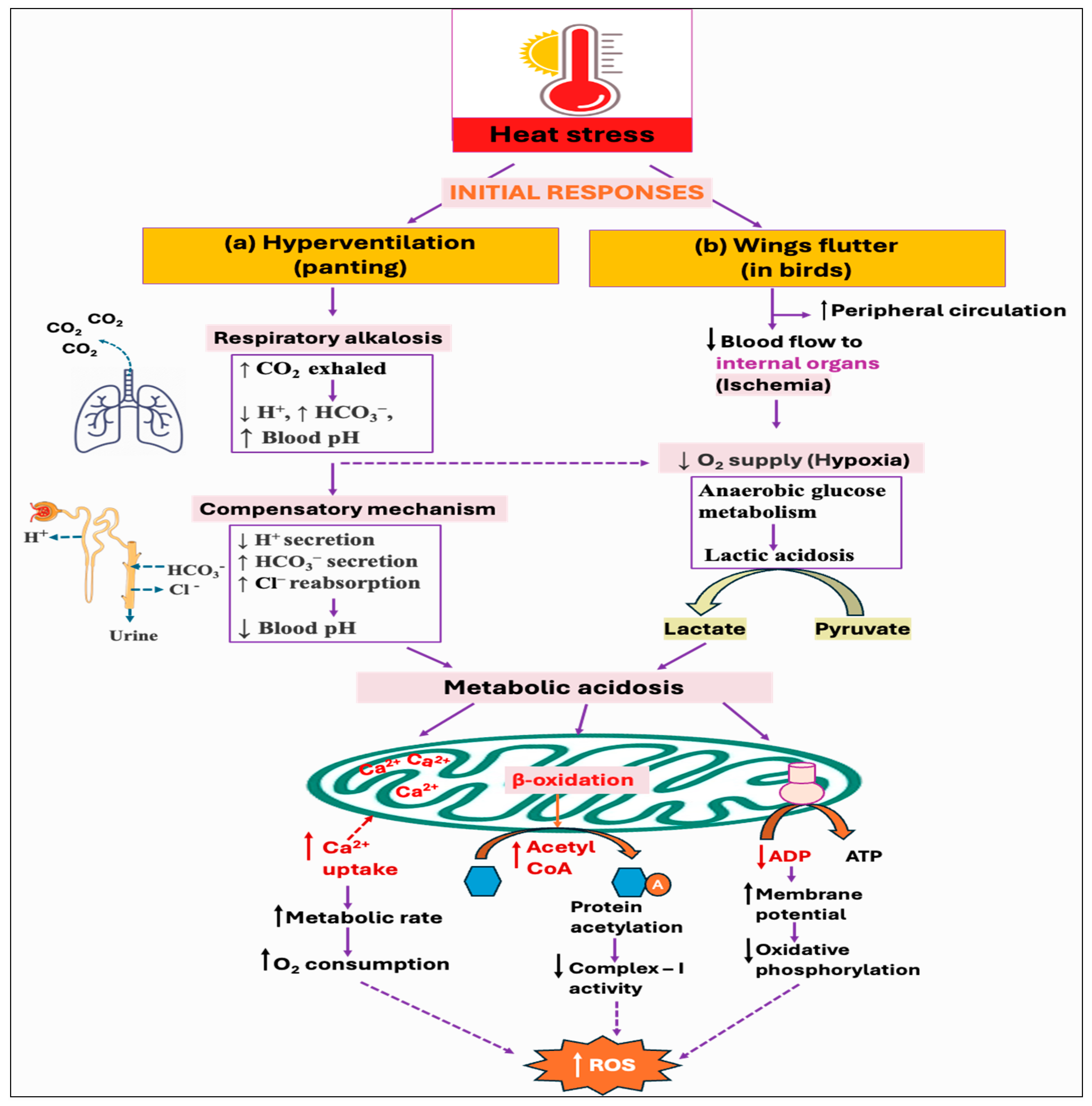
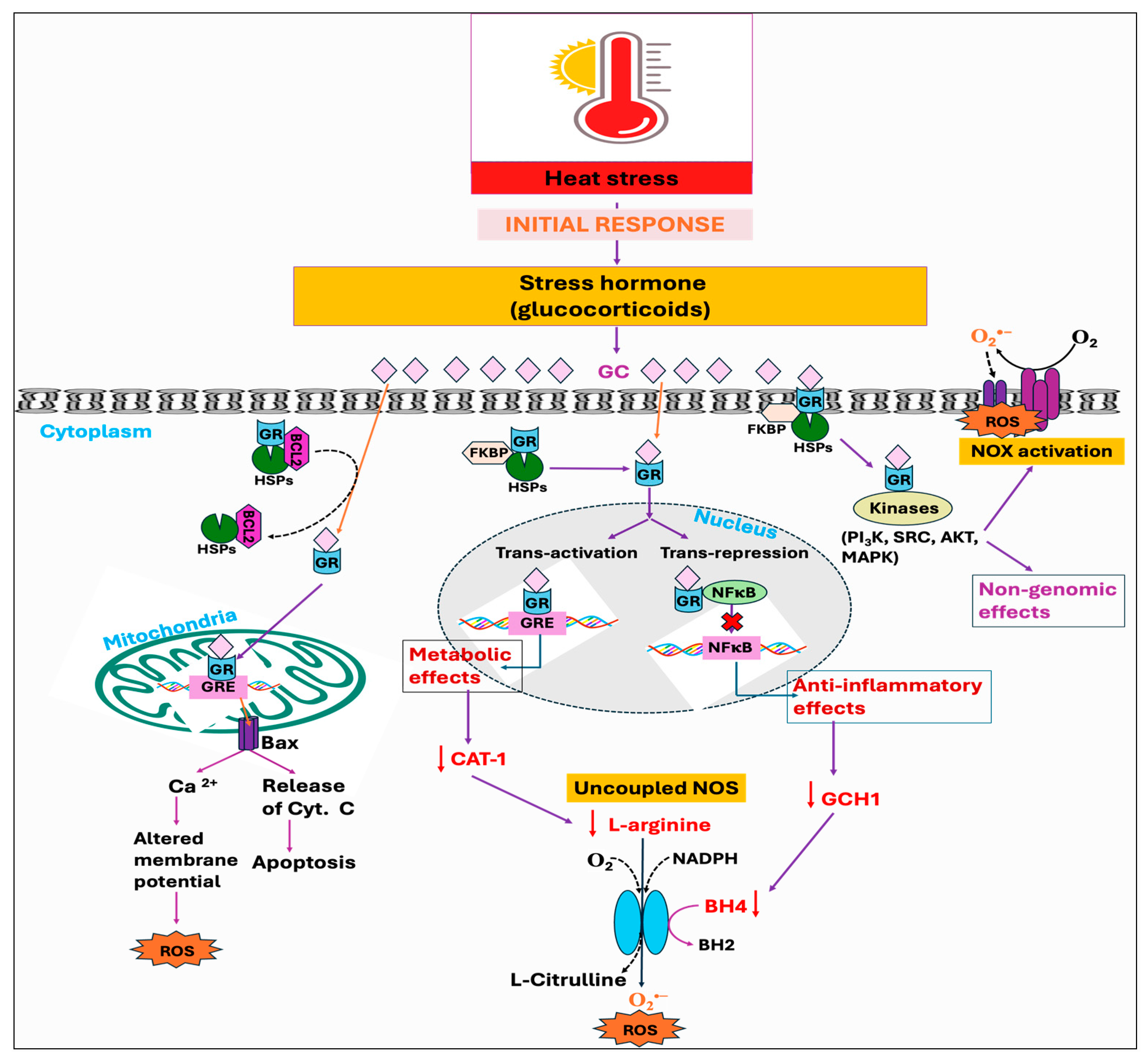
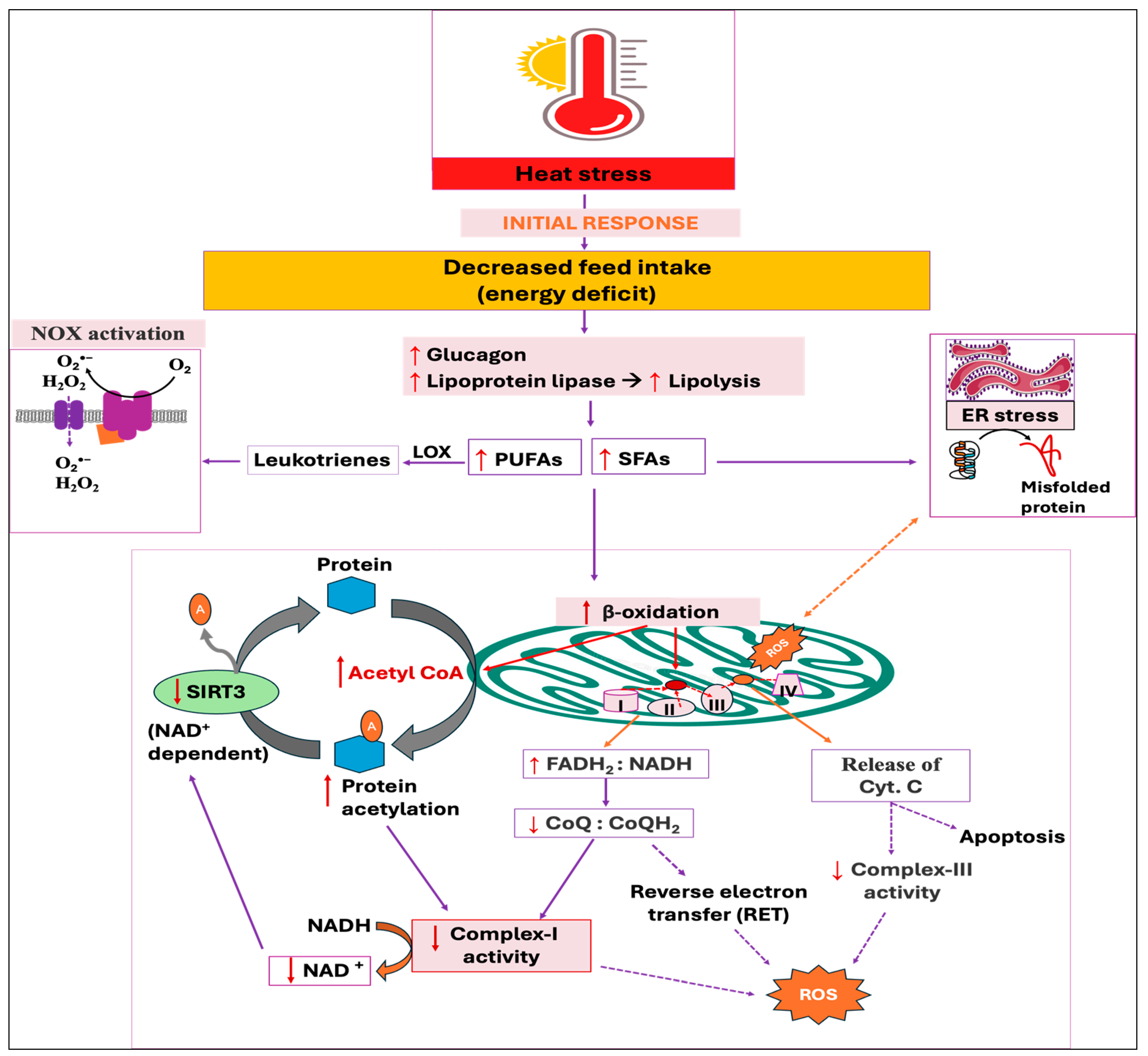
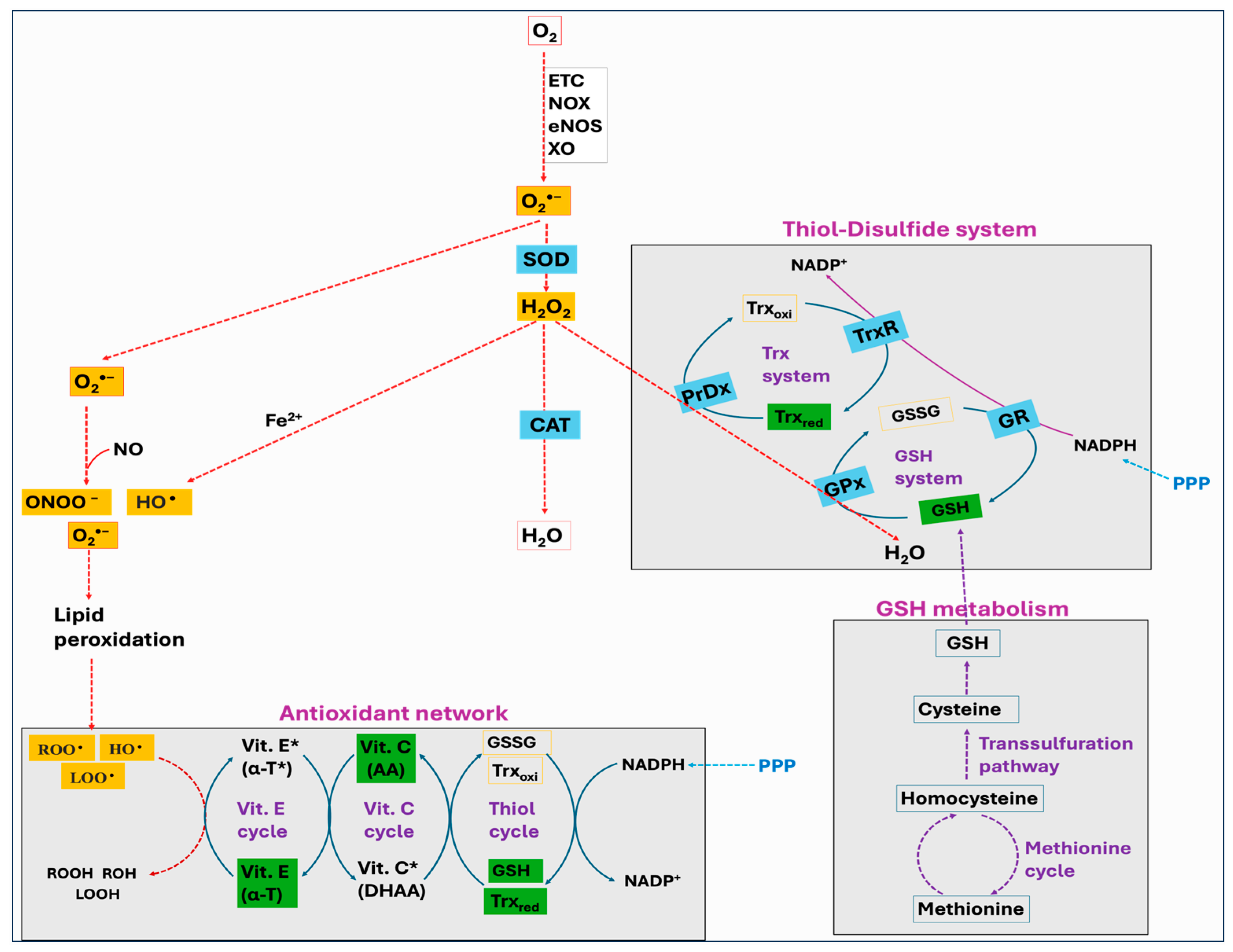
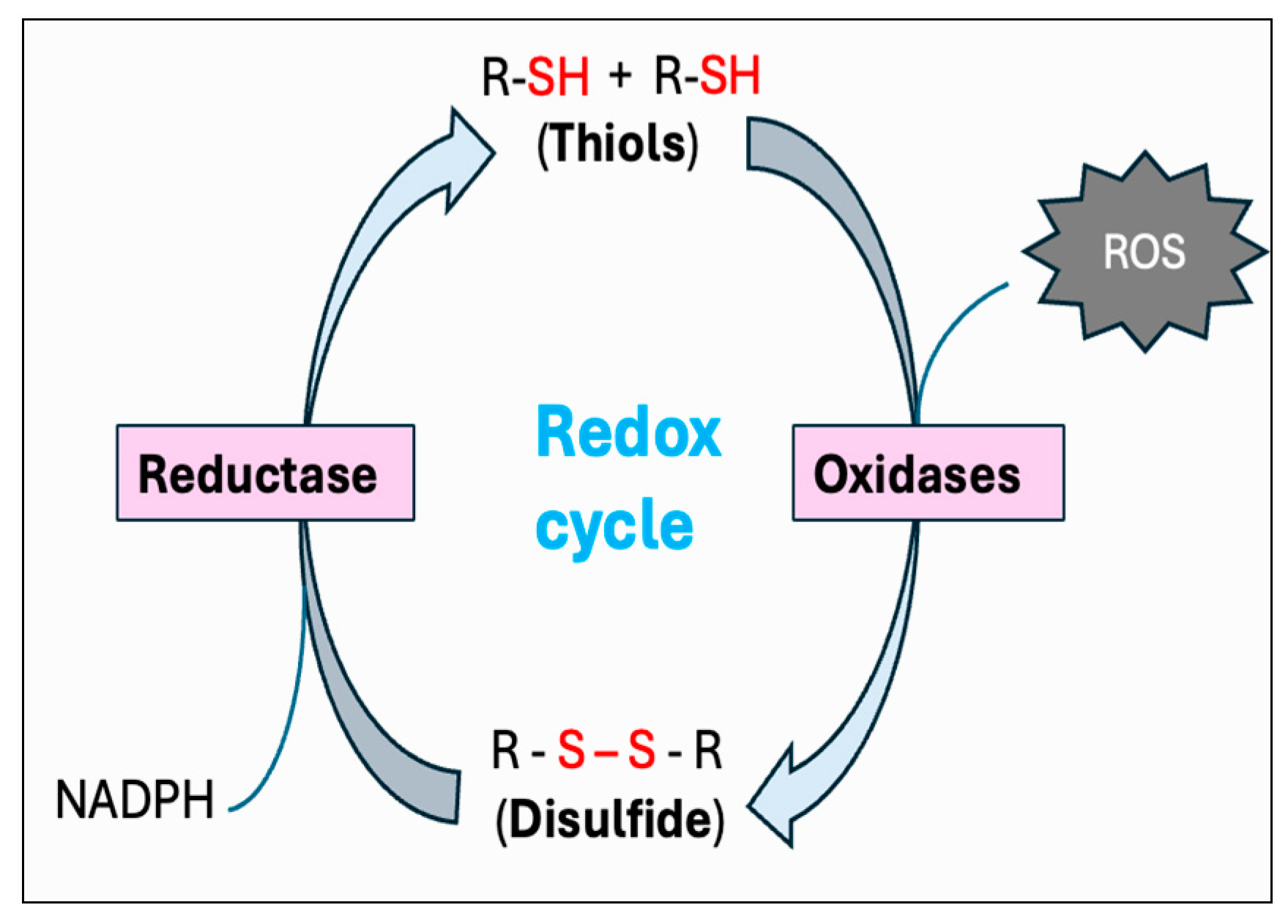
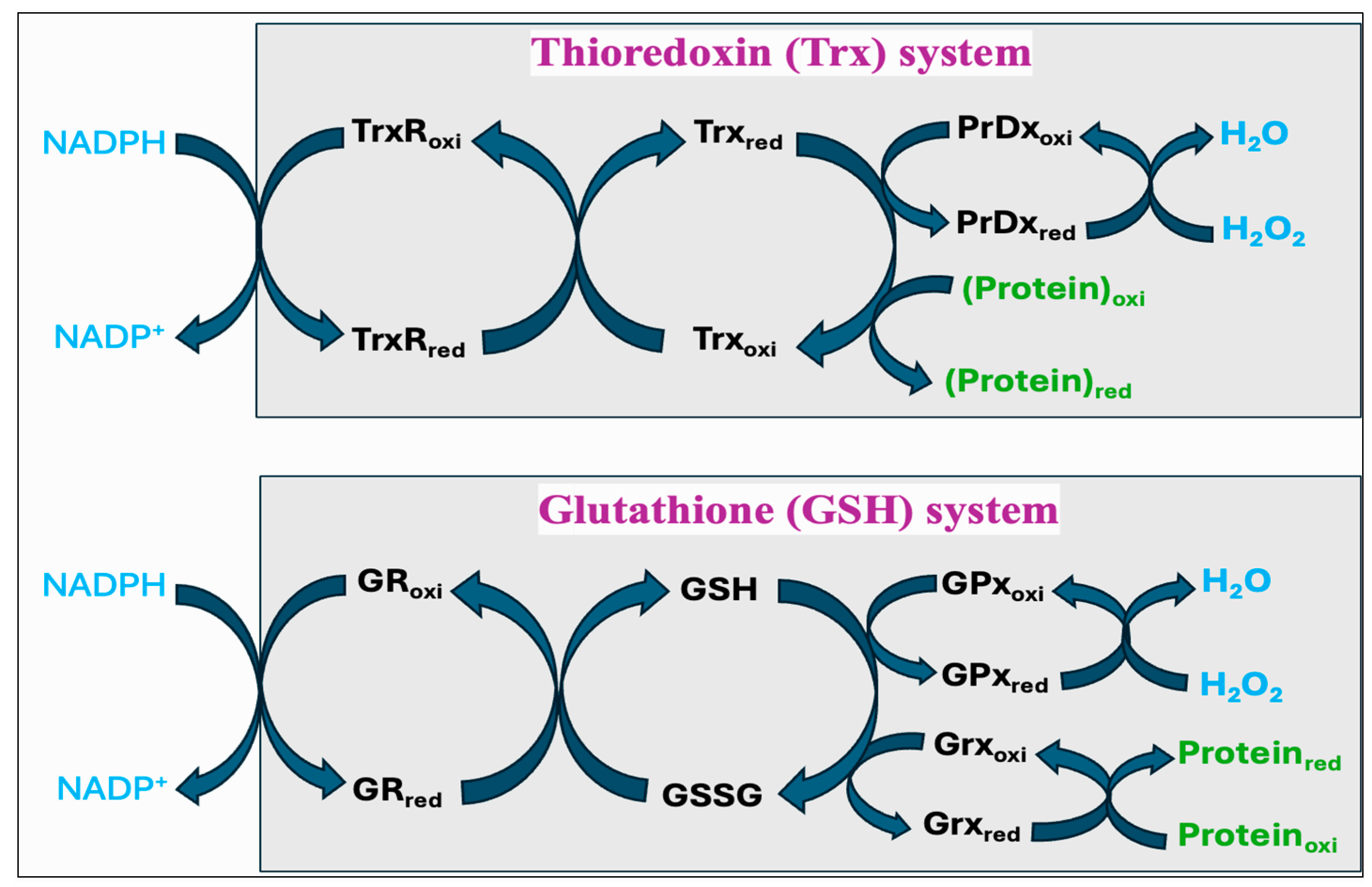
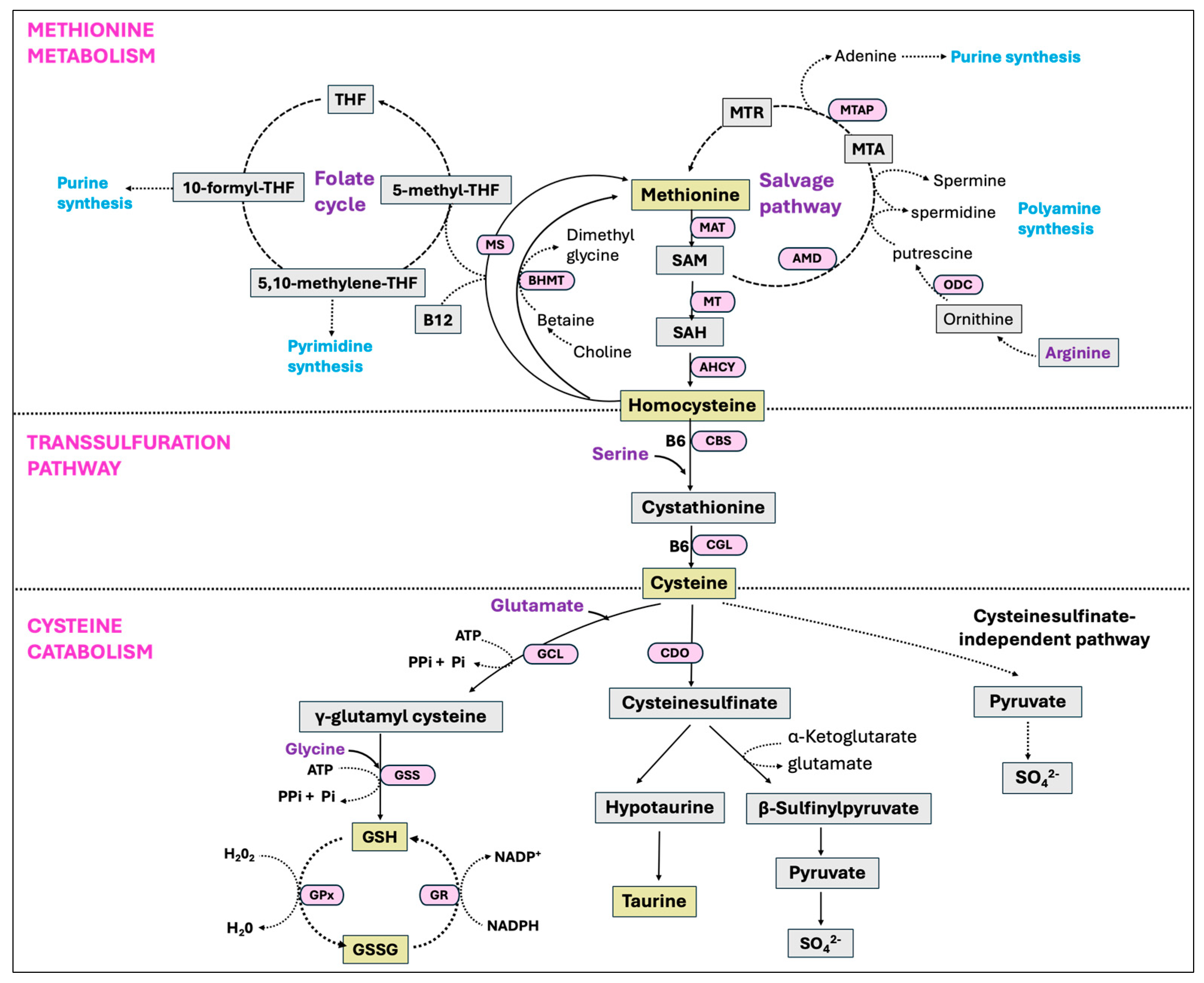
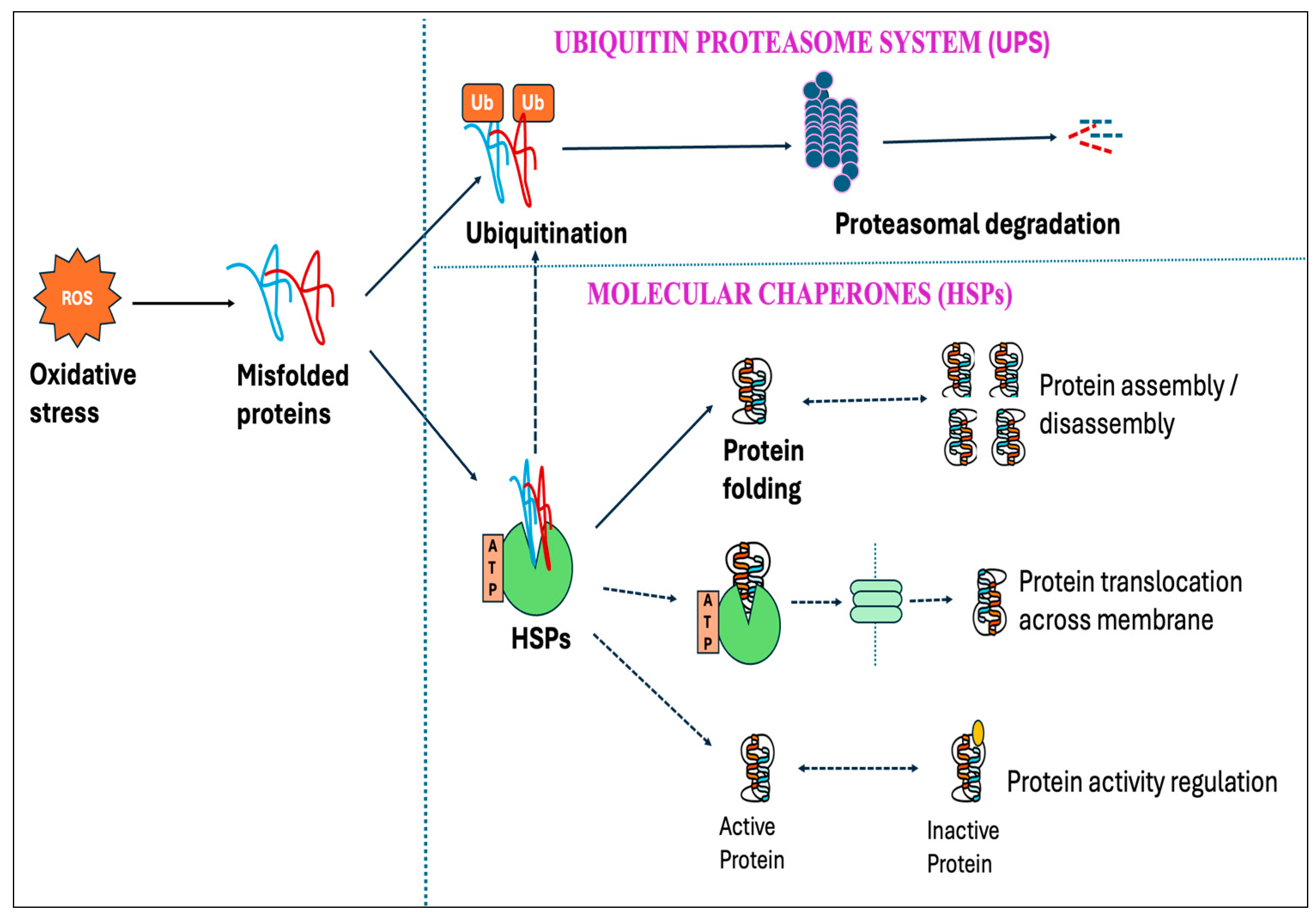
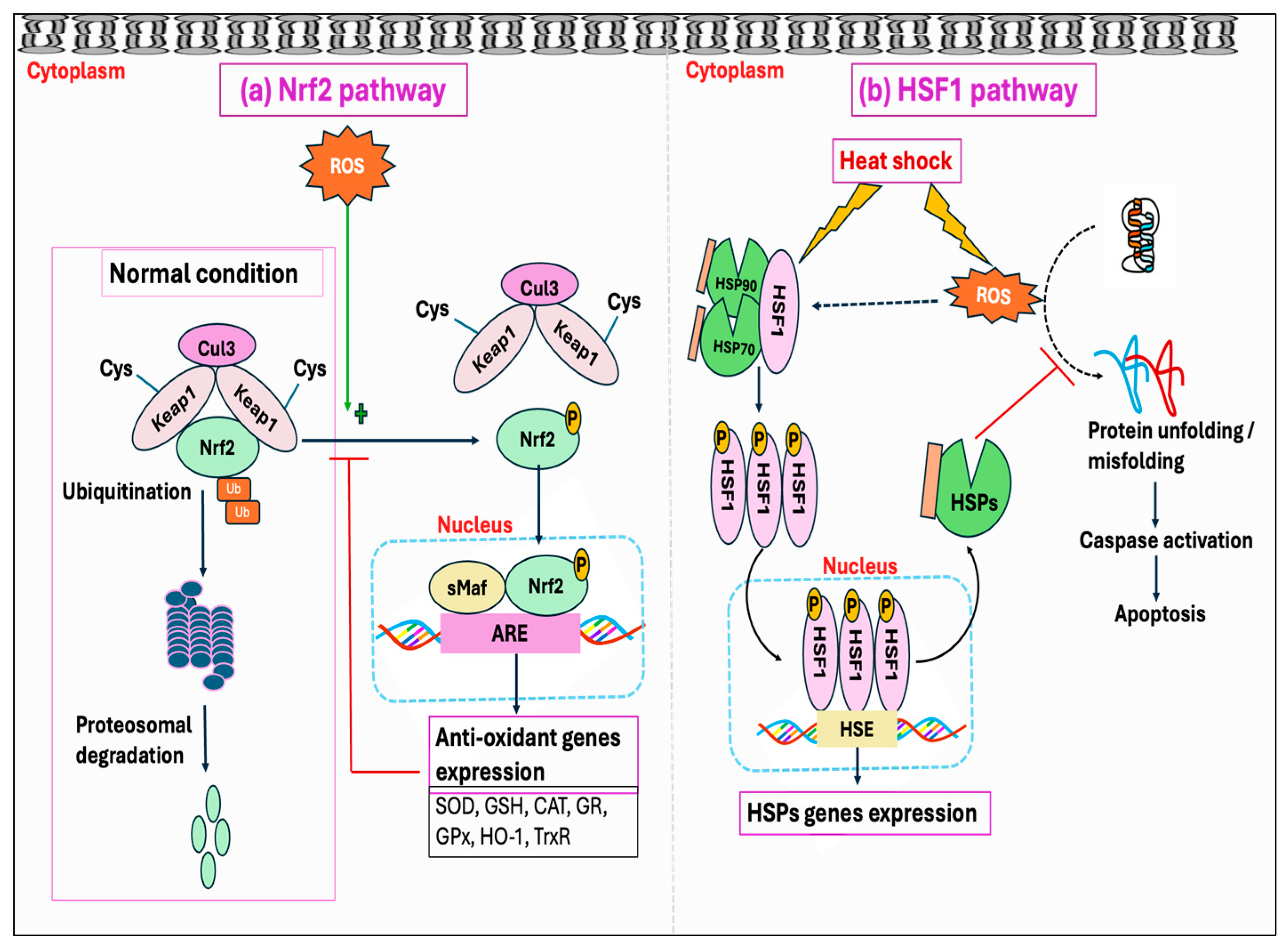
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).