
Allantoin Serves as a Novel Risk Factor for the Progression of MASLD

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Human Subjects
2.3. Individual-Level Data from UK Biobank
2.4. Observational Analysis
2.5. GWAS Summary Data
2.6. Animals
2.7. Recombinant Proteins
2.8. Dual-Luciferase Reporter Assay
2.9. Surface Plasmon Resonance (SPR)
2.10. Statistical Analysis
3. Results
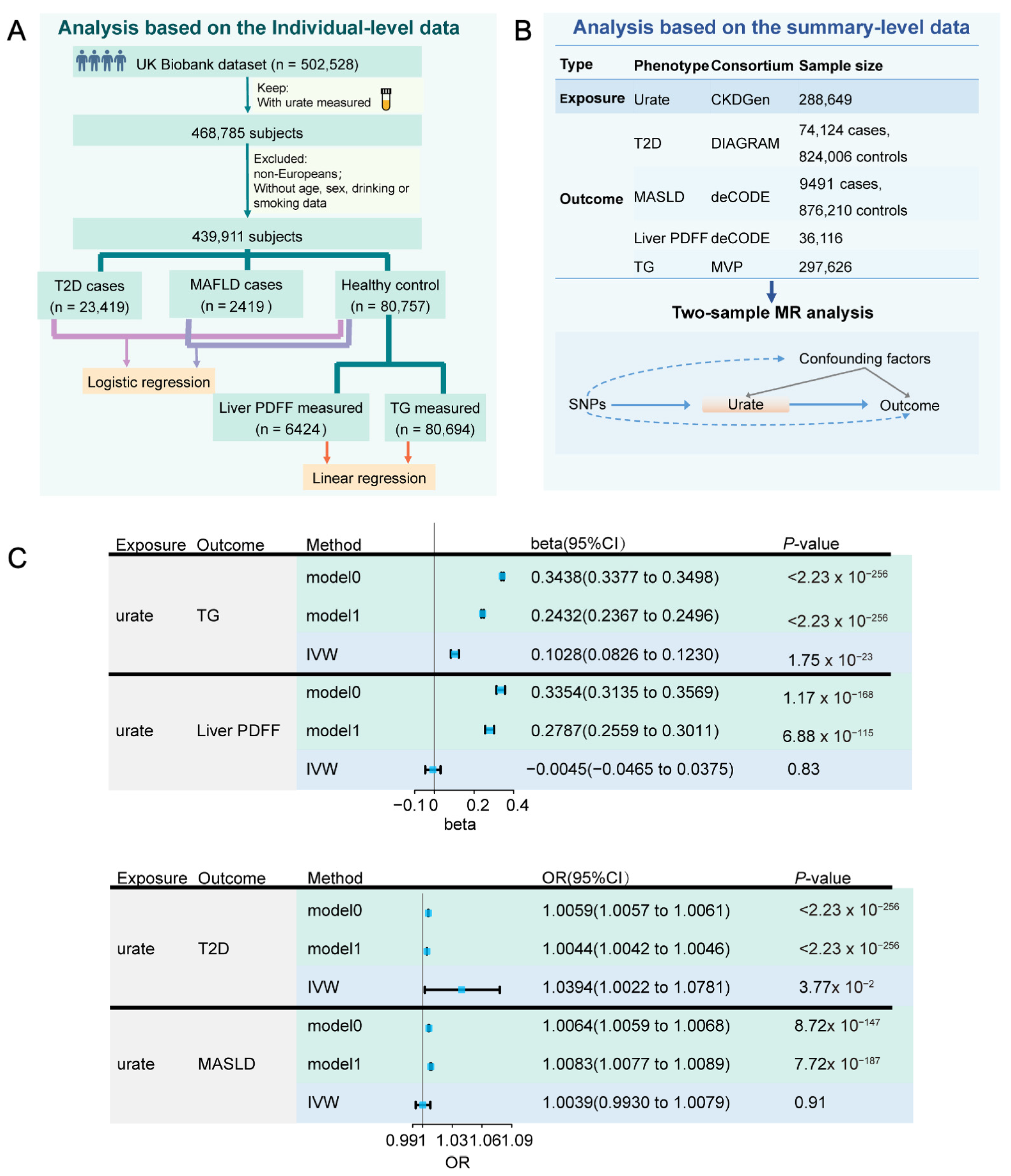
3.1. Close Association of UA with Risk of MASLD
3.2. Serum Allantoin Is Positively Correlated with DLP
3.3. Allantoin Increases Susceptibility of Glucose Intolerance in Mice Feeding High-Fat Diet
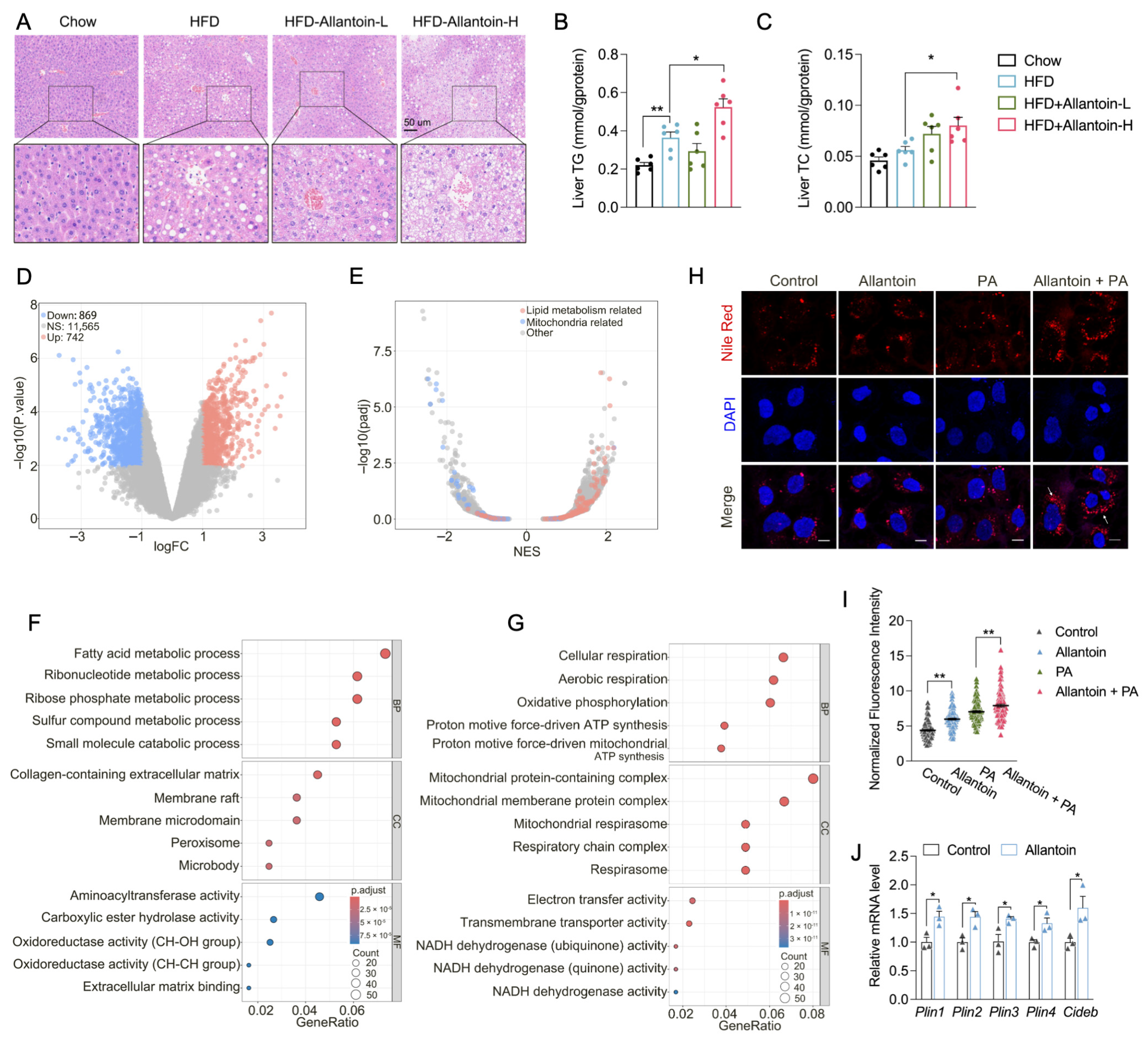
3.4. Allantoin Aggravates Hepatic Lipid Accumulation in Mice Feeding HFD
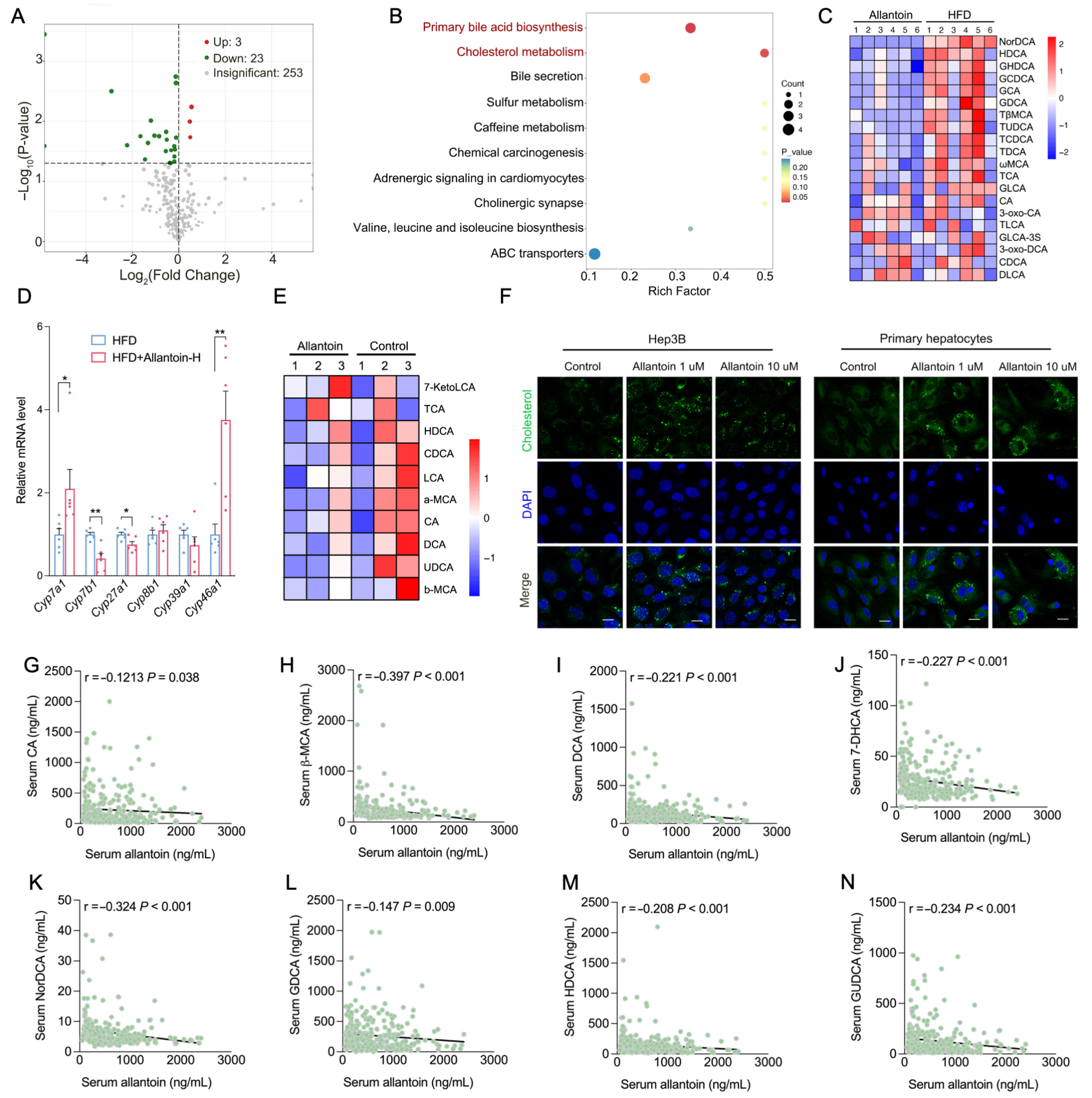
3.5. Allantoin Suppresses Hepatic Cholesterol Metabolism
3.6. Allantoin Is Potential Endogenous PPARα Antagonist
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DLP | Dyslipidemia |
| UA | Uric acid |
| MASLD | Metabolic dysfunction-associated steatotic liver disease |
| T2D | Type 2 diabetes |
| LDL | Low-density lipoprotein |
| ROS | Reactive oxygen species |
| PDFF | Proton density fat fraction |
| TC | Total cholesterol |
| TG | Triglyceride |
| PPARα | Peroxisome proliferator-activated receptor alpha |
References
- Paik, J.M.; Golabi, P.; Younossi, Y.; Mishra, A.; Younossi, Z.M. Changes in the Global Burden of Chronic Liver Diseases from 2012 to 2017: The Growing Impact of NAFLD. Hepatology 2020, 72, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Matchett, K.P.; Paris, J.; Teichmann, S.A.; Henderson, N.C. Spatial genomics: Mapping human steatotic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 646–660. [Google Scholar] [CrossRef] [PubMed]
- Scoditti, E.; Sabatini, S.; Carli, F.; Gastaldelli, A. Hepatic glucose metabolism in the steatotic liver. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 319–334. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Li, H.; Yu, X.H.; Ou, X.; Ouyang, X.P.; Tang, C.K. Hepatic cholesterol transport and its role in non-alcoholic fatty liver disease and atherosclerosis. Prog. Lipid Res. 2021, 83, 101109. [Google Scholar] [CrossRef]
- Li, Z.; Zheng, D.; Zhang, T.; Ruan, S.; Li, N.; Yu, Y.; Peng, Y.; Wang, D. The roles of nuclear receptors in cholesterol metabolism and reverse cholesterol transport in nonalcoholic fatty liver disease. Hepatol. Commun. 2024, 8, e0343. [Google Scholar] [CrossRef]
- Alvarez-Lario, B.; Macarron-Vicente, J. Uric acid and evolution. Rheumatology 2010, 49, 2010–2015. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Johnson, R.J. Uric acid: The oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608–619. [Google Scholar] [CrossRef]
- Kuwabara, M.; Borghi, C.; Cicero, A.F.G.; Hisatome, I.; Niwa, K.; Ohno, M.; Johnson, R.J.; Lanaspa, M.A. Elevated serum uric acid increases risks for developing high LDL cholesterol and hypertriglyceridemia: A five-year cohort study in Japan. Int. J. Cardiol. 2018, 261, 183–188. [Google Scholar] [CrossRef]
- Tan, M.Y.; Mo, C.Y.; Li, F.; Zhao, Q. The association between serum uric acid and hypertriglyceridemia: Evidence from the national health and nutrition examination survey (2007–2018). Front. Endocrinol. 2023, 14, 1215521. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Liang, A.; Shi, P.; Yuan, S.; Zhu, Y.; Fu, J.; Zheng, T.; Wen, Z.; Wu, X. Higher serum uric acid to HDL-cholesterol ratio is associated with onset of non-alcoholic fatty liver disease in a non-obese Chinese population with normal blood lipid levels. BMC Gastroenterol. 2022, 22, 196. [Google Scholar] [CrossRef]
- Liu, N.; Sun, Q.; Xu, H.; Yu, X.; Chen, W.; Wei, H.; Jiang, J.; Xu, Y.; Lu, W. Hyperuricemia induces lipid disturbances mediated by LPCAT3 upregulation in the liver. FASEB J. 2020, 34, 13474–13493. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Xu, C.; Lin, Y.; Lu, C.; Li, D.; Sang, J.; He, H.; Liu, X.; Li, Y.; Yu, C. Uric acid regulates hepatic steatosis and insulin resistance through the NLRP3 inflammasome-dependent mechanism. J. Hepatol. 2016, 64, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Halliwell, B. Action of biologically-relevant oxidizing species upon uric acid. Identification of uric acid oxidation products. Chem. Biol. Interact. 1990, 73, 235–247. [Google Scholar] [CrossRef]
- Kopcil, M.; Kandar, R. Screening method for the simultaneous determination of allantoin and uric acid from dried blood spots. J. Pharm. Biomed. Anal. 2023, 225, 115222. [Google Scholar] [CrossRef]
- Grootveld, M.; Halliwell, B. Measurement of allantoin and uric acid in human body fluids. A potential index of free-radical reactions in vivo? Biochem. J. 1987, 243, 803–808. [Google Scholar] [CrossRef]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxid. Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Kand’ar, R.; Zakova, P. Allantoin as a marker of oxidative stress in human erythrocytes. Clin. Chem. Lab. Med. 2008, 46, 1270–1274. [Google Scholar] [CrossRef]
- Lee, M.Y.; Lee, N.H.; Jung, D.; Lee, J.A.; Seo, C.S.; Lee, H.; Kim, J.H.; Shin, H.K. Protective effects of allantoin against ovalbumin (OVA)-induced lung inflammation in a murine model of asthma. Int. Immunopharmacol. 2010, 10, 474–480. [Google Scholar] [CrossRef]
- Araujo, L.U.; Grabe-Guimaraes, A.; Mosqueira, V.C.; Carneiro, C.M.; Silva-Barcellos, N.M. Profile of wound healing process induced by allantoin. Acta Cir. Bras. 2010, 25, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sun, Y.; Zhu, C.; Shen, X.; Sun, J.; Jing, T.; Jun, S.; Wang, C.; Yu, G.; Dong, X.; et al. Allantoin induces pruritus by activating MrgprD in chronic kidney disease. J. Cell. Physiol. 2023, 238, 813–828. [Google Scholar] [CrossRef]
- Bentley-Lewis, R.; Huynh, J.; Xiong, G.; Lee, H.; Wenger, J.; Clish, C.; Nathan, D.; Thadhani, R.; Gerszten, R. Metabolomic profiling in the prediction of gestational diabetes mellitus. Diabetologia 2015, 58, 1329–1332. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, L.; Dong, F.; Liu, Y.; Li, N.; Li, H.; Lei, H.; Hao, F.; Wang, Y.; Zhu, Y.; et al. Metabonomic Changes Associated with Atherosclerosis Progression for LDLR(-/-) Mice. J. Proteome Res. 2015, 14, 2237–2254. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Ling, X.; Wang, Q.; Huang, Z.; Guo, B.; Zhang, C.; Meng, M.; Feng, S.; Guo, Y.; Zheng, H.; et al. Integrated Gut Microbiota and Urine Metabolite Analyses of T2DM with NAFLD Rat Model. Appl. Biochem. Biotechnol. 2023, 195, 6478–6494. [Google Scholar] [CrossRef]
- Tin, A.; Marten, J.; Halperin Kuhns, V.L.; Li, Y.; Wuttke, M.; Kirsten, H.; Sieber, K.B.; Qiu, C.; Gorski, M.; Yu, Z.; et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 2019, 51, 1459–1474. [Google Scholar] [CrossRef]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef]
- Klarin, D.; Damrauer, S.M.; Cho, K.; Sun, Y.V.; Teslovich, T.M.; Honerlaw, J.; Gagnon, D.R.; DuVall, S.L.; Li, J.; Peloso, G.M.; et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet. 2018, 50, 1514–1523. [Google Scholar] [CrossRef]
- Sveinbjornsson, G.; Ulfarsson, M.O.; Thorolfsdottir, R.B.; Jonsson, B.A.; Einarsson, E.; Gunnlaugsson, G.; Rognvaldsson, S.; Arnar, D.O.; Baldvinsson, M.; Bjarnason, R.G.; et al. Multiomics study of nonalcoholic fatty liver disease. Nat. Genet. 2022, 54, 1652–1663. [Google Scholar] [CrossRef]
- Price, A.L.; Weale, M.E.; Patterson, N.; Myers, S.R.; Need, A.C.; Shianna, K.V.; Ge, D.; Rotter, J.I.; Torres, E.; Taylor, K.D.; et al. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet. 2008, 83, 132–135. [Google Scholar] [CrossRef]
- Lukkunaprasit, T.; Rattanasiri, S.; Ongphiphadhanakul, B.; McKay, G.J.; Attia, J.; Thakkinstian, A. Causal Associations of Urate With Cardiovascular Risk Factors: Two-Sample Mendelian Randomization. Front. Genet. 2021, 12, 687279. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Preidis, G.A.; Kim, K.H.; Moore, D.D. Nutrient-sensing nuclear receptors PPARalpha and FXR control liver energy balance. J. Clin. Investig. 2017, 127, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Li, K.; Peng, X.; Kan, Y.; Li, H.; Zhu, Y.; Wang, Z.; Li, Z.; Liu, H.Y.; Cai, D. Nuclear Receptor PPARalpha as a Therapeutic Target in Diseases Associated with Lipid Metabolism Disorders. Nutrients 2023, 15, 4772. [Google Scholar] [CrossRef]
- Yang, P.S.; Wu, H.T.; Chung, H.H.; Chen, C.T.; Chi, C.W.; Yeh, C.H.; Cheng, J.T. Rilmenidine improves hepatic steatosis through p38-dependent pathway to higher the expression of farnesoid X receptor. Naunyn Schmiedebergs Arch. Pharmacol. 2012, 385, 51–56. [Google Scholar] [CrossRef]
- Ames, B.N.; Cathcart, R.; Schwiers, E.; Hochstein, P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: A hypothesis. Proc. Natl. Acad. Sci. USA 1981, 78, 6858–6862. [Google Scholar] [CrossRef]
- Bowman, G.L.; Shannon, J.; Frei, B.; Kaye, J.A.; Quinn, J.F. Uric acid as a CNS antioxidant. J. Alzheimer’s Dis. 2010, 19, 1331–1336. [Google Scholar] [CrossRef]
- Aerqin, Q.; Jia, S.S.; Shen, X.N.; Li, Q.; Chen, K.L.; Ou, Y.N.; Huang, Y.Y.; Dong, Q.; Chen, S.F.; Yu, J.T. Serum Uric Acid Levels in Neurodegenerative Disorders: A Cross-Sectional Study. J. Alzheimer’s Dis. 2022, 90, 761–773. [Google Scholar] [CrossRef]
- Copur, S.; Demiray, A.; Kanbay, M. Uric acid in metabolic syndrome: Does uric acid have a definitive role? Eur. J. Intern. Med. 2022, 103, 4–12. [Google Scholar] [CrossRef]
- Katsiki, N.; Dimitriadis, G.D.; Mikhailidis, D.P. Serum Uric Acid and Diabetes: From Pathophysiology to Cardiovascular Disease. Curr. Pharm. Des. 2021, 27, 1941–1951. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Loria, P.; Leonardi, F.; Borsatti, A.; Neri, P.; Pulvirenti, M.; Verrone, A.M.; Bagni, A.; Bertolotti, M.; Ganazzi, D.; et al. Fasting insulin and uric acid levels but not indices of iron metabolism are independent predictors of non-alcoholic fatty liver disease. A case-control study. Dig. Liver Dis. 2002, 34, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Sirota, J.C.; McFann, K.; Targher, G.; Johnson, R.J.; Chonchol, M.; Jalal, D.I. Elevated serum uric acid levels are associated with non-alcoholic fatty liver disease independently of metabolic syndrome features in the United States: Liver ultrasound data from the National Health and Nutrition Examination Survey. Metabolism 2013, 62, 392–399. [Google Scholar] [CrossRef]
- Wei, F.; Li, J.; Chen, C.; Zhang, K.; Cao, L.; Wang, X.; Ma, J.; Feng, S.; Li, W.D. Higher Serum Uric Acid Level Predicts Non-alcoholic Fatty Liver Disease: A 4-Year Prospective Cohort Study. Front. Endocrinol. 2020, 11, 179. [Google Scholar] [CrossRef]
- Baba, T.; Amasaki, Y.; Soda, M.; Hida, A.; Imaizumi, M.; Ichimaru, S.; Nakashima, E.; Seto, S.; Yano, K.; Akahoshi, M. Fatty liver and uric acid levels predict incident coronary heart disease but not stroke among atomic bomb survivors in Nagasaki. Hypertens. Res. 2007, 30, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fu, Y.; Liu, Y.; Zhang, X.; Li, H.; Tian, L.; Zhuo, L.; Liu, M.; Cui, J. Serum Uric Acid Levels and Nonalcoholic Fatty Liver Disease: A 2-Sample Bidirectional Mendelian Randomization Study. J. Clin. Endocrinol. Metab. 2022, 107, e3497–e3503. [Google Scholar] [CrossRef]
- Raut, S.K.; Khullar, M. Oxidative stress in metabolic diseases: Current scenario and therapeutic relevance. Mol. Cell. Biochem. 2023, 478, 185–196. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouche, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Regnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Hunt, M.C.; Yang, Y.Z.; Eggertsen, G.; Carneheim, C.M.; Gafvels, M.; Einarsson, C.; Alexson, S.E. The peroxisome proliferator-activated receptor alpha (PPARalpha) regulates bile acid biosynthesis. J. Biol. Chem. 2000, 275, 28947–28953. [Google Scholar] [CrossRef]
- Zhong, J.; He, X.; Gao, X.; Liu, Q.; Zhao, Y.; Hong, Y.; Zhu, W.; Yan, J.; Li, Y.; Li, Y.; et al. Hyodeoxycholic acid ameliorates nonalcoholic fatty liver disease by inhibiting RAN-mediated PPARalpha nucleus-cytoplasm shuttling. Nat. Commun. 2023, 14, 5451. [Google Scholar] [CrossRef]
- Ma, P.; Patching, S.G.; Ivanova, E.; Baldwin, J.M.; Sharples, D.; Baldwin, S.A.; Henderson, P.J.F. Allantoin transport protein, PucI, from Bacillus subtilis: Evolutionary relationships, amplified expression, activity and specificity. Microbiology 2016, 162, 823–836. [Google Scholar] [CrossRef]
- Pelissier, H.C.; Tegeder, M. PvUPS1 plays a role in source-sink transport of allantoin in French bean (Phaseolus vulgaris). Funct. Plant Biol. 2007, 34, 282–291. [Google Scholar] [CrossRef]
- Sun, H.L.; Wu, Y.W.; Bian, H.G.; Yang, H.; Wang, H.; Meng, X.M.; Jin, J. Function of Uric Acid Transporters and Their Inhibitors in Hyperuricaemia. Front. Pharmacol. 2021, 12, 667753. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Fairley, S.; Lowy-Gallego, E.; Perry, E.; Flicek, P. The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 2019, 48, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Spiller, W.; Del Greco, M.F.; Sheehan, N.; Thompson, J.; Minelli, C.; Davey Smith, G. Improving the visualization, interpretation and analysis of two-sample summary data Mendelian randomization via the Radial plot and Radial regression. Int. J. Epidemiol. 2018, 47, 2100. [Google Scholar] [CrossRef]
- Kamat, M.A.; Blackshaw, J.A.; Young, R.; Surendran, P.; Burgess, S.; Danesh, J.; Butterworth, A.S.; Staley, J.R. PhenoScanner V2: An expanded tool for searching human genotype-phenotype associations. Bioinformatics 2019, 35, 4851–4853. [Google Scholar] [CrossRef]
- Hemani, G.; Tilling, K.; Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017, 13, e1007081. [Google Scholar] [CrossRef]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef]
- Hartwig, F.P.; Davey Smith, G.; Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Hemani, G.; Bowden, J.; Small, D.S. Statistical inference in two-sample summary-data Mendelian randomization using robust adjusted profile score. Ann. Stat. 2020, 48, 1742–1769. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef]
- Hemani, G.; Bowden, J.; Davey Smith, G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum. Mol. Genet. 2018, 27, R195–R208. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, W.; Wang, X.; Feng, Z.; Sun, C.; Wu, H.; Zeng, M.; Gao, T.; Cao, K.; Xu, J.; Zou, X.; et al. Allantoin Serves as a Novel Risk Factor for the Progression of MASLD. Antioxidants 2025, 14, 500. https://doi.org/10.3390/antiox14050500
Lv W, Wang X, Feng Z, Sun C, Wu H, Zeng M, Gao T, Cao K, Xu J, Zou X, et al. Allantoin Serves as a Novel Risk Factor for the Progression of MASLD. Antioxidants. 2025; 14(5):500. https://doi.org/10.3390/antiox14050500
Chicago/Turabian StyleLv, Weiqiang, Xueqiang Wang, Zhaode Feng, Cunxiao Sun, Hansen Wu, Mengqi Zeng, Tianlin Gao, Ke Cao, Jie Xu, Xuan Zou, and et al. 2025. "Allantoin Serves as a Novel Risk Factor for the Progression of MASLD" Antioxidants 14, no. 5: 500. https://doi.org/10.3390/antiox14050500
APA StyleLv, W., Wang, X., Feng, Z., Sun, C., Wu, H., Zeng, M., Gao, T., Cao, K., Xu, J., Zou, X., Yang, T., Li, H., Chen, L., Liu, J., Dong, S., & Feng, Z. (2025). Allantoin Serves as a Novel Risk Factor for the Progression of MASLD. Antioxidants, 14(5), 500. https://doi.org/10.3390/antiox14050500