Nonclinical Development of BCG Replacement Vaccine Candidates

Abstract
:1. Introduction
2. Experimental Section
2.1. Construction of VPM1002
2.2. VPM1002 Manufacturing
2.3. VPM1002 Protection Study in Mice
2.4. VPM1002 Guinea Pig Toxicology Study
2.5. VPM1002 Newborn Rabbit Safety Study
2.6. Construction of AERAS-422
2.7. AERAS-422 Manufacturing
2.8. Immunogenicity of AERAS-422 in Non-Human Primates (NHPs)
2.9. Mouse Protection Study of AERAS-422
2.10. AERAS-422 Safety Studies
3. Results and Discussion
3.1. Novel Recombinant BCG—VPM1002


3.2. Novel Recombinant BCG—AERAS-422

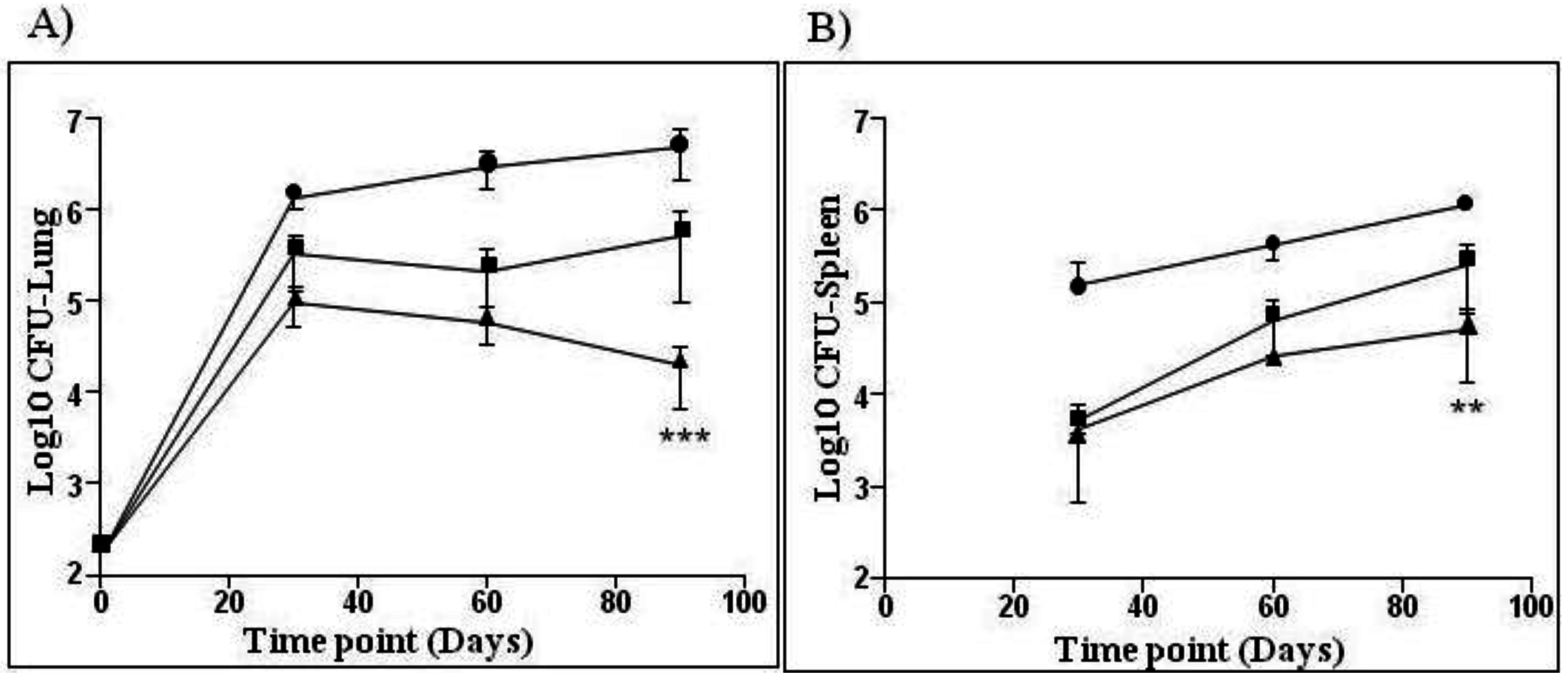
 —BCG SSI and
—BCG SSI and  —AERAS-422. Bars show the Mean ± SD.
—BCG SSI and —AERAS-422. Bars show the Mean ± SD.
—AERAS-422. Bars show the Mean ± SD.
—BCG SSI and —AERAS-422. Bars show the Mean ± SD.
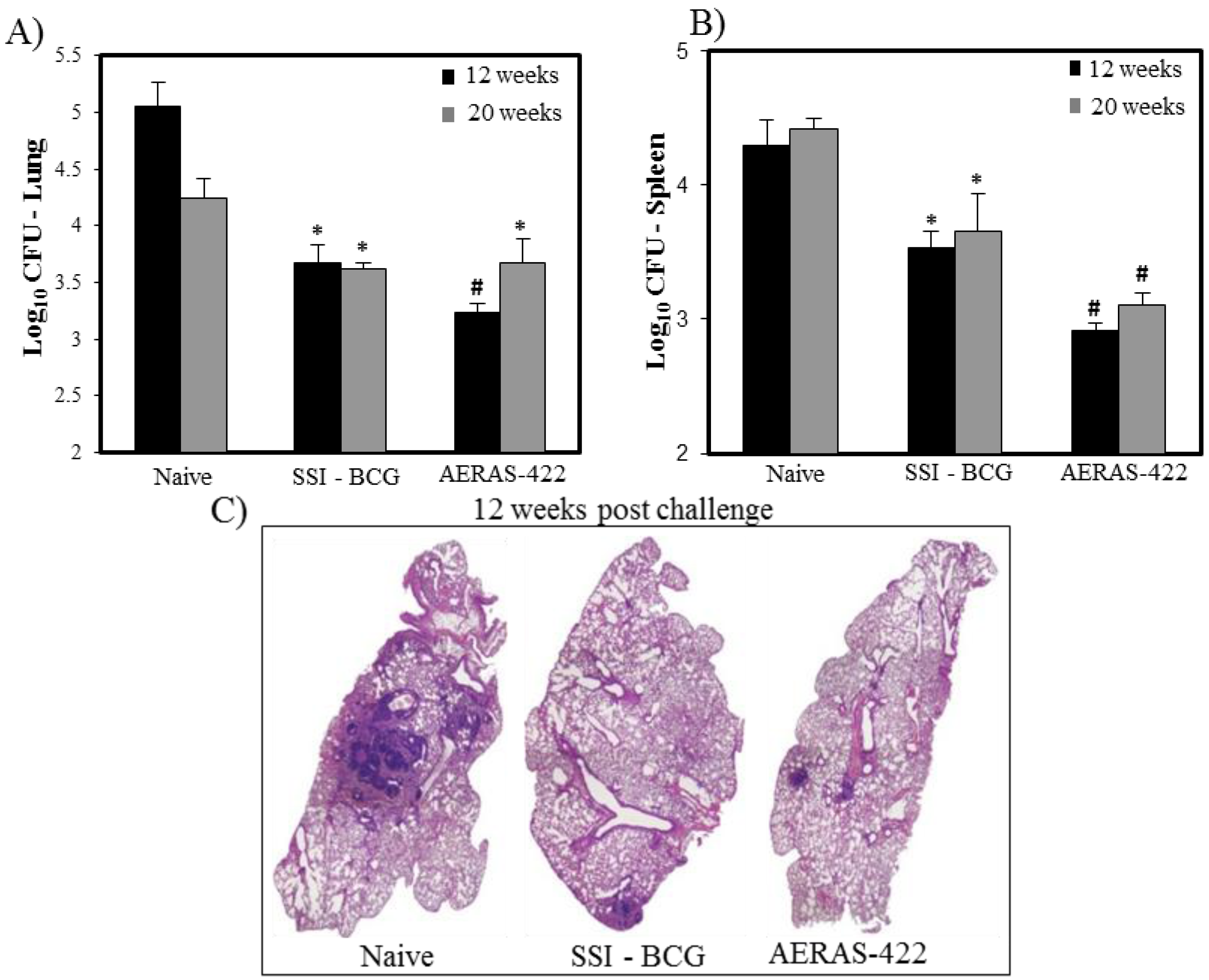
 ) weeks post-challenge. Statistical analysis was performed using the unpaired t test and the symbols represent the following: * = significantly better than naive at p < 0.05 and # = significantly better than both naive and BCG at p < 0.05. (C) Histopathology at 12 weeks post-challenge demonstrating fewer granuloma-like lesions and more open alveolar space in lungs of mice vaccinated with both BCG Danish-SSI 1331 and AERAS-422.
) weeks post-challenge. Statistical analysis was performed using the unpaired t test and the symbols represent the following: * = significantly better than naive at p < 0.05 and # = significantly better than both naive and BCG at p < 0.05. (C) Histopathology at 12 weeks post-challenge demonstrating fewer granuloma-like lesions and more open alveolar space in lungs of mice vaccinated with both BCG Danish-SSI 1331 and AERAS-422.
) weeks post-challenge. Statistical analysis was performed using the unpaired t test and the symbols represent the following: * = significantly better than naive at p < 0.05 and # = significantly better than both naive and BCG at p < 0.05. (C) Histopathology at 12 weeks post-challenge demonstrating fewer granuloma-like lesions and more open alveolar space in lungs of mice vaccinated with both BCG Danish-SSI 1331 and AERAS-422.
) weeks post-challenge. Statistical analysis was performed using the unpaired t test and the symbols represent the following: * = significantly better than naive at p < 0.05 and # = significantly better than both naive and BCG at p < 0.05. (C) Histopathology at 12 weeks post-challenge demonstrating fewer granuloma-like lesions and more open alveolar space in lungs of mice vaccinated with both BCG Danish-SSI 1331 and AERAS-422.

3.3. New Standards for Assessing the Quality, Safety and Efficacy of Recombinant BCG Vaccines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | Comments | New TB vaccines * | Traditional BCG vaccine ** |
|---|---|---|---|
| Identity | For master, working seed and final lots | Multiplex PCR, sequencing | Microbiologic methods |
| Safety | For bulk and/or final lots | Tests for attenuation, persistence, lack of reversion to virulence | General safety test in mice and guinea pigs |
| Safety | Meet GMO Standards | Evidence for lack of shedding of live organisms in animals | NA |
| Freedom from virulent Mtb | For master, working seed, bulk and/or final lots | Multiplex PCR for live vaccine identity and animal safety studies | Guinea pig assay |
| Antibiotic sensitivity | Evidence of susceptibility to first line drugs for TB; removal of antibiotic resistance selection markers | MGIT analyses (liquid culture) | Solid media culture analyses |
| Viability | For bulk and final lots | ATP assay live/dead ratio | Solid media culture |
| Potency | For bulk and final lots | Immune-biological assay (to be defined) | Viability |
| Stability | For bulk and final lots | Immune-biological assay (to be defined) | Viability/Moisture. Thermal & real time stability. Intradermal skin test in guinea pigs |
| Toxicology | For investigational lots | Immune-relevant toxicology test (to be defined) | Necropsy analyses in rabbits |
| Lot release tests | New methods may include fermentation process and synthetic media used to culture mycobacteria | Tests for residual contaminants from fermentation | Evidence for no TSA—containing culture media |
| Preclinical studies | To assess safety, immunogenicity & efficacy in animal models | Survival in animal models of immunosuppression. Protection in guinea pigs and/or mice. Safety studies in NHP, (for live attenuated Mtb vaccines). Identification of immunological markers | NA |
| Validation of facilities | Manufacturing facilities for live TB vaccines | PCR of individual products and cleaning, validation when campaigning products other than TB vaccines; Certain live attenuated vaccines may require dedicated facilities | Dedicated facilities, equipment and staff |
4. Concluding Remarks
4.1. Challenges for Recombinant BCG Vaccines
4.2. Potential Benefits of New BCG Vaccines
- new well-characterized products manufactured by state-of-the-art technologies
- shown to be safe and effective in contemporary clinical trials compared to currently used traditional BCG vaccines
- may serve as excellent prime vaccines for novel booster TB vaccines currently under development.
References
- Calmette, A.; Guérin, C.; Boquet, A.; Négre, L. La Vaccination Préventive Contre la Tuberculose par le “BCG”; Masson: Paris, France, 1927; p. 250. [Google Scholar]
- Bekker, A.; Du Preez, K.; Schaaf, H.S.; Cotton, M.F.; Hesseling, A.C. High tuberculosis exposure among neonates in a high tuberculosis and human immunodeficiency virus burden setting. Int. J. Tuberc. Lung Dis. 2012, 16, 1040–1046. [Google Scholar]
- Brennan, M.J.; Stone, M.R.; Evans, T. A rational vaccine pipeline for tuberculosis [State of the art series. New tools. Number 5 in the series]. Int. J. Tuberc. Lung Dis. 2012, 16, 1566–1573. [Google Scholar]
- Hess Miko, D.; Catic, A.; Lehmensiek, V.; Russell, D.G.; Kaufmann, S.H. Mycobacterium bovis Bacille Calmette-Guérin strains secreting listeriolysin of Listeria monocytogenes. Proc. Natl. Acad. Sci. USA 1998, 95, 5299–5304. [Google Scholar] [CrossRef]
- Grode, L.; Seiler, P.; Baumann, S.; Hess, J.; Brinkmann, V.; Nasser Eddine, A.; Mann, P.; Goosmann, C.; Bandermann, S.; Smith, D.; et al. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guérin mutants that secrete listeriolysin. J. Clin. Invest. 2005, 115, 2472–2479. [Google Scholar]
- Horwitz, M.A.; Harth, G.; Dillon, B.J.; Masleša-Galić, S. Recombinant bacillus calmette-guerin (BCG) vaccines expressing the Mycobacterium tuberculosis 30-kDa major secretory protein induce greater protective immunity against tuberculosis than conventional BCG vaccines in a highly susceptible animal model. Proc. Natl. Acad. Sci. USA 2000, 97, 13853–13858. [Google Scholar]
- Hoft, D.F.; Blazevic, A.; Abate, G.; Hanekom, W.A.; Kaplan, G.; Soler, J.H.; Weichold, F.; Geiter, L.; Sadoff, J.C.; Horwitz, M.A. A new recombinant bacille Calmette-Guérin vaccine safely induces significantly enhanced tuberculosis-specific immunity in human volunteers. J. Infect. Dis. 2008, 198, 1491–1501. [Google Scholar]
- Sun, R.; Skeiky, Y.A.; Izzo, A.; Dheenadhayalan, V.; Imam, Z.; Penn, E.; Stagliano, K.; Haddock, S.; Mueller, S.; Fulkerson, J.; et al. Novel recombinant BCG expressing perfringolysin O and the over-expression of key immunodominant antigens; pre-clinical characterization, safety and protection against challenge with Mycobacterium tuberculosis. Vaccine 2009, 27, 4412–4423. [Google Scholar]
- WHO Expert Committee on Biological Standardization. Thirty-sixth Report. World Health Organ. Tech. Rep. Ser. 1987, 745, 1–149.
- Laddy, D.J.; Yan, J.; Khan, A.S.; Andersen, H.; Cohn, A.; Greenhouse, J.; Lewis, M.; Manischewitz, J.; King, L.R.; Golding, H.; et al. Electroporation of synthetic DNA antigens offers protection in nonhuman primates challenged with highly pathogenic avian influenza virus. J. Virol. 2009, 83, 4624–4630. [Google Scholar] [CrossRef]
- Hokey, D.A.; Johnson, F.B.; Smith, J.; Weber, J.L.; Yan, J.; Hirao, L.; Boyer, J.D.; Lewis, M.G.; Makedonas, G.; Betts, M.R.; et al. Activation drives PD-1 expression during vaccine-specific proliferation and following lentiviral infection in macaques. Eur. J. Immunol. 2008, 38, 1435–1445. [Google Scholar] [CrossRef]
- Sambandamurthy, V.K.; Wang, X.; Chen, B.; Russell, R.G.; Derrick, S.; Collins, F.M.; Morris, S.L.; Jacobs, W.R., Jr. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat. Med. 2002, 8, 1171–1174. [Google Scholar] [CrossRef]
- Jeon, B.Y.; Derrick, S.C.; Kolibab, K.; Yang, A.; Lim, J.; Morris, S.L. Immunization of mice with BCG vaccine protects against aerosol infections with nine different Mycobacterium tuberculosis strains. Infect. Immun. 2008, 76, 5173–5180. [Google Scholar] [CrossRef]
- Directorate for the Quality of Medicines of the Council of Europe (EDQM), BCG vaccine, freeze-dried. 01/2008:0163. In European Pharmacopoeia; Cedex: Strasbourg, France, 2008; pp. 759–761.
- Ho, M.M.; Southern, J.; Kang, H.N.; Knezevic, I. WHO Informal Consultation on standardization and evaluation of BCG vaccines Geneva, Switzerland 22–23 September 2009. Vaccine 2010, 28, 6945–6950. [Google Scholar] [CrossRef]
- Glomski, I.J.; Gedde, M.M.; Tsang, A.W.; Swanson, J.A.; Portnoy, D.A. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity andprevent damage to infected host cells. J. Cell Biol. 2002, 156, 1029–1038. [Google Scholar] [CrossRef]
- Bavdek, A.; Kostanjšek, R.; Antonini, V.; Lakey, J.H.; Dalla Serra, M.; Gilbert, R.J.; Anderluh, G. pH dependence of listeriolysin O aggregation and pore-forming ability. FEBS J. 2012, 279, 126–141. [Google Scholar] [CrossRef]
- Decatur, A.L.; Portnoy, D.A. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science 2000, 290, 992–995. [Google Scholar] [CrossRef]
- Schnupf, P.; Portnoy, D.A.; Decatur, A.L. Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: Role of the PEST-like sequence. Cell. Microbiol. 2006, 8, 353–364. [Google Scholar] [CrossRef]
- Van der Wel, N.; Hava, D.; Houben, D.; Fluitsma, D.; van Zon, M.; Pierson, J.; Brenner, M.; Peters, P.J. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 2007, 129, 1287–1289. [Google Scholar] [CrossRef]
- Farinacci, M.; Weber, S.; Kaufmann, S.H. The recombinant tuberculosis vaccine rBCG ΔureC::hly(+) induces apoptotic vesicles for improved priming of CD4(+) and CD8(+) T cells. Vaccine 2012, 30, 7608–7614. [Google Scholar]
- Bardarov, S.; Bardarov, S., Jr.; Pavelka, M.S., Jr.; Sambandamurthy, V.; Larsen, M.; Tufariello, J.; Chan, J.; Hatfull, G.; Jacobs, W.R., Jr. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 2002, 148, 3007–3017. [Google Scholar]
- Capuano, S.V., 3rd; Croix, D.A.; Pawar, S.; Zinovik, A.; Myers, A.; Lin, P.L.; Bissel, S.; Fuhrman, C.; Klein, E.; Flynn, J.L. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect. Immun. 2003, 71, 5831–5844. [Google Scholar]
- Langermans, J.A.; Andersen, P.; van Soolingen, D.; Vervenne, R.A.; Frost, P.A.; van derLaan, T.; van Pinxteren, L.A.; van den Hombergh, J.; Kroon, S.; Peekel, I.; et al. Divergent effect of bacillus Calmette-Guérin (BCG) vaccination on Mycobacterium tuberculosis infection in highly related macaque species: Implications for primate models in tuberculosis vaccine research. Proc. Natl. Acad. Sci. USA 2001, 98, 11497–11502. [Google Scholar] [CrossRef]
- Roberts, A.D.; Cooper, A.M. Murine models of tuberculosis. Methods Microbiol. 2002, 32, 433–461. [Google Scholar] [CrossRef]
- Smith, D.W.; Balasubramanian, V.; Wiegeshaus, E. A guinea pig model of experimental airborne tuberculosis for evaluation of the response to chemotherapy: The effect on bacilli in the initial phase of treatment. Tubercle 1991, 72, 223–231. [Google Scholar] [CrossRef]
- Baldwin, S.L.; D’Souza, C.; Roberts, A.D.; Kelly, B.P.; Frank, A.A.; Lui, M.A.; Ulmer, J.B.; Huygen, K.; McMurray, D.M.; Orme, I.M. Evaluation of new vaccines in the mouse and guinea pig model of tuberculosis. Infect. Immun. 1998, 66, 2951–2959. [Google Scholar]
- Martin, C.; Williams, A.; Hernandez-Pando, R.; Cardona, P.J.; Gormley, E.; Bordat, Y.; Soto, C.Y.; Clark, S.O.; Hatch, G.J.; Aguilar, D.; et al. The live Mycobacterium tuberculosis phoP mutant strain is more attenuated than BCG and confers protective immunity against tuberculosis in mice and guinea pigs. Vaccine 2006, 24, 3408–3419. [Google Scholar]
- McShane, H.; Jacobs, W.R.; Fine, P.E.; Reed, S.G.; McMurray, D.N.; Behr, M.; Williams, A.; Orme, I.M. BCG: Myths, realities, and the need for alternative vaccine strategies. Tuberculosis (Edinb.) 2012, 92, 283–288. [Google Scholar] [CrossRef]
- Desel, C.; Dorhoi, A.; Bandermann, S.; Grode, L.; Eisele, B.; Kaufmann, S.H. Recombinant BCG ΔureC hly+ induces superior protection over parental BCG by stimulating a balanced combination of type 1 and type 17 cytokine responses. J. Infect. Dis. 2011, 204, 1573–1584. [Google Scholar] [CrossRef]
- Ottenhoff, T.H.; Ellner, J.J.; Kaufmann, S.H. Ten challenges for TB biomarkers. Tuberculosis 2012, 92, S17–S20. [Google Scholar]
- Brennan, M.J.; Clagett, B.; Fitzgerald, H.; Chen, V.; Williams, A.; Izzo, A.A.; Barker, L.F. Preclinical evidence for implementing a prime-boost vaccine strategy for tuberculosis. Vaccine 2012, 30, 2811–2823. [Google Scholar]
- Kaufmann, S.H.; Gengenbacher, M. Recombinant live vaccine candidates against tuberculosis. Curr. Opin. Biotechnol. 2012, 23, 900–997. [Google Scholar] [CrossRef]
- Trunz, B.B.; Fine, P.; Dye, C. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: A meta-analysis and assessment of cost-effectiveness. Lancet 2006, 367, 1173–1180. [Google Scholar] [CrossRef]
- Tuberculosis Research Centre (ICMR), Chennai. Fifteen year follow up of trial of BCG vaccines in south India for tuberculosis prevention. Indian J. Med. Res. 1999, 110, 56–69.
- Comstock, G.W. Identification of an effective vaccine against tuberculosis. Am. Rev. Respir. Dis. 1988, 138, 479–480. [Google Scholar]
- Brennan, M.J.; Thole, J. Tuberculosis vaccines: A strategic blueprint for the next decade. Tuberculosis (Edinb.) 2012, 92, S6–S13. [Google Scholar] [CrossRef]
- Kaufmann, S.H. Tuberculosis vaccine development: Strength lies in tenacity. Trends Immunol. 2012, 33, 373–379. [Google Scholar] [CrossRef]
- Ritz, N.; Hanekom, W.A.; Robins-Browne, R.; Britton, W.J.; Curtis, N. Influence of BCG vaccine strain on the immune response and protection against tuberculosis. FEMS Microbiol. Rev. 2008, 32, 821–841. [Google Scholar]
- Talbot, E.A.; Perkins, M.D.; Silva, S.F.; Frothingham, R. Disseminated bacille Calmette-Guerin disease after vaccination: Case report and review. Clin. Infect. Dis. 1997, 24, 1139–1146. [Google Scholar]
- Hengster, P.; Schnapka, J.; Fille, M.; Menardi, G. Occurrence of suppurative lymphadenitis after a change of BCG vaccine. Arch. Dis. Child. 1992, 67, 952–955. [Google Scholar]
- Scriba, T.J.; Tameris, M.; Smit, E.; van der Merwe, L.; Hughes, E.J.; Kadira, B.; Mauff, K.; Moyo, S.; Brittain, N.; Lawrie, A.; et al. A Phase IIa Trial of the New Tuberculosis Vaccine, MVA85A, in HIV- and/or Mycobacterium tuberculosis-infected. Adults Am. J. Respir. Crit. Care Med. 2012, 185, 769–778. [Google Scholar] [CrossRef]
- Magalhaes, I.; Sizemore, D.R.; Ahmed, R.K.; Mueller, S.; Wehlin, L.; Scanga, C.; Weichold, F.; Schirru, G.; Pau, M.G.; Goudsmit, J.; et al. rBCG induces strong antigen-specific T cell responses in rhesus macaques in a prime-boost setting with an adenovirus 35 tuberculosis vaccine vector. PLoS One 2008, 3, e3790. [Google Scholar] [CrossRef]
- Tchilian, E.Z.; Desel, C.; Forbes, E.K.; Bandermann, S.; Sander, C.R.; Hill, A.V.; McShane, H.; Kaufmann, S.H. Immunogenicity and protective efficacy of prime-boost regimens with recombinant (delta) ureC hly+ Mycobacterium bovis BCG and modified vaccinia virus ankara expressing M. tuberculosis antigen 85A against murine tuberculosis. Infect. Immun. 2009, 77, 622–631. [Google Scholar] [CrossRef]
- Clinical study of one of the rBCG vaccines discussed in this article, AERAS-422, has been discontinued due to a safety signal observed in the first phase 1 study of this vaccine. A manuscript describing this trial is currently under preparation.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Velmurugan, K.; Grode, L.; Chang, R.; Fitzpatrick, M.; Laddy, D.; Hokey, D.; Derrick, S.; Morris, S.; McCown, D.; Kidd, R.; et al. Nonclinical Development of BCG Replacement Vaccine Candidates. Vaccines 2013, 1, 120-138. https://doi.org/10.3390/vaccines1020120
Velmurugan K, Grode L, Chang R, Fitzpatrick M, Laddy D, Hokey D, Derrick S, Morris S, McCown D, Kidd R, et al. Nonclinical Development of BCG Replacement Vaccine Candidates. Vaccines. 2013; 1(2):120-138. https://doi.org/10.3390/vaccines1020120
Chicago/Turabian StyleVelmurugan, Kamalakannan, Leander Grode, Rosemary Chang, Megan Fitzpatrick, Dominick Laddy, David Hokey, Steven Derrick, Sheldon Morris, David McCown, Reginald Kidd, and et al. 2013. "Nonclinical Development of BCG Replacement Vaccine Candidates" Vaccines 1, no. 2: 120-138. https://doi.org/10.3390/vaccines1020120
APA StyleVelmurugan, K., Grode, L., Chang, R., Fitzpatrick, M., Laddy, D., Hokey, D., Derrick, S., Morris, S., McCown, D., Kidd, R., Gengenbacher, M., Eisele, B., Kaufmann, S. H. E., Fulkerson, J., & Brennan, M. J. (2013). Nonclinical Development of BCG Replacement Vaccine Candidates. Vaccines, 1(2), 120-138. https://doi.org/10.3390/vaccines1020120