Exercise and Mitochondrial Dynamics: Keeping in Shape with ROS and AMPK

Abstract
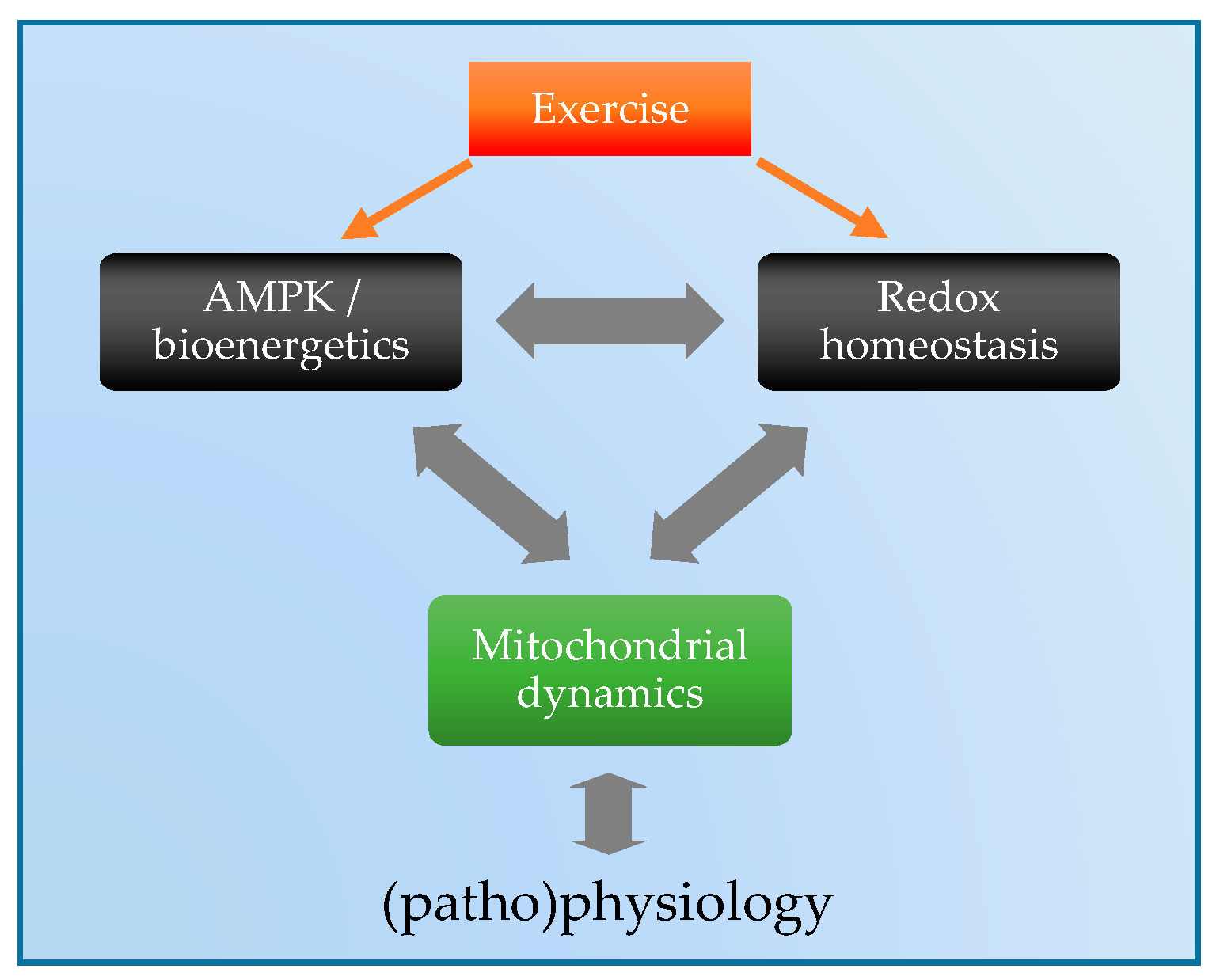
:1. Introduction
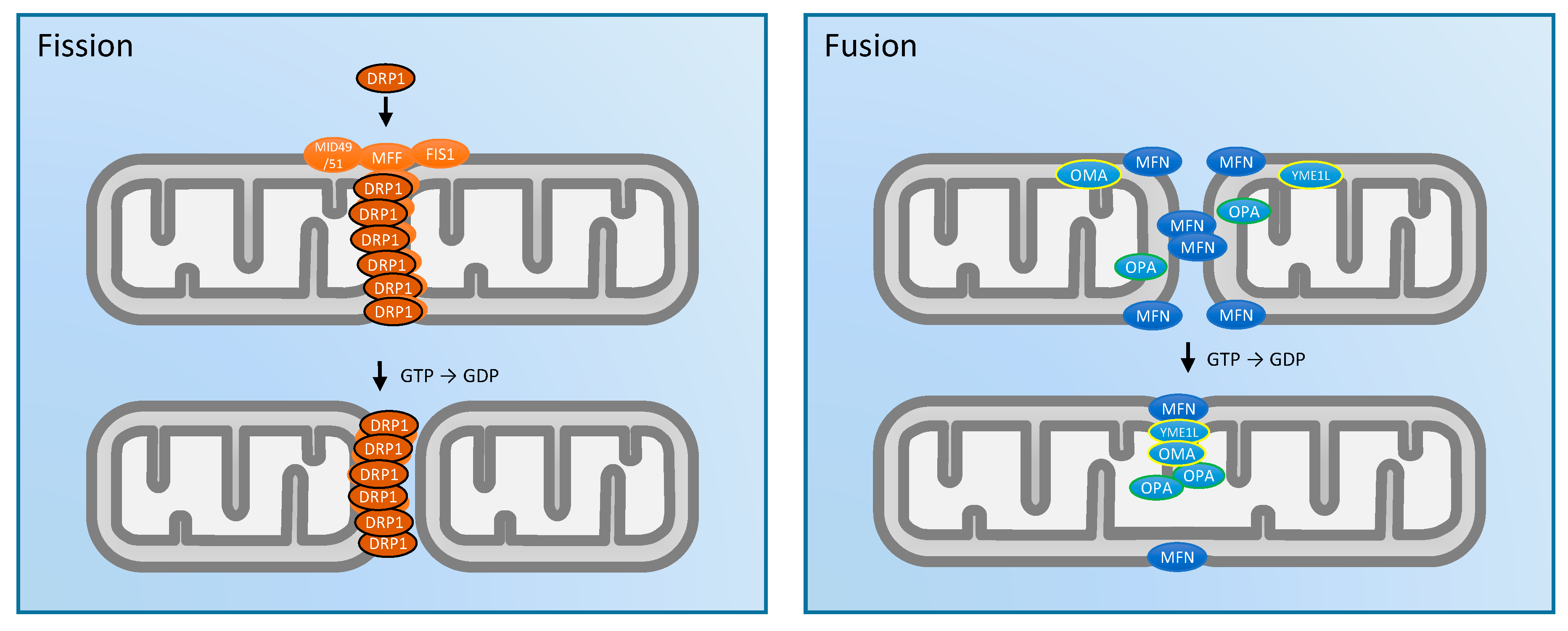
2. Mitochondrial Morphology and Dynamics
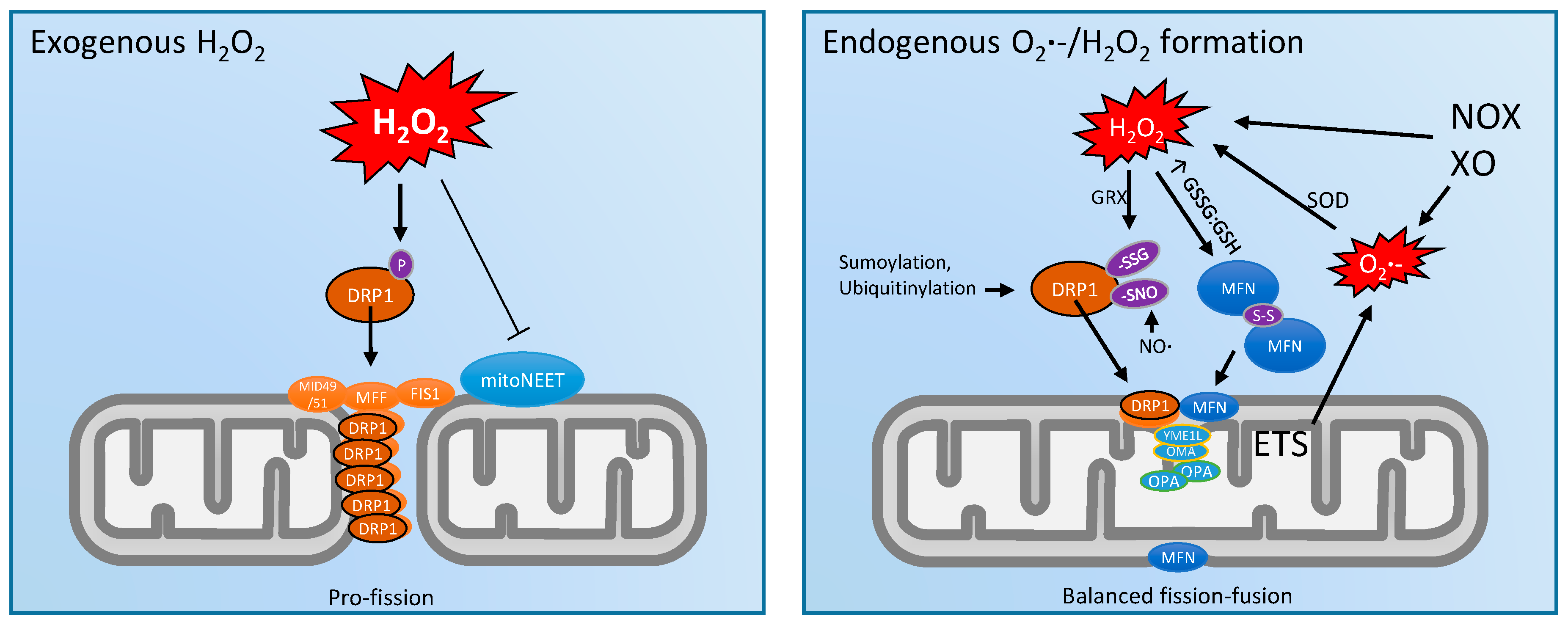
3. Mitochondrial Dynamics and ROS
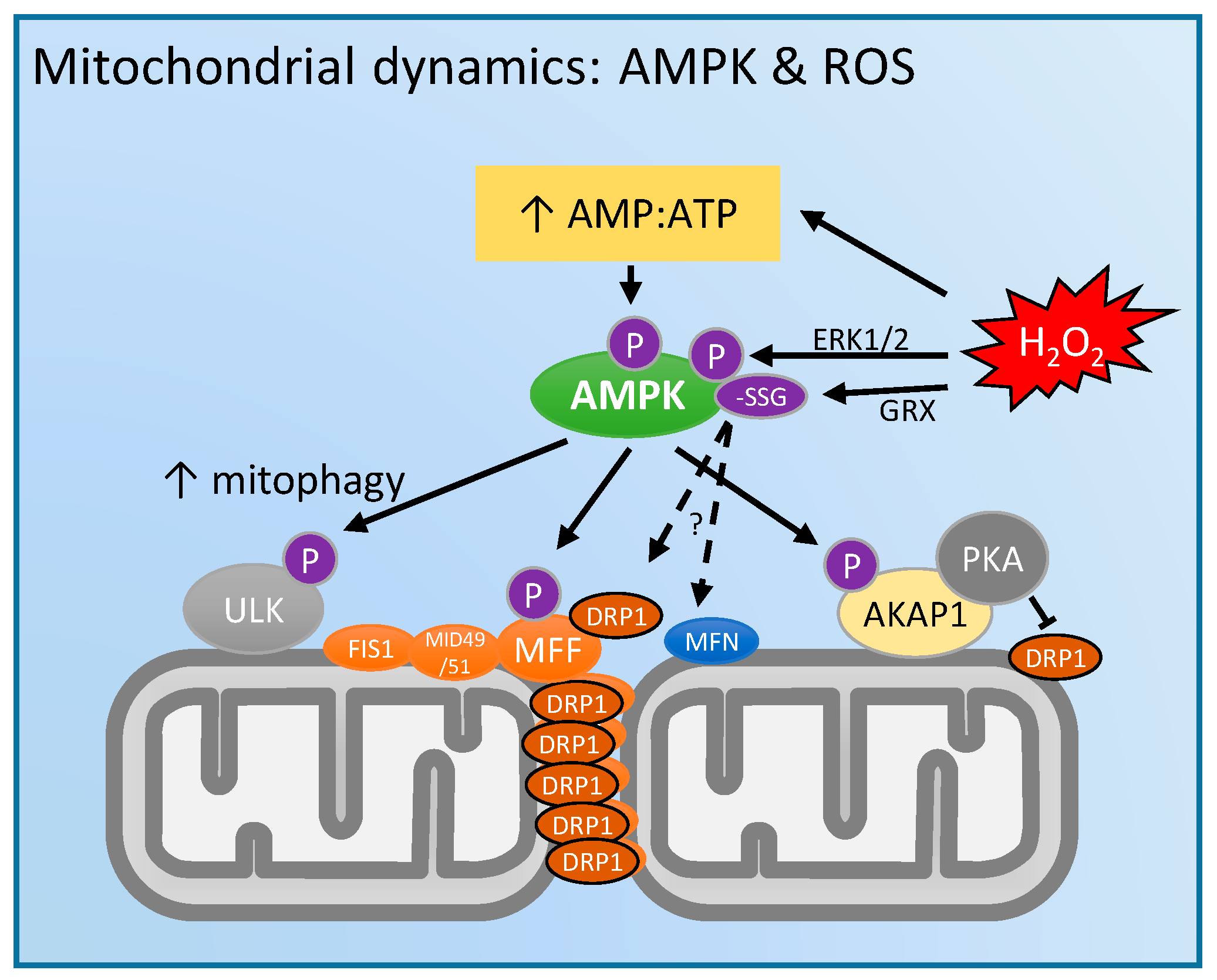
4. Mitochondrial Dynamics and 5'-Adenosine Monophosphate (AMP)-Activated Protein Kinase (AMPK): Regulation by ROS?
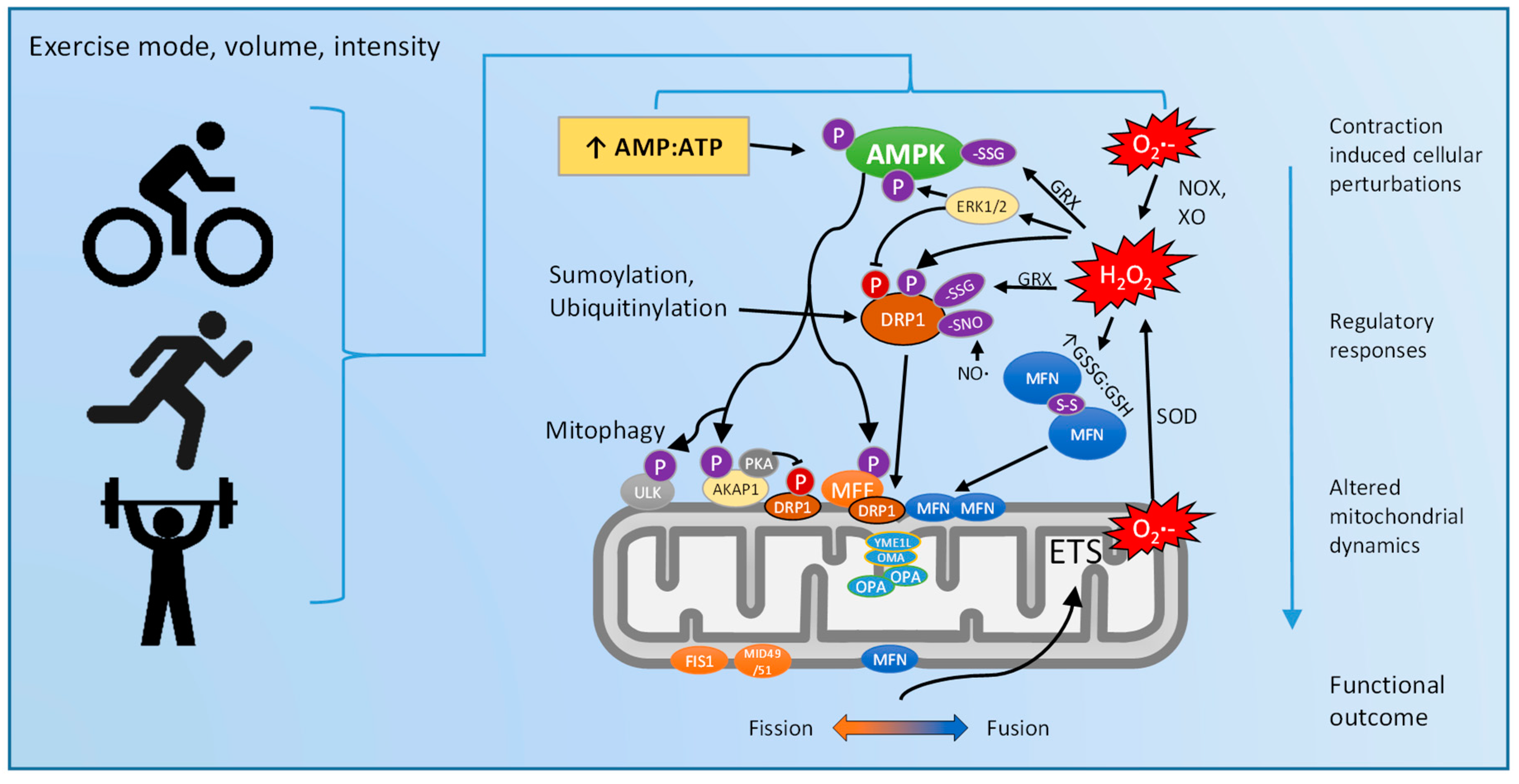
5. Exercise Regulation of Mitochondrial Dynamics: Via ROS and/or AMPK?
6. Future Research and Novel Methodologies
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hordern, M.D.; Dunstan, D.W.; Prins, J.B.; Baker, M.K.; Singh, M.A.F.; Coombes, J.S. Exercise prescription for patients with type 2 diabetes and pre-diabetes: A position statement from Exercise and Sport Science Australia. J. Sci. Med. Sport 2012, 15, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Saltin, B. Exercise as medicine-evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand. J. Med. Sci. Sports 2015, 25, 1–72. [Google Scholar] [CrossRef] [PubMed]
- Neufer, P.D.; Bamman, M.M.; Muoio, D.M.; Bouchard, C.; Cooper, D.M.; Goodpaster, B.H.; Booth, F.W.; Kohrt, W.M.; Gerszten, R.E.; Mattson, M.P. Understanding the cellular and molecular mechanisms of physical activity-induced health benefits. Cell Metab. 2015, 22, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Colberg, S.R.; Sigal, R.J.; Yardley, J.E.; Riddell, M.C.; Dunstan, D.W.; Dempsey, P.C.; Horton, E.S.; Castorino, K.; Tate, D.F. Physical activity/exercise and diabetes: A position statement of the American Diabetes Association. Diabetes Care 2016, 39, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Hallal, P.C.; Andersen, L.B.; Bull, F.C.; Guthold, R.; Haskell, W.; Ekelund, U. Lancet Physical Activity Series Working Group. Global physical activity levels: Surveillance progress, pitfalls, and prospects. Lancet 2012, 380, 247–257. [Google Scholar] [CrossRef]
- Garber, C.E.; Blissmer, B.; Deschenes, M.R.; Franklin, B.A.; Lamonte, M.J.; Lee, I.-M.; Nieman, D.C.; Swain, D.P.; American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: Guidance for prescribing exercise. Med. Sci. Sports Exerc. 2011, 43, 1334–1359. [Google Scholar] [CrossRef] [PubMed]
- Mercken, E.M.; Carboneau, B.A.; Krzysik-Walker, S.M.; de Cabo, R. Of mice and men: The benefits of caloric restriction, exercise, and mimetics. Ageing Res. Rev. 2012, 11, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Wall, C.E.; Ruth, T.Y.; Atkins, A.R.; Downes, M.; Evans, R.M. Nuclear receptors and AMPK: Can exercise mimetics cure diabetes? J. Mol. Endocrinol. 2016, 57, R49–R58. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Laher, I. Exercise Mimetics: Running Without a Road Map. Clin. Pharmacol. Ther. 2017, 101, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Hawley, J.A.; Holloszy, J.O. Exercise: It’s the real thing! Nutr. Rev. 2009, 67, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Joyner, M.J.; Green, D.J. Exercise protects the cardiovascular system: Effects beyond traditional risk factors. J. Physiol. 2009, 587, 5551–5558. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, F.; Larson, K.; Bogardus, C.; Ravussin, E. Skeletal muscle metabolism is a major determinant of resting energy expenditure. J. Clin. Investig. 1990, 86, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R.; Hoppeler, H. Exercise-induced maximal metabolic rate scales with muscle aerobic capacity. J. Exp. Biol. 2005, 208, 1635–1644. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.P.; Foletta, V.C.; Snow, R.J.; Wadley, G.D. Skeletal muscle mitochondria: A major player in exercise, health and disease. Biochim. Biophys. Acta 2014, 1840, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O. Biochemical adaptations in muscle effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [PubMed]
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial cristae: Where beauty meets functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Lizana, L.; Bauer, B.; Orwar, O. Controlling the rates of biochemical reactions and signaling networks by shape and volume changes. Proc. Natl. Acad. Sci. USA 2008, 105, 4099–4104. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Hartnell, L.M.; Malide, D.; Yu, Z.-X.; Combs, C.A.; Connelly, P.S.; Subramaniam, S.; Balaban, R.S. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 2015, 523, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; White, K.; Turnbull, D.M. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: A quantitative three-dimensional electron microscopy study. J. Appl. Physiol. 2013, 114, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Longo, D.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Ryan, M.T. Mitochondrial fusion: Reaching the end of mitofusin’s tether. J. Cell Biol. 2016, 215, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Kar, R.; Singha, P.K.; Venkatachalam, M.A.; McEwen, D.G.; Saikumar, P. Inhibition of mitochondrial division through covalent modification of Drp1 protein by 15 deoxy-Δ12,14-prostaglandin J2. Biochem. Biophys. Res. Commun. 2010, 395, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease–The long and short of it. J. Cell Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Vernay, A.; Marchetti, A.; Sabra, A.; Jauslin, T.N.; Rosselin, M.; Scherer, P.E.; Demaurex, N.; Orci, L.; Cosson, P. MitoNEET-dependent formation of intermitochondrial junctions. Proc. Natl. Acad. Sci. USA 2017, 114, 8277–8282. [Google Scholar] [CrossRef] [PubMed]
- Colca, J.R.; McDonald, W.G.; Waldon, D.J.; Leone, J.W.; Lull, J.M.; Bannow, C.A.; Lund, E.T.; Mathews, W.R. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am J. Physiol. Endocrinol. Metab. 2004, 286, E252–E260. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; van der Laan, M.; Amati, P.; Capaldi, R.A.; Caudy, A.A.; Chacinska, A.; Darshi, M.; Deckers, M.; Hoppins, S.; Icho, T. Uniform nomenclature for the mitochondrial contact site and cristae organizing system. J. Cell Biol. 2014, 204, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Kozjak-Pavlovic, V. The MICOS complex of human mitochondria. Cell Tissue Res. 2017, 367, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Mun, J.Y.; Lee, T.H.; Kim, J.H.; Yoo, B.H.; Bahk, Y.Y.; Koo, H.S.; Han, S.S. Caenorhabditis elegans mitofilin homologs control the morphology of mitochondrial cristae and influence reproduction and physiology. J. Cell. Physiol. 2010, 224, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Guarani, V.; McNeill, E.M.; Paulo, J.A.; Huttlin, E.L.; Fröhlich, F.; Gygi, S.P.; Van Vactor, D.; Harper, J.W. QIL1 is a novel mitochondrial protein required for MICOS complex stability and cristae morphology. eLife 2015, 4, e06265. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Mourier, A.; Yamada, J.; McCaffery, J.M.; Nunnari, J. MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. eLife 2015, 4, e07739. [Google Scholar] [CrossRef] [PubMed]
- Ikon, N.; Ryan, R.O. Cardiolipin and Mitochondrial Cristae Organization; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Wilkens, V.; Kohl, W.; Busch, K. Restricted diffusion of OXPHOS complexes in dynamic mitochondria delays their exchange between cristae and engenders a transitory mosaic distribution. J. Cell Sci. 2013, 126, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Norton, M.; Ng, A.C.-H.; Baird, S.; Dumoulin, A.; Shutt, T.; Mah, N.; Andrade-Navarro, M.A.; McBride, H.M.; Screaton, R.A. ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci. Signal. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.E.; Turnbull, D.M.; Eisner, V.; Hajnóczky, G.; Picard, M. Mitochondrial Nanotunnels. Trends Cell Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sun, L.; Ji, S.; Zhao, T.; Zhang, W.; Xu, J.; Zhang, J.; Wang, Y.; Wang, X.; Franzini-Armstrong, C. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc. Natl. Acad. Sci. USA 2013, 110, 2846–2851. [Google Scholar] [CrossRef] [PubMed]
- Ichishita, R.; Tanaka, K.; Sugiura, Y.; Sayano, T.; Mihara, K.; Oka, T. An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J. Biochem. 2008, 143, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Smith, S.B.; Yoon, Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J. Biol. Chem. 2017, 292, 7115–7130. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Talbert, E.E.; Adhihetty, P.J. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J. Physiol. 2011, 589, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Jendrach, M.; Mai, S.; Pohl, S.; Vöth, M.; Bereiter-Hahn, J. Short-and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion 2008, 8, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Hussien, R.; Brooks, G.A. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic. Biol. Med. 2010, 49, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Hood, D.A. Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts. Am. J. Physiol. Cell Physiol. 2014, 306, C1176–C1183. [Google Scholar] [CrossRef] [PubMed]
- Debattisti, V.; Gerencser, A.A.; Saotome, M.; Das, S.; Hajnóczky, G. ROS Control Mitochondrial Motility through p38 and the Motor Adaptor Miro/Trak. Cell Rep. 2017, 21, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox homeostasis and mitochondrial dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Bhatnagar, A. Protein S-glutathiolation: Redox-sensitive regulation of protein function. J. Mol. Cell. Cardiol. 2012, 52, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012, 13, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Thaher, O.; Wolf, C.; Dey, P.N.; Pouya, A.; Wüllner, V.; Tenzer, S.; Methner, A. The thiol switch C684 in Mitofusin-2 mediates redox-induced alterations of mitochondrial shape and respiration. Neurochem. Int. 2017. [Google Scholar] [CrossRef] [PubMed]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Sabouny, R.; Fraunberger, E.; Geoffrion, M.; Ng, A.; Baird, S.; Screaton, R.; Milne, R.; McBride, H.M.; Shutt, T. The Keap1-Nrf2 stress response pathway promotes mitochondrial hyperfusion through degradation of the mitochondrial fission protein Drp1. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Cannata, S.M.; Rotilio, G.; Ciriolo, M.R. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid. Redox Signal. 2013, 18, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Borda-d’Água, B.; Medina-Gómez, G.; Lelliott, C.J.; Paz, J.C.; Rojo, M.; Palacín, M.; Vidal-Puig, A.; Zorzano, A. Mitochondrial fusion is increased by the nuclear coactivator PGC-1β. PLoS ONE 2008, 3, e3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, J.; Choi, C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS ONE 2011, 6, e23211. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Rev. Physiol. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- John, G.B.; Shang, Y.; Li, L.; Renken, C.; Mannella, C.A.; Selker, J.M.; Rangell, L.; Bennett, M.J.; Zha, J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol. Biol. Cell 2005, 16, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Shealinna, X.G. The Putative Drp1 Inhibitor mdivi-1 is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed]
- Glytsou, C.; Calvo, E.; Cogliati, S.; Mehrotra, A.; Anastasia, I.; Rigoni, G.; Raimondi, A.; Shintani, N.; Loureiro, M.; Vazquez, J. Optic atrophy 1 is epistatic to the core MICOS component MIC60 in mitochondrial cristae shape control. Cell Rep. 2016, 17, 3024–3034. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Ducommun, S.; Deak, M.; Sumpton, D.; Ford, R.J.; Galindo, A.N.; Kussmann, M.; Viollet, B.; Steinberg, G.R.; Foretz, M.; Dayon, L. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: Identification of mitochondrial fission factor as a new AMPK substrate. Cell. Signal. 2015, 27, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J. AMPK phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-L.; Kim, S.-J.; Lee, K.-T.; Kim, J.; Mu, J.; Birnbaum, M.J.; Kim, S.S.; Ha, J. The regulation of AMP-activated protein kinase by H2O2. Biochem. Biophys. Res. Commun. 2001, 287, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M. Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Auciello, F.R.; Ross, F.A.; Ikematsu, N.; Hardie, D.G. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett. 2014, 588, 3361–3366. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Oka, S.-I.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Wu, M.; Liu, X.; Huang, Y.; Zhang, D.; Wang, Y.; Yan, L.-J.; Shi, D. Glutaredoxins concomitant with optimal ROS activate AMPK through S-glutathionylation to improve glucose metabolism in type 2 diabetes. Free Radic. Biol. Med. 2016, 101, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.C.; Wilson, R.J.; Yan, Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J. 2015, 30, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.; Hoppel, F.; Macek, C.; Messner, H.; Faulhaber, M.; Kobel, C.; Parson, W.; Burtscher, M.; Schocke, M.; Gnaiger, E. Similar qualitative and quantitative changes of mitochondrial respiration following strength and endurance training in normoxia and hypoxia in sedentary humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1078–R1087. [Google Scholar] [CrossRef] [PubMed]
- Penman, K.A. Ultrastructural changes in human striated muscle using three methods of training. Res. Q. Am. Assoc. Health Phys. Educ. Recreat. 1969, 40, 764–772. [Google Scholar]
- Gollnick, P.D.; King, D.W. Effect of exercise and training on mitochondria of rat skeletal muscle. Am. J. Physiol. Leg. Content 1969, 216, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Gollnick, P.; King, D. The immediate and chronic effect of exercise on the number and structure of skeletal muscle mitochondria. In Biochemistry of Exercise; Karger Publishers: Basel, Switzerland, 1969; Volume 3, pp. 239–244. [Google Scholar]
- Kiessling, K.-H.; Piehl, K.; Lundquist, C.-G. Effect of physical training on ultrastructural features in human skeletal muscle. In Muscle Metabolism during Exercise; Springer: Berlin/Heidelberg, Germany, 1971; pp. 97–101. [Google Scholar]
- Kirkwood, S.; Packer, L.; Brooks, G. Effects of endurance training on a mitochondrial reticulum in limb skeletal muscle. Arch. Biochem. Biophys. 1987, 255, 80–88. [Google Scholar] [CrossRef]
- Cartoni, R.; Léger, B.; Hock, M.B.; Praz, M.; Crettenand, A.; Pich, S.; Ziltener, J.L.; Luthi, F.; Dériaz, O.; Zorzano, A. Mitofusins 1/2 and ERRα expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005, 567, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.G.; Lally, J.; Holloway, G.P.; Heigenhauser, G.J.; Bonen, A.; Spriet, L.L. Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J. Physiol. 2010, 588, 4795–4810. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, Y.; Ogasawara, R.; Tamura, Y.; Fujita, S.; Hatta, H. Effect of electrical stimulation-induced resistance exercise on mitochondrial fission and fusion proteins in rat skeletal muscle. Appl. Physiol. Nutr. Metab. 2015, 40, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- MacInnis, M.J.; Zacharewicz, E.; Martin, B.J.; Haikalis, M.E.; Skelly, L.E.; Tarnopolsky, M.A.; Murphy, R.M.; Gibala, M.J. Superior mitochondrial adaptations in human skeletal muscle after interval compared to continuous single-leg cycling matched for total work. J. Physiol. 2017, 595, 2955–2968. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.R.; Suer, M.K.; Wolff, C.A.; Harber, M.P. Markers of human skeletal muscle mitochondrial biogenesis and quality control: Effects of age and aerobic exercise training. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2013, 69, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Kang, C.; Dickman, J.R.; Koenig, R.; Awoyinka, I.; Zhang, Y.; Ji, L.L. Training-induced mitochondrial adaptation: Role of peroxisome proliferator-activated receptor γ coactivator-1α, nuclear factor-κB and β-blockade. Exp. Physiol. 2013, 98, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Marton, O.; Koltai, E.; Takeda, M.; Koch, L.G.; Britton, S.L.; Davies, K.J.; Boldogh, I.; Radak, Z. Mitochondrial biogenesis-associated factors underlie the magnitude of response to aerobic endurance training in rats. Pflüg. Arch. Eur. J. Physiol. 2015, 467, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Garnier, A.; Fortin, D.; Zoll, J.; N’Guessan, B.; Mettauer, B.; Lampert, E.; Veksler, V.; Ventura-Clapier, R. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB J. 2005, 19, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Wyckelsma, V.L.; Levinger, I.; McKenna, M.J.; Formosa, L.E.; Ryan, M.T.; Petersen, A.C.; Anderson, M.J.; Murphy, R.M. Preservation of skeletal muscle mitochondrial content in older adults: Relationship between mitochondria, fibre type and high-intensity exercise training. J. Physiol. 2017, 595, 3345–3359. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Jiang, N.; Liu, H.; Liu, X.; Liu, D.; Zhao, F.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. BBA Gen. Subj. 2010, 1800, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Gentil, B.J.; McManus, M.J.; White, K.; Louis, K.S.; Gartside, S.E.; Wallace, D.C.; Turnbull, D.M. Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J. Appl. Physiol. 2013, 115, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Jamart, C.; Naslain, D.; Gilson, H.; Francaux, M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J. Physiol. Endocrinol. Metab. 2013, 305, E964–E974. [Google Scholar] [CrossRef] [PubMed]
- Kruse, R.; Pedersen, A.J.; Kristensen, J.M.; Petersson, S.J.; Wojtaszewski, J.F.; Højlund, K. Intact initiation of autophagy and mitochondrial fission by acute exercise in skeletal muscle of patients with type 2 diabetes. Clinic. Science 2017, 131, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Fealy, C.E.; Mulya, A.; Lai, N.; Kirwan, J.P. Exercise training decreases activation of the mitochondrial fission protein dynamin-related protein-1 in insulin-resistant human skeletal muscle. J. Appl. Physiol. 2014, 117, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Caffin, F.; Prola, A.; Piquereau, J.; Novotova, M.; David, D.; Garnier, A.; Fortin, D.; Alavi, M.; Veksler, V.; Ventura-Clapier, R. Altered skeletal muscle mitochondrial biogenesis but improved endurance capacity in trained OPA1-deficient mice. J. Physiol. 2013, 591, 6017–6037. [Google Scholar] [CrossRef] [PubMed]
- Weir, H.J.; Yao, P.; Huynh, F.K.; Escoubas, C.C.; Goncalves, R.L.; Burkewitz, K.; Laboy, R.; Hirschey, M.D.; Mair, W.B. Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Franco, A.; Fleischer, J.A.; Zhang, L.; Dorn, G.W., II. Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Coronado, M.; Fajardo, G.; Nguyen, K.; Zhao, M.; Kooiker, K.B.; Hu, D.-Q.; Reddy, S.; Sandoval, E.; Stotland, A.; Gottlieb, R.A. Physiologic Mitochondrial Fragmentation is a Normal Cardiac Adaptation to Increased Energy Demand. Circ. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Brandes, R.; Schendel, T.; Trocha, S.; Miller, R.; Weiner, M. Energy use by contractile and noncontractile processes in skeletal muscle estimated by 31P-NMR. Am. J. Physiol. Cell Physiol. 1994, 266, C825–C831. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Varuzhanyan, G.; Pham, A.H.; Chan, D.C. Mitochondrial dynamics is a distinguishing feature of skeletal muscle fiber types and regulates organellar compartmentalization. Cell Metab. 2015, 22, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Hartnell, L.M.; Combs, C.A.; Fenmou, A.; Sun, J.; Murphy, E.; Subramaniam, S.; Balaban, R.S. Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell Rep. 2017, 19, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Legros, F.; Lombès, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 2002, 13, 4343–4354. [Google Scholar] [CrossRef] [PubMed]
- Mattenberger, Y.; James, D.I.; Martinou, J.-C. Fusion of mitochondria in mammalian cells is dependent on the mitochondrial inner membrane potential and independent of microtubules or actin. FEBS Lett. 2003, 538, 53–59. [Google Scholar] [CrossRef]
- Brand, M.D.; Chien, L.-F.; Ainscow, E.K.; Rolfe, D.F.; Porter, R.K. The causes and functions of mitochondrial proton leak. Biochim. Biophys. Acta Bioenerg. 1994, 1187, 132–139. [Google Scholar] [CrossRef]
- Nobes, C.; Brown, G.C.; Olive, P.N.; Brand, M.D. Non-ohmic proton conductance of the mitochondrial inner membrane in hepatocytes. J. Biol. Chem. 1990, 265, 12903–12909. [Google Scholar] [PubMed]
- Plecitá-Hlavatá, L.; Ježek, P. Integration of superoxide formation and cristae morphology for mitochondrial redox signaling. Int. J. Biochem. Cell Biol. 2016, 80, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Greggio, C.; Jha, P.; Kulkarni, S.S.; Lagarrigue, S.; Broskey, N.T.; Boutant, M.; Wang, X.; Alonso, S.C.; Ofori, E.; Auwerx, J. Enhanced respiratory chain supercomplex formation in response to exercise in human skeletal muscle. Cell Metab. 2017, 25, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Kemp, B.E. AMPK in health and disease. Rev. Physiol. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Lantier, L.; Fentz, J.; Mounier, R.; Leclerc, J.; Treebak, J.T.; Pehmøller, C.; Sanz, N.; Sakakibara, I.; Saint-Amand, E.; Rimbaud, S. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J. 2014, 28, 3211–3224. [Google Scholar] [CrossRef] [PubMed]
- Trewin, A.J.; Lundell, L.S.; Perry, B.D.; Patil, K.V.; Chibalin, A.V.; Levinger, I.; McQuade, L.R.; Stepto, N.K. Effect of N-acetylcysteine infusion on exercise induced modulation of insulin sensitivity, and signaling pathways in human skeletal muscle. Am J. Physiol. Endocrinol. Metab. 2015, 309, E388–E397. [Google Scholar] [CrossRef] [PubMed]
- Merrill, R.A.; Strack, S. Mitochondria: A kinase anchoring protein 1, a signaling platform for mitochondrial form and function. Int. J. Biochem. Cell Biol. 2014, 48, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Scimia, M.C.; Wilkinson, D.; Trelles, R.D.; Wood, M.R.; Bowtell, D.; Dillin, A.; Mercola, M.; Ze’ev, A.R. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 2011, 44, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Morales-Alamo, D.; Calbet, J.A. AMPK signaling in skeletal muscle during exercise: Role of reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2016, 98, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.M.; Young, I.S.; McEneny, J.; Lawrenson, L.; Kim, J.; Barden, J.; Richardson, R.S. Regulation of free radical outflow from an isolated muscle bed in exercising humans. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1689–H1699. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.; Haack, K.; Franchek, K.; Valberg, P.; Kobzik, L.; West, M. Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J. Appl. Physiol. 1992, 73, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.; Reid, M. Muscle-derived ROS and thiol regulation in muscle fatigue. J. Appl. Physiol. 2008, 104, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.; Quintanilha, A.; Brooks, G.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Wadley, G.D.; Nicolas, M.A.; Hiam, D.; McConell, G.K. Xanthine oxidase inhibition attenuates skeletal muscle signaling following acute exercise but does not impair mitochondrial adaptations to endurance training. Am J. Physiol. Endocrinol. Metab. 2013, 304, E853–E862. [Google Scholar] [CrossRef] [PubMed]
- Trewin, A.J.; Levinger, I.; Parker, L.; Shaw, C.S.; Serpiello, F.R.; Anderson, M.J.; McConell, G.K.; Hare, D.L.; Stepto, N.K. Acute exercise alters skeletal muscle mitochondrial respiration and H2O2 emission in response to hyperinsulinemic-euglycemic clamp in middle-aged obese men. PLoS ONE 2017, 12, e0188421. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.J.; Schiffer, T.A.; Ørtenblad, N.; Zinner, C.; Morales-Alamo, D.; Willis, S.J.; Calbet, J.A.; Holmberg, H.-C.; Boushel, R. High-intensity sprint training inhibits mitochondrial respiration through aconitase inactivation. FASEB J. 2016, 30, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U.; Wittig, I. Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Higdon, A.N.; Dranka, B.P.; Darley-Usmar, V.M. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Cabo, H.; Ferrando, B.; Viña, J. Redox modulation of mitochondriogenesis in exercise. Does antioxidant supplementation blunt the benefits of exercise training? Free Radic. Biol. Med. 2015, 86, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Jensen, M.B.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of Superoxide- H2O2 Production at Site I Q of Mitochondrial Complex I Protect against Stem Cell Hyperplasia and Ischemia-Reperfusion Injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Trewin, A.J.; Petersen, A.C.; Billaut, F.; McQuade, L.R.; McInerney, B.V.; Stepto, N.K. N-acetylcysteine alters substrate metabolism during high-intensity cycle exercise in well-trained humans. Appl. Physiol. Nutr. Metab. 2013, 38, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.A.; Morrison, D.; McConell, G.K.; Wadley, G.D. Muscle redox signalling pathways in exercise. Role of antioxidants. Free Radic. Biol. Med. 2016, 98, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Strobel, N.A.; Peake, J.M.; Matsumoto, A.; Marsh, S.A.; Coombes, J.S.; Wadley, G.D. Antioxidant supplementation reduces skeletal muscle mitochondrial biogenesis. Med. Sci. Sports Exerc. 2011, 43, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.W.; Brand, M.D.; Petrosyan, S.; Ashok, D.; Elorza, A.A.; Ferrick, D.A.; Murphy, A.N. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS ONE 2011, 6, e21746. [Google Scholar] [CrossRef] [PubMed]
- Krumschnabel, G.; Fontana-Ayoub, M.; Sumbalova, Z.; Heidler, J.; Gauper, K.; Fasching, M.; Gnaiger, E. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Mitochondrial Med. 2015, 1264, 245–261. [Google Scholar]
- Husson, S.J.; Liewald, J.F.; Schultheis, C.; Stirman, J.N.; Lu, H.; Gottschalk, A. Microbial light-activatable proton pumps as neuronal inhibitors to functionally dissect neuronal networks in C. elegans. PLoS ONE 2012, 7, e40937. [Google Scholar] [CrossRef] [PubMed]
- Tkatch, T.; Greotti, E.; Baranauskas, G.; Pendin, D.; Roy, S.; Nita, L.I.; Wettmarshausen, J.; Prigge, M.; Yizhar, O.; Shirihai, O.S. Optogenetic control of mitochondrial metabolism and Ca2+ signaling by mitochondria-targeted opsins. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, A.; Takagi, S. An optogenetic application of proton pump ArchT to C. elegans cells. Neurosci. Res. 2013, 75, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Fan, L.Z.; Li, P.; Shen, K.; Lin, M.Z. Optical control of cell signaling by single-chain photoswitchable kinases. Science 2017, 355, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Wojtovich, A.P.; Foster, T.H. Optogenetic control of ROS production. Redox Biol. 2014, 2, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Wojtovich, A.P.; Wei, A.Y.; Sherman, T.A.; Foster, T.H.; Nehrke, K. Chromophore-assisted light inactivation of mitochondrial electron transport chain complex II in Caenorhabditis elegans. Sci. Rep. 2016, 6, 29695. [Google Scholar] [CrossRef] [PubMed]
- Trewin, A.J.; Bahr, L.; Wojtovich, A.P. Mitochondrial Membrane Sidedness Dependent Effects of ROS Generation in the Complex-II Microdomain using Optogenetics. Free Radic. Biol. Med. 2017, 112. [Google Scholar] [CrossRef]
- Hung, C.H.-L.; Cheng, S.S.-Y.; Cheung, Y.-T.; Wuwongse, S.; Zhang, N.Q.; Ho, Y.-S.; Lee, S.M.-Y.; Chang, R.C.-C. A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol. 2018, 14, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Schneider, J.F.; Degrossoli, A.; Lupilova, N.; Dick, T.P.; Leichert, L.I. Systematic in vitro assessment of responses of roGFP2-based probes to physiologically relevant oxidant species. Free Radic. Biol. Med. 2017, 106, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Laranjeiro, R.; Harinath, G.; Burke, D.; Braeckman, B.P.; Driscoll, M. Single swim sessions in C. elegans induce key features of mammalian exercise. BMC Biol. 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.-S.; Kuo, W.-J.; Lee, C.-L.; Chu, I.-H.; Chen, C.-S. Exercise in an electrotactic flow chamber ameliorates age-related degeneration in Caenorhabditis elegans. Sci. Rep. 2016, 6, 28064. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N. Mitochondrial function, biology, and role in disease. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author, Year [Reference] | Species/Model | Acute Exercise Stimulus | Summary Skeletal Muscle mRNA and/or Protein Responses | Evidence for Pro-Fission Responses | Evidence for Pro-Fusion Responses |
|---|---|---|---|---|---|
| Cartoni et al. 2005 [95] | Human (well trained cyclists); SAOS2 cell culture | 45 min ∼80% VO2peak cycling | ↑ mfn1/2 mRNA (24 h post), via ERRα and PGC1α. ↔ MFN2 protein abundance 0–24 h post exercise | n/a | ↑ mfn1/2 mRNA (24 h post) |
| Ding et al. 2010 [104] | Rat | 2.5 h ∼75% VO2peak treadmill running | ↑ mfn1 mRNA, but ↓ MFN1 protein (0–24 h post); ↑ mfn2 mRNA 24 h post. ↑ fis1 mRNA and FIS1 protein 0–24 h post | ↑ fis1 mRNA, ↑ FIS1 and ↓ MFN1 protein 0–24 h post | ↑ mfn1 mRNA (0–24 h post); ↑ mfn2 mRNA 24 h post. |
| Perry et al. 2010 [96] | Human | 1 h ∼90% VO2peak cycling, high intensity intervals | ↔ MFN1/2 , FIS1 or DRP1 protein abundance 4 h post exercise | n/d | n/d |
| Picard et al. 2013 [105] | Mice | 3 h voluntary running (∼1.8 km covered) | ↑ intermitochondrial contacts; ↔ morphology or MFN2 and OPA1 protein abundance | n/d | ↑ intermitochondrial contacts |
| Jamart et al. 2013 [106] | Mice | 1.5 h low-intesntiy (~55% VO2 max) treadmill running | ↑ DRP1 Ser616 phosphorylation, ↔ dnm1l mRNA, ↔ mfn1/2 mRNA ~5 min post-exercise | ↑ DRP1 Ser616 phosphorylation | n/d |
| Kitaoka et al. 2015 [97] | Rat | ~1 h ‘resistance exercise’ electrical stimulation isometric contraction | ↑ DRP1 Ser616 phosphorylation 0 h post exercise. ↔ DRP1, FIS1, MFN1/2, OPA1 protein 0–24 h post contraction | ↑ DRP1 Ser616 phosphorylation 0 h post exercise | n/d |
| Kruse et al. 2017 [107] | Human (healthy controls and obese+T2DM) | 1 h (70% VO2max) cycling | Healthy subjects 0 h post-exercise mRNA: ↑ mfn2, ↔ opa1, ↔ dnm1l, ↔ fis1; 3 h post-exercise mRNA: ↔ mfn2, ↔ opa1, ↓ dnm1l, ↔ fis1; Post-exercise protein content: ↑ MFN2 ↔ OPA1, DRP1; ↑ DRP1 Ser616 phosphorylation. Obese-T2DM subjects similar, except ↔ MFN2 post-ex protein content | ↑ DRP1 Ser616 phosphorylation | ↑ mfn2 mRNA 0 h post; ↓ dnm1l mRNA 3 h post; ↑ MFN2 post-exercise protein content |
| Author, Year [Reference] | Species/Model | Exercise Training Protocol | Summary Skeletal Muscle mRNA and/or Protein Responses | Evidence for Pro-Fission Responses | Evidence for Pro-Fusion Responses |
|---|---|---|---|---|---|
| Kirkwood et al. 1987 [94] | Rat | 10 weeks, 5 day/week, 10–120 min/day moderate-high intensity treadmill running | ↑ mitochondrial volume density % in vastus lateralis (VL) and soleus. ↓ mitochondrial surface:volume ratio in deep VL yet ↔ in superficial VL or soleus | n/d | ↓ mitochondrial surface: volume |
| Perry et al. 2010 [96] | Human | 2 weeks, 3–4 day/week, 1 h/day high intensity interval cycling exercise ∼90% VO2peak | ↑ MFN1 , FIS1 and DRP1 protein; ↔ MFN2 protein after 2 week training | ↑ FIS1 and DRP1 protein | ↑ MFN1 |
| Konopka et al. 2013 [99] | Human | 12 weeks moderate intensity cycling exercise training | ↑ MFN1/2 and FIS1 total protein | ↑ FIS1 protein | ↑ MFN1/2 protein |
| Feng et al. 2013 [100] | Rat | 4 weeks treadmill training | ↓ MFN2 protein in mitochondrial fraction, ↔ in total homogenate | ↓ MFN2 in mito fraction | n/d |
| Iqbal et al. 2013 [57] | Rat | 7 day, 3 h/day electrical stimulation | ↑ thickness of the subsarcolemma (SS) mitochondrial layer by 58%. Intermyofibrillar (IMF) mitochondria 75% larger and more reticular. Protein in SS mitochondria: ↑ OPA1 (36%) and MFN2 (53%); ↓ DRP1 (13%), ↔ FIS1. Whole homogenate similar changes (therefore, not due to IMF) | n/d | ↑ MFN2 and OPA1, ↓ DRP1 protein |
| Fealy et al. 2014 [108] | Human | 12 weeks, 5 h/week, ~80% Hrmax | ↔ DRP1 total protein, ↓ basal DRP1 Ser616 phosphorylation. ↑ opa1 and dnm1l mRNA (basal) | ↑ dnm1l mRNA | ↑ opa1 mRNA |
| Kitaoka et al. 2015 [97] | Rat | 4 weeks ‘resistance exercise’ electrical stimulation isometric contraction | ↑ OPA1 and MFN1/2 protein | n/d | ↑ OPA1 and MFN1/2 protein |
| Marton et al. 2015 [101] | Rat | 3 months treadmill running training | ↑ FIS1, ↓ MFN1 protein content | ↑ FIS1, ↓ MFN1 protein | |
| MacInnis et al. 2017 [98] | Human | 2 weeks, 3 day/week single-leg cycling moderate and high intensity in either leg | ↑ MFN2 protein in whole homogenate, but ↔ in type I or type II fibers analysed separately | n/a | ↑ MFN2 protein |
| Wyckelsma et al. 2017 [103] | Human (older) | 12 weeks, ~2 h/week cycling ~90% Hrmax | ↓ MFN2 protein in type II fibers, but ↔ in type I or whole homogenate. ↔ MID49 in whole homogenate | ↓ MFN2 protein in type II fibers | n/d |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trewin, A.J.; Berry, B.J.; Wojtovich, A.P. Exercise and Mitochondrial Dynamics: Keeping in Shape with ROS and AMPK. Antioxidants 2018, 7, 7. https://doi.org/10.3390/antiox7010007
Trewin AJ, Berry BJ, Wojtovich AP. Exercise and Mitochondrial Dynamics: Keeping in Shape with ROS and AMPK. Antioxidants. 2018; 7(1):7. https://doi.org/10.3390/antiox7010007
Chicago/Turabian StyleTrewin, Adam J., Brandon J. Berry, and Andrew P. Wojtovich. 2018. "Exercise and Mitochondrial Dynamics: Keeping in Shape with ROS and AMPK" Antioxidants 7, no. 1: 7. https://doi.org/10.3390/antiox7010007
APA StyleTrewin, A. J., Berry, B. J., & Wojtovich, A. P. (2018). Exercise and Mitochondrial Dynamics: Keeping in Shape with ROS and AMPK. Antioxidants, 7(1), 7. https://doi.org/10.3390/antiox7010007