
Nitazoxanide Modulates Mitochondrial Function and Inflammatory Metabolism in Chondrocytes from Patients with Osteoarthritis via AMPK/mTORC1 Signaling
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Chondrocyte Isolation from Patients
2.3. Cell Culture and Stimulation
2.4. Cell Viability Assay
2.5. Quantitative Real-Time PCR (qPCR)
2.6. Western Blotting
2.7. Oxygen Consumption Rate (OCR)
2.8. Extracellular Acidification Rate (ECAR)
2.9. Glucose Uptake Assay
2.10. Mitochondrial Content Measurement
2.11. siRNA Transfection
2.12. Immunofluorescence Imaging
2.13. Statistical Analysis
3. Results
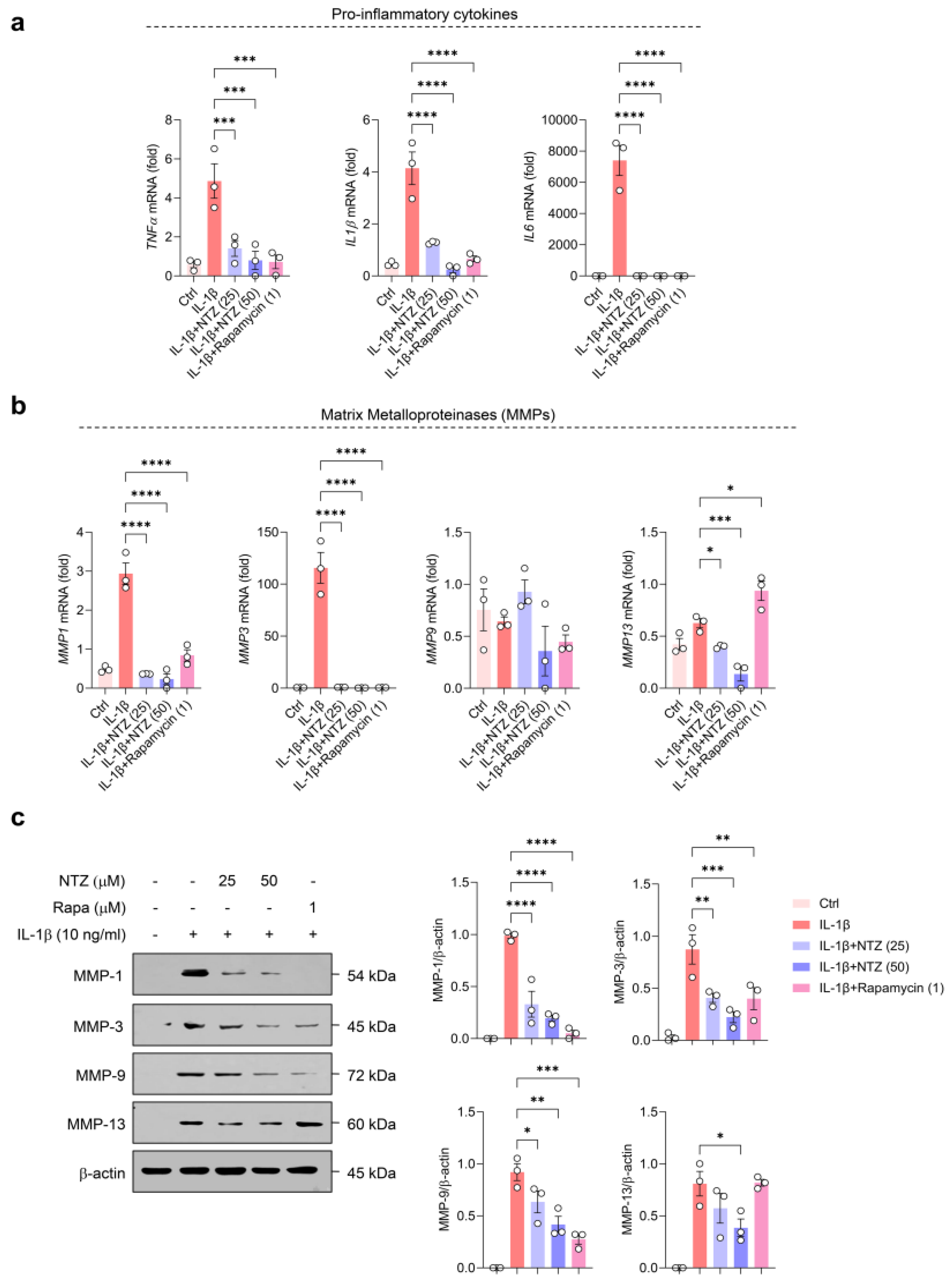
3.1. NTZ Attenuates IL-1β-Induced Inflammatory Responses and MMP Expression in OA Chondrocytes
3.2. NTZ Inhibits IL-1β-Induced Glycolysis-Biased Metabolic Changes in OA Chondrocytes
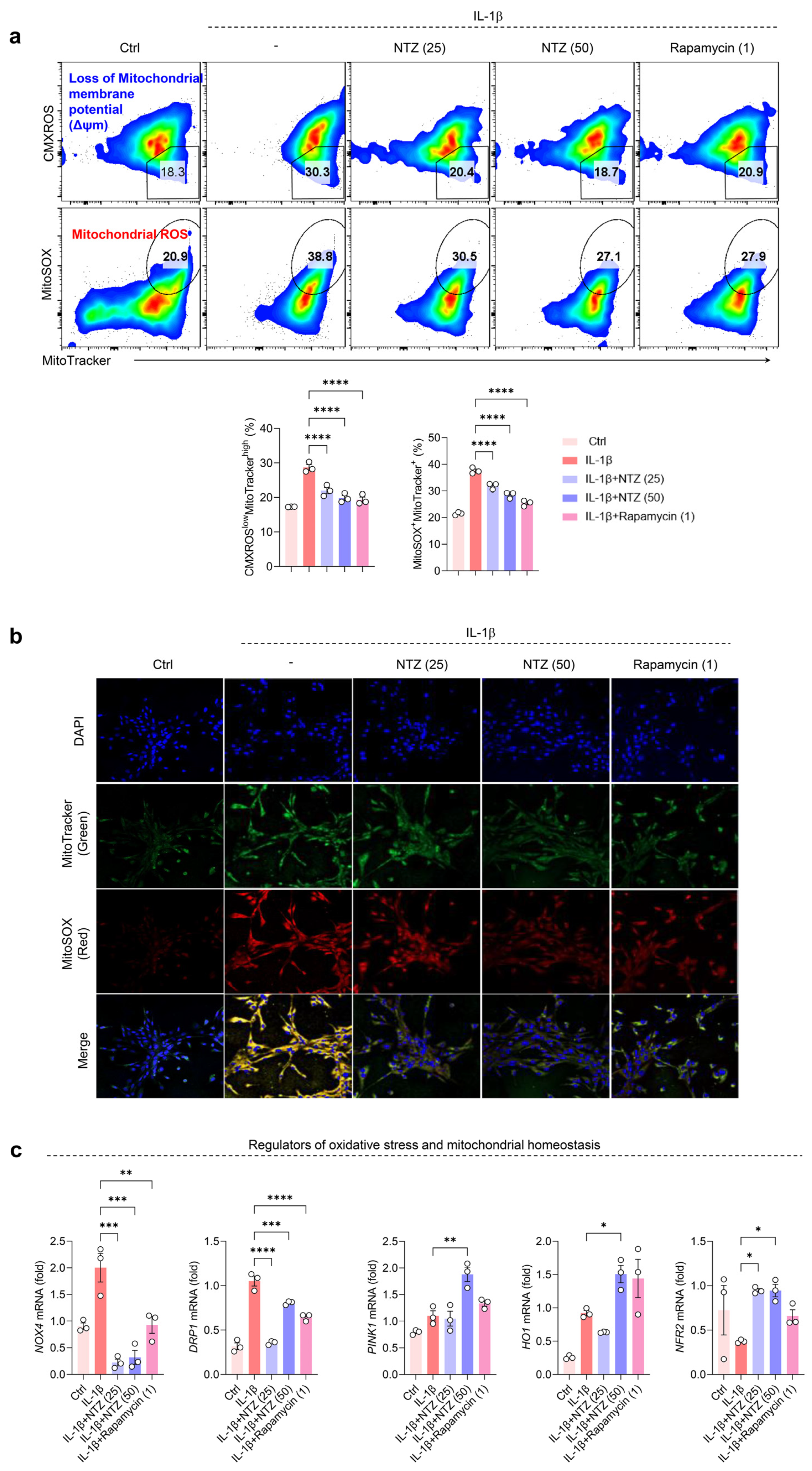
3.3. NTZ Restores Impaired Mitochondrial Function and Suppresses Oxidative Stress in IL-1β-Induced OA Chondrocytes
3.4. NTZ Attenuates mTORC1 Pathway Activation and Concurrently Stimulates AMPK Signaling in Chondrocytes Under IL-1β-Induced Inflammatory Stress
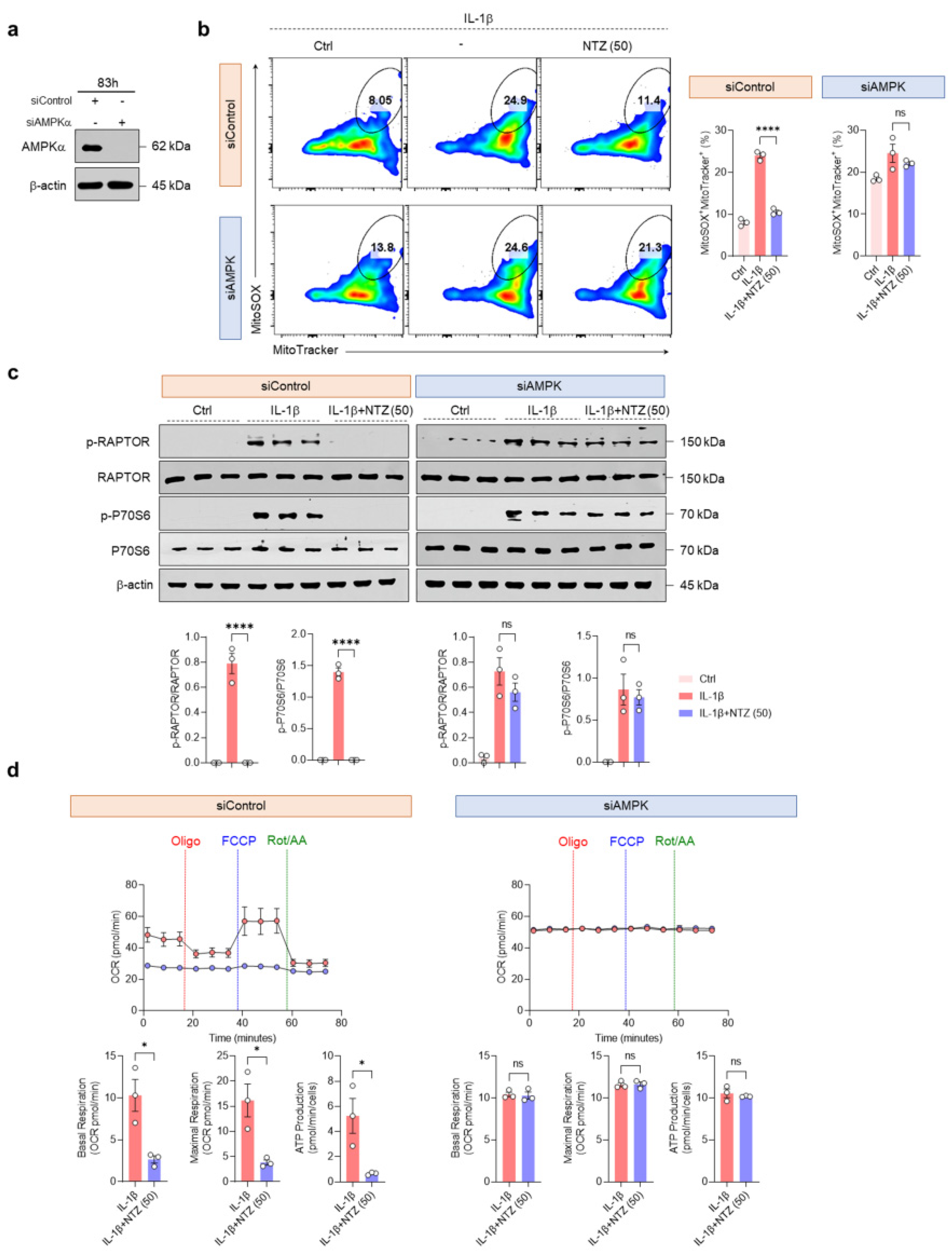
3.5. AMPK Activation by NTZ Is an Essential Regulatory Mechanism of mTORC1 Inhibition and Mitochondrial Protective Effects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OA | osteoarthritis |
| NTZ | nitazoxanide |
| MMPs | matrix metalloproteinases |
| TNF-α | tumor necrosis factor-α |
| IL | interleukin |
| ROS | reactive oxygen species |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mTOR complex 1 |
| FDA | Food and Drug Administration |
| SF DMEM | serum-free Dulbecco’s modified Eagle medium |
| FBS | fetal bovine serum |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| OCR | oxygen consumption rate |
| ECAR | extracellular acidification rate |
| PBS | phosphate-buffered saline |
| DPBS | Dulbecco’s PBS |
| DAPI | diamidino-2-phenylindole |
| qPCR | quantitative real-time PCR |
| OXPHOS | oxidative phosphorylation |
| DMOAD | disease-modifying osteoarthritis drug |
| FLS | fibroblast-like synoviocytes |
References
- GBD 2021 Osteoarthritis Collaborators. Global, regional, and national burden of osteoarthritis, 1990–2020 and projections to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet Rheumatol. 2023, 5, e508–e522. [Google Scholar] [CrossRef] [PubMed]
- Moulin, D.; Sellam, J.; Berenbaum, F.; Guicheux, J.; Boutet, M.A. The role of the immune system in osteoarthritis: Mechanisms, challenges and future directions. Nat. Rev. Rheumatol. 2025, 21, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Coras, R.; Torres, A.; Lane, N.E.; Guma, M. Synovial inflammation in osteoarthritis progression. Nat. Rev. Rheumatol. 2022, 18, 258–275. [Google Scholar] [CrossRef] [PubMed]
- Favero, M.; El-Hadi, H.; Belluzzi, E.; Granzotto, M.; Porzionato, A.; Sarasin, G.; Rambaldo, A.; Iacobellis, C.; Cigolotti, A.; Fontanella, C.G.; et al. Infrapatellar fat pad features in osteoarthritis: A histopathological and molecular study. Rheumatology 2017, 56, 1784–1793. [Google Scholar] [CrossRef]
- Driban, J.B.; Davis, J.E.; Lu, B.; Price, L.L.; Ward, R.J.; MacKay, J.W.; Eaton, C.B.; Lo, G.H.; Barbe, M.F.; Zhang, M.; et al. Accelerated Knee Osteoarthritis Is Characterized by Destabilizing Meniscal Tears and Preradiographic Structural Disease Burden. Arthritis Rheumatol. 2019, 71, 1089–1100. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.P. Osteoarthritis. Nat. Rev. Dis. Primers 2016, 2, 16072. [Google Scholar] [CrossRef]
- Kan, S.; Duan, M.; Liu, Y.; Wang, C.; Xie, J. Role of Mitochondria in Physiology of Chondrocytes and Diseases of Osteoarthritis and Rheumatoid Arthritis. Cartilage 2021, 13, 1102S–1121S. [Google Scholar] [CrossRef]
- Yao, Q.; Wu, X.; Tao, C.; Gong, W.; Chen, M.; Qu, M.; Zhong, Y.; He, T.; Chen, S.; Xiao, G. Osteoarthritis: Pathogenic signaling pathways and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 56. [Google Scholar] [CrossRef]
- Sun, K.; Luo, J.; Guo, J.; Yao, X.; Jing, X.; Guo, F. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: A narrative review. Osteoarthr. Cartil. 2020, 28, 400–409. [Google Scholar] [CrossRef]
- Latourte, A.; Kloppenburg, M.; Richette, P. Emerging pharmaceutical therapies for osteoarthritis. Nat. Rev. Rheumatol. 2020, 16, 673–688. [Google Scholar] [CrossRef]
- Amireddy, N.; Puttapaka, S.N.; Vinnakota, R.L.; Ravuri, H.G.; Thonda, S.; Kalivendi, S.V. The unintended mitochondrial uncoupling effects of the FDA-approved anti-helminth drug nitazoxanide mitigates experimental parkinsonism in mice. J. Biol. Chem. 2017, 292, 15731–15743. [Google Scholar] [CrossRef] [PubMed]
- Isac, E.; Picanco, G.A.; Costa, T.L.; Lima, N.F.; Alves, D.; Fraga, C.M.; Lino Junior, R.S.; Vinaud, M.C. In vitro nitazoxanide exposure affects energetic metabolism of Taenia crassiceps. Exp. Parasitol. 2020, 208, 107792. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zeng, S.; Qiu, Q.; Xiao, Y.; Shi, M.; Zou, Y.; Yang, X.; Xu, H.; Liang, L. Niclosamide induces apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes. Int. Immunopharmacol. 2016, 31, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.H.; Li, F.F.; Li, W.F.; Zhao, H.; Jiang, M.; Yu, Y.Y.; Dong, Y.C.; Zhang, Y.X.; Li, P.; Bu, W.J.; et al. Repurposing nitazoxanide as a novel anti-atherosclerotic drug based on mitochondrial uncoupling mechanisms. Br. J. Pharmacol. 2023, 180, 62–79. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, H.E.; Lee, S.T.; Park, J.; Nam, K.H.; Park, J.Y.; Choi, J.K. The Repurposing of Nitazoxanide for Psoriasis Treatment Exerts Therapeutic Effects through Skin Metabolic Reprogramming. J. Investig. Dermatol. 2025; in press. [Google Scholar] [CrossRef]
- Li, C.; Wang, F.; Han, Y.; Zhai, J.; Jin, Y.; Liu, R.; Niu, Y.; Yao, Z.; Zhao, J. Nitazoxanide reduces inflammation and bone erosion in mice with collagen-induced arthritis via inhibiting the JAK2/STAT3 and NF-kappaB pathways in fibroblast-like synoviocytes. Biomed. Pharmacother. 2024, 171, 116195. [Google Scholar] [CrossRef]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef]
- Arra, M.; Abu-Amer, Y. Cross-talk of inflammation and chondrocyte intracellular metabolism in osteoarthritis. Osteoarthr. Cartil. 2023, 31, 1012–1021. [Google Scholar] [CrossRef]
- Mobasheri, A.; Rayman, M.P.; Gualillo, O.; Sellam, J.; van der Kraan, P.; Fearon, U. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017, 13, 302–311. [Google Scholar] [CrossRef]
- Wu, X.; Fan, X.; Crawford, R.; Xiao, Y.; Prasadam, I. The Metabolic Landscape in Osteoarthritis. Aging Dis. 2022, 13, 1166–1182. [Google Scholar] [CrossRef]
- Guo, P.; Alhaskawi, A.; Adel Abdo Moqbel, S.; Pan, Z. Recent development of mitochondrial metabolism and dysfunction in osteoarthritis. Front. Pharmacol. 2025, 16, 1538662. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, Z.; Sheng, P.; Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 2021, 66, 101249. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.; Yu, H.; Lu, K.; Ruan, C.; Ding, C.; Tong, L.; Zhao, X.; Chen, D. AMPK Signaling in Energy Control, Cartilage Biology, and Osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 696602. [Google Scholar] [CrossRef] [PubMed]
- Henry, O.C.; O’Neill, L.A.J. Metabolic Reprogramming in Stromal and Immune Cells in Rheumatoid Arthritis and Osteoarthritis: Therapeutic Possibilities. Eur. J. Immunol. 2025, 55, e202451381. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Huang, Y.; Li, J.; Yi, D.; Liao, J.; Xiao, J.; Xiao, G.; Tong, L.; Huang, W.; Di, C. AMPK activator decelerates osteoarthritis development by inhibition of β-catenin signaling in chondrocytes. J. Orthop. Transl. 2023, 38, 158–166. [Google Scholar] [CrossRef]
- Rosa, S.C.; Goncalves, J.; Judas, F.; Mobasheri, A.; Lopes, C.; Mendes, A.F. Impaired glucose transporter-1 degradation and increased glucose transport and oxidative stress in response to high glucose in chondrocytes from osteoarthritic versus normal human cartilage. Arthritis Res. Ther. 2009, 11, R80. [Google Scholar] [CrossRef]
- Vaamonde-Garcia, C.; Riveiro-Naveira, R.R.; Valcarcel-Ares, M.N.; Hermida-Carballo, L.; Blanco, F.J.; Lopez-Armada, M.J. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. 2012, 64, 2927–2936. [Google Scholar] [CrossRef]
- Reed, K.N.; Wilson, G.; Pearsall, A.; Grishko, V.I. The role of mitochondrial reactive oxygen species in cartilage matrix destruction. Mol. Cell. Biochem. 2014, 397, 195–201. [Google Scholar] [CrossRef]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef]
- Carames, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Song, D.; Ni, J.; Ding, M.; Huang, J.; Yan, M. AMPK: Implications in osteoarthritis and therapeutic targets. Am. J. Transl. Res. 2020, 12, 7670–7681. [Google Scholar] [PubMed]
- Mei, R.; Lou, P.; You, G.; Jiang, T.; Yu, X.; Guo, L. 17β-Estradiol Induces Mitophagy Upregulation to Protect Chondrocytes via the SIRT1-Mediated AMPK/mTOR Signaling Pathway. Front. Endocrinol. 2020, 11, 615250. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Pan, J.; Li, J.; Zeng, C.; Qi, W.; Shao, Y.; Liu, X.; Liu, L.; Xiao, G.; Zhang, H.; et al. Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR. Aging 2020, 12, 1087–1103. [Google Scholar] [CrossRef]
- Zhuang, H.; Ren, X.; Zhang, Y.; Li, H.; Zhou, P. β-Hydroxybutyrate enhances chondrocyte mitophagy and reduces cartilage degeneration in osteoarthritis via the HCAR2/AMPK/PINK1/Parkin pathway. Aging Cell 2024, 23, e14294. [Google Scholar] [CrossRef]
- Terkeltaub, R.; Yang, B.; Lotz, M.; Liu-Bryan, R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1β and tumor necrosis factor α. Arthritis Rheumatol. 2011, 63, 1928–1937. [Google Scholar] [CrossRef]
- Amireddy, N.; Dulam, V.; Kaul, S.; Pakkiri, R.; Kalivendi, S.V. The mitochondrial uncoupling effects of nitazoxanide enhances cellular autophagy and promotes the clearance of α-synuclein: Potential role of AMPK-JNK pathway. Cell Signal. 2023, 109, 110769. [Google Scholar] [CrossRef]
- Fan, L.; Qiu, X.X.; Zhu, Z.Y.; Lv, J.L.; Lu, J.; Mao, F.; Zhu, J.; Wang, J.Y.; Guan, X.W.; Chen, J.; et al. Nitazoxanide, an anti-parasitic drug, efficiently ameliorates learning and memory impairments in AD model mice. Acta Pharmacol. Sin. 2019, 40, 1279–1291. [Google Scholar] [CrossRef]
- Weng, T.C.; Weng, T.S.; Lai, C.C.; Chao, C.M.; Wang, J.H. Clinical outcomes, virological efficacy and safety of nitazoxanide in the treatment of patients with COVID-19: A systematic review and meta-analysis of randomized controlled trials. Expert Rev. Anti-Infect. Ther. 2022, 20, 1615–1622. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.E.; Lee, J.Y.; Son, G.-Y.; Park, J.-Y.; Kim, K.B.; Choi, C.-M.; Moon, Y.J.; Choi, J.K. Nitazoxanide Modulates Mitochondrial Function and Inflammatory Metabolism in Chondrocytes from Patients with Osteoarthritis via AMPK/mTORC1 Signaling. Antioxidants 2025, 14, 512. https://doi.org/10.3390/antiox14050512
Kim HE, Lee JY, Son G-Y, Park J-Y, Kim KB, Choi C-M, Moon YJ, Choi JK. Nitazoxanide Modulates Mitochondrial Function and Inflammatory Metabolism in Chondrocytes from Patients with Osteoarthritis via AMPK/mTORC1 Signaling. Antioxidants. 2025; 14(5):512. https://doi.org/10.3390/antiox14050512
Chicago/Turabian StyleKim, Ha Eun, Jong Yeong Lee, Ga-Yeon Son, Jun-Young Park, Ki Bum Kim, Chul-Min Choi, Young Jae Moon, and Jin Kyeong Choi. 2025. "Nitazoxanide Modulates Mitochondrial Function and Inflammatory Metabolism in Chondrocytes from Patients with Osteoarthritis via AMPK/mTORC1 Signaling" Antioxidants 14, no. 5: 512. https://doi.org/10.3390/antiox14050512
APA StyleKim, H. E., Lee, J. Y., Son, G.-Y., Park, J.-Y., Kim, K. B., Choi, C.-M., Moon, Y. J., & Choi, J. K. (2025). Nitazoxanide Modulates Mitochondrial Function and Inflammatory Metabolism in Chondrocytes from Patients with Osteoarthritis via AMPK/mTORC1 Signaling. Antioxidants, 14(5), 512. https://doi.org/10.3390/antiox14050512