4.1. Discussions
The proposed method is different from the current diagnostic methods in several ways. First, our new method (S4) theoretically leads to finding the smallest number of genes with clear signature patterns which are interpretable. Second, our method directly deals with heterogeneous populations and performs natural clustering and classifications of samples into their respective groups. Third, our proposed method can describe gene-gene interactions and gene-subtypes interactions.
Simultaneously observing the same set of five genes for two different datasets has not been reported in published literature. In our opinion, those published genes by many other studies are more like at the surface level (biologically directly related to the disease) based on the analysis methods used, and the new set of five genes in this work is at the deep level or the root level, where genes are not directly related to the diseases by the present biological knowledge. Furthermore, many reported key genes are based on their individualized expression value changes and significance, i.e., not based on gene-gene interactions. As a result, treatments are palliative, and the disease is difficult to cure. The findings in our new research are based on nonlinear and competing risk factors interactions, which is an advanced gene-gene interaction mechanism. Our proposition is that the biomedical discovery of new variants of COVID-19 is only the surface level of the virus (diseases). More profound, underlying “competing factors” of the virus need to be studied. Metaphorically, an expert in hydraulic engineering finds a dam with cracks and treats them on the surface. However, the reservoir has an interconnected water dynamic below the surface that will further impact other points of the dam. As a result, it will cause new cracks unless there is a fundamental treatment solution with the entire structure in mind. Similarly, scientists may observe the variants (rather passively) and develop vaccines in response to the variants. However, knowledge of the virus’s deeper advanced genomic architecture that will systematically cause other mutations is limited. Traditional methods in statistics, machine learning, and AI are limited to understanding COVID-19 from surface-level observations. However, our innovative method has achieved significant results in identifying and understanding COVID-19 genomic signatures.
The newly identified genes and their combinations may be used as new biomarkers. In our opinion, traditional methods (e.g., PCR, serology) are directly associated with the disease symptoms, i.e., they do not provide pathological characteristics; they are a onefold indicator. On the other hand, the new classifier is a multifold indicator that can further divide the disease into subgroups (variants may be another word). In addition, the new classifier shows gene-gene interactions and advanced (or root) structures.
This work has verified that when all component classifiers simultaneously classify a group of patients as COVID-19-positive, these patients are ICU patients (
Section 3.4), which definitely points out the advanced genomic structure of COVID-19 disease.
In the literature and the current practice, tremendous efforts have been made to study COVID-19 genomic sequences, variants and their impacts, and vaccine effectiveness. However, the progress on the pathological causes of COVID-19 and the functional effects of genes is still limited. In terms of computational medicine, our new work is the first to accurately define the functional effects of five critical genes and lead to the mathematical and biological equivalence between five genes and COVID-19 status. Furthermore, this paper introduces an advanced machine learning algorithm that identifies five essential genes, which further determine three genomic signature patterns and seven subtypes of COVID-19 with high accuracy. The final classifiers are expressed by explicit mathematical equations which are interpretable and can guide medical practice. In addition, new graphical diagnostic tools are introduced. Besides the striking advance in studying genomic signature patterns of COVID-19, our work also sheds new light on computational medicine, genetic studies, informatics, algorithm and machine learning, and statistics.
We realized readers would ask about the model overfitting and robustness. Please note that our model is fitted to three different datasets and has reached the highest accuracy. Each dataset has its heterogeneous patterns (subgroups). Datasets are measured at different scales. It is hard for the existing models to simultaneously fit such datasets and get satisfactory accuracy, not to mention the interpretability of the fitted models. Using three such datasets naturally serves as cross-validation and robust checking. It turns out our new approach is robust.
In many scenarios, a 100% accuracy may be thought of as “too good to be true”. However, “too good to be true” may also be dangerous to use to guide science discovery and innovation. In many applied sciences, the truths can be simple but not straightforward. Complicating or aggravating the problem can mask the nature of the problem. Blindly insisting on “too good to be true” may miss ample opportunities of finding the truth. In contrast, we know it is hard to see the forest through the trees.
One may argue that the dataset we used in this analysis is not large enough as it has only 126 samples with 19,472 predictors in the first two datasets and 34 samples with 64,083 genes in the third dataset. It is, of course, preferable to have a large dataset. Nevertheless, we argue that the conclusions and inferences are trustworthy with a convincing accuracy on three different datasets that show nonlinear and heterogeneous relationships.
On the other hand, if an approach cannot gain a satisfactory performance with a small dataset, applying it to a large dataset can be a wrong strategy as it may lead to wrong or suboptimal conclusions.
A natural question is whether or not the high accuracy is by chance. Note that each of our component competing classifiers has reached 100% specificity, which may be true with a probability smaller than by chance. In addition, when all three signature patterns are satisfied, all classified patients are lab-confirmed ICU patients, which indicates it cannot be by chance.
We have used analogies to interpret our modeling strategy. We note that our proposed method is still in its primary stage of development, just started in 2021. Researchers are still not very familiar with the method. Once the proposed method becomes a standard method, the introduction of the method and analogies will no longer be needed.
4.2. Conclusions
As discussed in
Section 1 and
Section 3, the three signature patterns and seven subtypes maintain the most important biological informatics of COVID-19. This set of genes is the only set that leads to accurately classifying hospitalized COVID-19 patients, including ICU patients, into their respective groups. Unless a different new discovery of other advanced structures of genes other than these five genes can be obtained, and such a new discovery (if exists) can fully explain the three signature patterns and seven subtypes discussed in this paper, these five critical genes and their derived three signature patterns and seven subtypes remain the most informative findings.
When a model is fitted to the whole dataset and leads to (nearly) perfect accuracy, it will uniformly work for partitioned data as long as the partition is balanced to all heterogeneous subgroups. This is the case in our analysis. On the other hand, we have not seen any published papers that used the “standard” procedure to lead to accurate prediction.
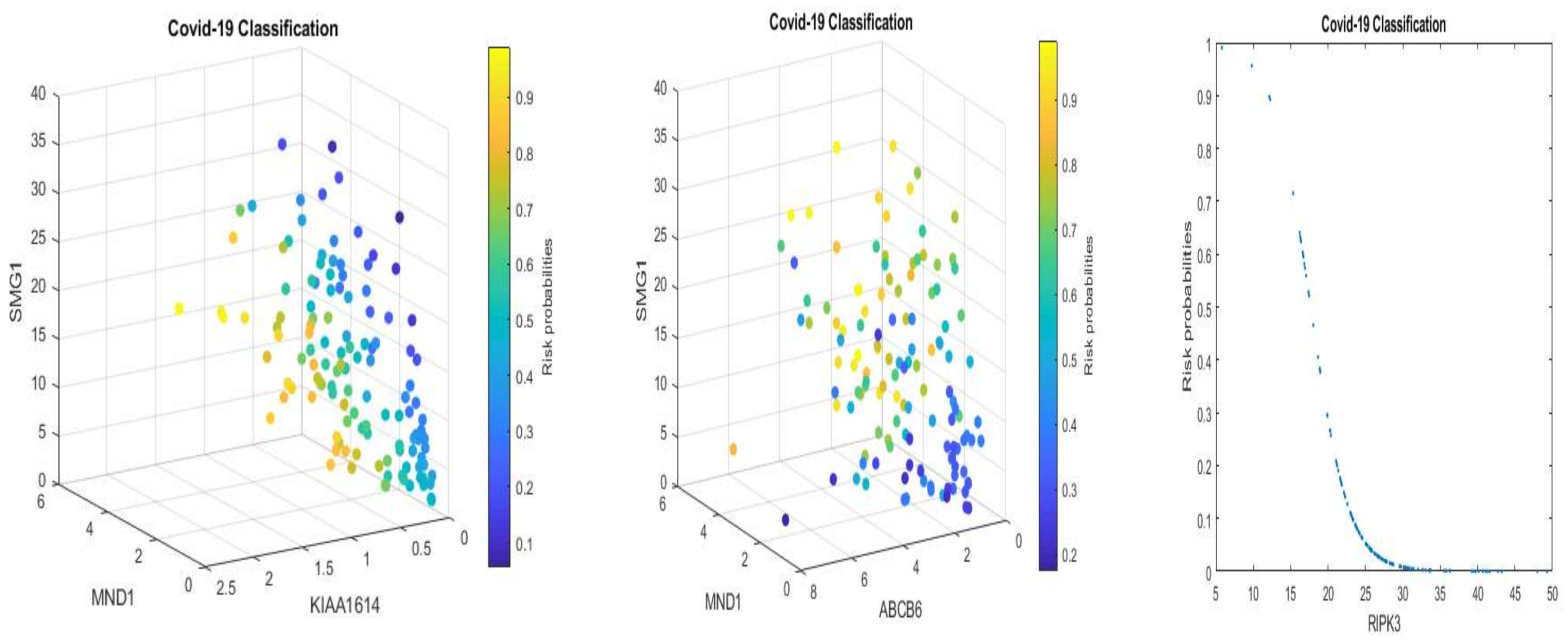
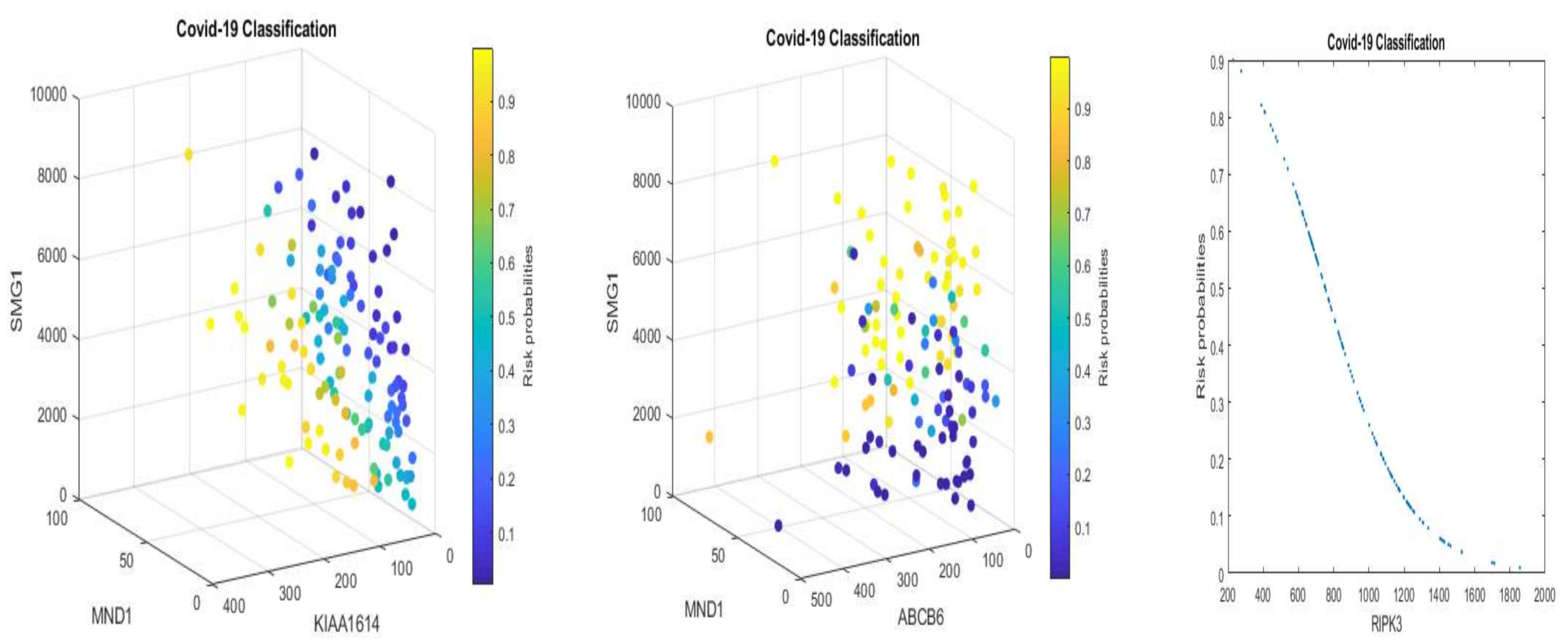
We note that multiplying a constant to Equations (5) and (6) will not change the classification results and the shapes in
Figure 1 and
Figure 2 with a convincing accuracy being achieved. However, the color strengths will be changed. Such a phenomenon justifies the use of signatures to describe the advanced gene-gene interaction structures. Using this idea, we can express (6) into the following equivalent forms:
Equation (11), after applying scale changes, shares similar signatures as in Equation (5). Considering the nonlinear relationships between TPM data and EC data, this observation again proves our proposed competing factor classifiers are robust.
Our findings can be used to develop precision test kits for testing COVID-19 and to evaluate the function and performance of already implemented vaccines, i.e., used as new antibody indexes. Interpretable and implementable formulas are given in the paper. After a COVID-19 case is confirmed, personalized treatments can be implemented. For example, increasing or decreasing levels of critical genes based on the identified COVID-19 subtype can be crucial to the patients’ recovery. Using the relationship determined in the findings, antiviral drugs can be developed. Mathematically, the new objective function of Equation (4) is a mixture of combinatorial optimization and continuous optimization. It is a new type of classification benchmark. It is expected that this new classification formula will motivate research in statistics, computational mathematics, computer science, and many applied sciences. The findings can motivate many new types of research in COVID-19 studies and other studies, e.g., cancer studies. Many finished studies can re-start new investigations using the new methodology.
4.3. Future Perspectives
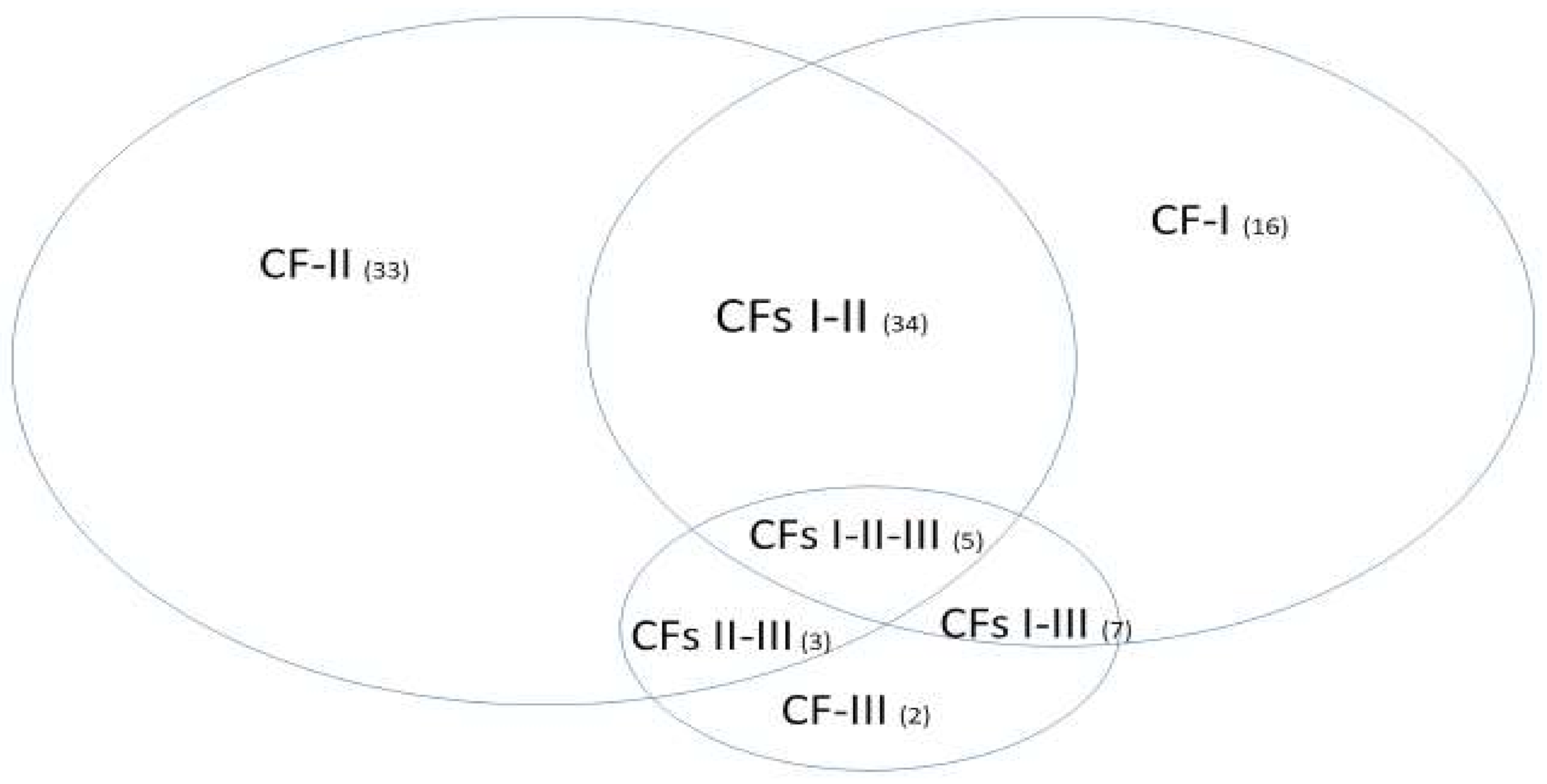
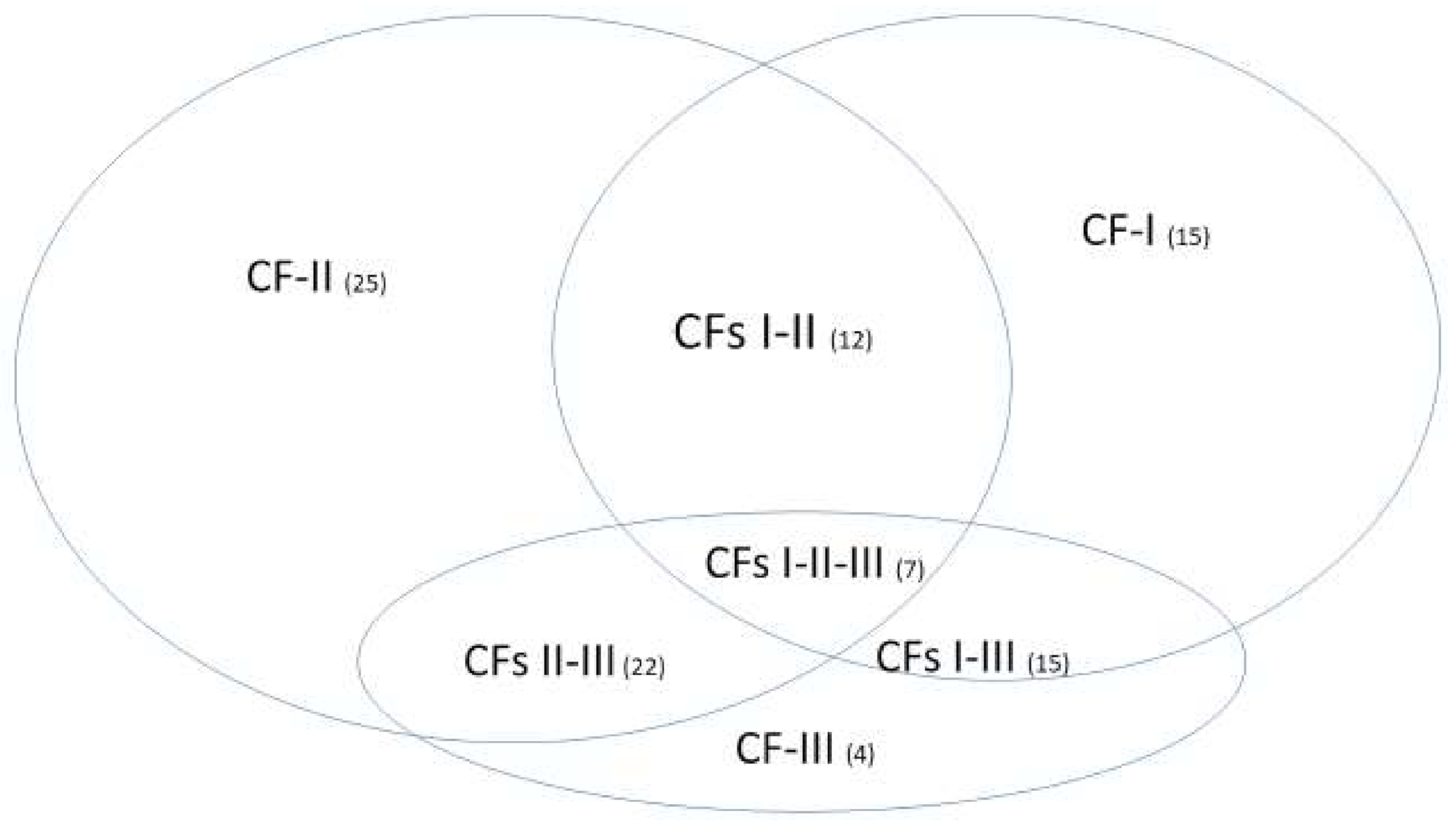
With the mathematical equivalence between Equations (6) and (11), different numbers of patients in different subtypes and in the control will not change the classification results. With the specificities of each individual classifier (CF-i) reaching 100%, the signature patterns (not presented in
Section 3.3) of patients in the control will be the same, i.e., just one type. This phenomenon shows that the identified five critical genes are COVID-19 specific. One can apply our method to study critical genes of disease types (also other respiratory diseases) in the control, i.e., assign the related samples in the treatment group and specify some other types of diseases or normal cases as the control.
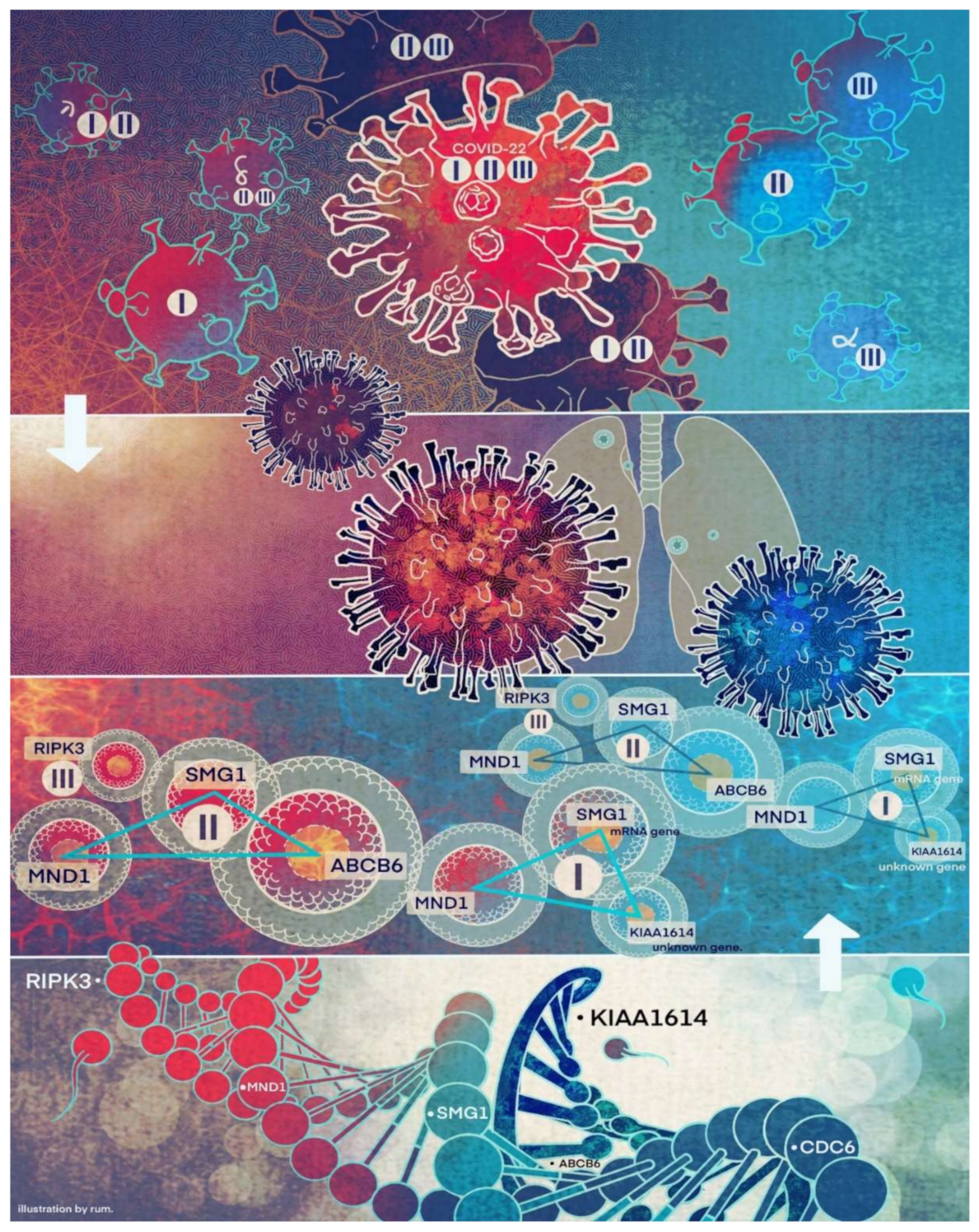
We want to hypothesize that the discovered COVID-19 variants (alpha, beta, delta, lambda, mu, omicron, etc.) may be connected to different signature strengths in our discovered signature patterns. COVID-22 in
Figure 4 means that SARS-CoV-2 variants in 2022 can be combinations of several subtypes (variants), i.e., they are no longer the same types as in 2019. Mathematically, hyperplanes in geometry formed by Equations (5) and (11) contain a subspace that can be further partitioned into subspaces. Therefore, we hypothesize that these variants may fall into separable subspaces. After obtaining new data with variants information, this hypothesis can be tested, or additional genes may be involved. For example, in the breast cancer study mentioned earlier, triple-negative breast cancer was accurately separated from other types of breast cancer using three genes identified by the S4 classifier. Furthermore, the discovered genomic signature patterns and COVID-19 subtypes are intrinsic no matter what variants have been identified or will be identified. Given our proven mathematical and biological equivalences, if these innate signature patterns and subtypes cannot be treated and fully studied now, they will cause trouble in the future again. In addition, with available data related to various variants, our study approach may be able to reveal the causes of higher transmission or mortality of specific variants.
Using classifiers CF-I, -II, and -III as new biomarkers, we can study other potential genes that are highly correlated with these biomarkers. For example, based on the first and second datasets, the most highly correlated five genes to each new biomarker lead to a total of fifteen genes: DBN1, LY6G6C, TMEM54, MTMR1, SNORC, ANP32E, ATAD2, SMC2, ZWILCH, SMC4, C6orf47, STRADA, LRSAM1, UNC93B1, and SASH3, which were listed in the Introduction.
Finally, in our analyses, we also found the gene CDC6 (cell division cycle 6) can be informative. Its combination with ZNF282 (zinc finger protein 282) can lead to 97.62% accuracy (98% sensitivity, 96.15% specificity), and its combination with both ZNF282 and CEP72 (centrosomal protein 72) can lead to 98.41% accuracy (99% sensitivity, 96.15% specificity). We found the high expression level of CDC6 increases risk while the high expression levels of ZNF282 and CEP72 decrease risk. Although they did not lead to the best accuracy as those five genes identified in
Section 3 did, such a performance is already better than other published gene combinations. From the literature, the gene CDC6 is a protein essential for the initiation of DNA replication, while ZNF282 is known to bind U5RE (U5 repressive element) of HLTV-1 (human T cell leukemia virus type 1) with a repressive effect, but little is known of its expression and function otherwise. The gene CEP72 coded protein is localized to the centrosome, a non-membraneous organelle that functions as animal cells’ major microtubule-organizing center. Zhang (2022) hypothesizes that CDC6 is a protein essential for the initiation of RNA replication of COVID-19 [
19].









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}